
Introduction
Hepatocellular carcinoma (HCC) is one of the most malignant tumors and the third leading cause of cancer-related deaths around the world [1, 2]. Hepatocarcinogenesis is a complex and multi-factorial process and at this time, some molecular pathways have been identified that contribute to the development, progression, angiogenesis, and metastasis of HCC. The tumor microenvironment is of interest in cancer research and is now recognized as a critical contributor to cancer progression. Infiltrating stromal and immune cells in the tumor microenvironment contribute to cancer biology as well [3].
Hepatic stellate cells (HSCs) are important stromal cells that are activated during liver injury, inflammation, infection, trauma, or by other pathogens [4, 5]. HSCs have immunomodulatory activity and prolong islet allografts survival [6, 7]. We reported that activated HSCs can induce immune-suppressing cells, specifically regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs) in tissues of tumor-bearing mice [8-10]. MDSCs are heterogeneous immature myeloid cells comprised of myeloid progenitors and precursors of macrophages, granulocytes, and dendritic cells (DCs) [11, 12]. Murine MDSCs co-express CD11b and Gr-1 [11] and their heterogeneity is also based on expression of Gr-1: granulocytic-MDSCs (G-MDSCs, CD11b+Ly6G+Ly6Cint/low), and monocytic-MDSCs (Mo-MDSCs, CD11b+ Ly6G-Ly6Chigh) [13, 14]. Höchst’s group reported that activated HSCs can recruit and transform peripheral blood monocytes into de novo MDSCs [15] but how HSCs induce MDSC expansion and activation is unclear.
Recently, Qian and colleagues reported that HSCs induced MDSCs via soluble factors secreted by HSCs and expanded these data by revealing that complement component 3 (C3) is critical for inducing MDSC expansion and protecting islet allografts [16]. However, HSCs deficient in C3 did not completely lose their capacity to induce MDSCs, implying the involvement of other factors that may synergize with C3 to promote MDSC induction. Vascular endothelial growth factor (VEGF), granulocyte macrophage colony-stimulating factor (GM-CSF), and granulocyte colony-stimulating factor (G-CSF) promote MDSC activity in cancer [11, 17] but Qian et al has proved that these factors do not involved in induction of MDSC [7] , so additional factors are required for induction of MDSCs by HSCs.
Prostaglandin E2 (PGE2) is a pro-inflammatory mediator produced by cancer, stromal, and infiltrating myeloid cells and acts on G-protein-coupled receptors (GPCRs) including EP1-EP4 [18]. Cyclooxygenase (COX)-2 is chiefly believed to be key to influencing the rate of PGE2 production during immune response [19]. A positive feedback loop between PGE2 and COX-2 determines the redirection of the development of CD1a+ DCs to CD1a-CD14+CD80-CD83- monocytic MDSCs [20]. Furthermore, Kalinski’s group reported that addition of PGE2 to GM-CSF/IL-4-supplemented monocytic precursor cultures generated numerous MDSCs [21]. Silencing COX-2 in 4T1 tumor cells reduced CD11b+Gr-1+ MDSC accumulation in mouse spleens [11]. Moreover, PGE2 can be produced by HSCs [22-24], which suggested the hypothesis that HSCs induce expansion of MDSCs via secreted PGE2. For this reason, bone marrow (BM) cells were cultured with HSC-conditioned medium (HSC-CM) plus SC-236, a COX2 inhibitor. Then, the effect of SC-236-treated HSCs on MDSC expansion and tumor growth was assessed.
Results
Incubation of BM cells with conditioned media from activated HSCs induced MDSCs
First, BALB/c BM cells were cultured with HSC-CM and cell surface marker expression by various myeloid cell types after HSC-CM treatment was measured. Figure 1A shows BM co-cultured with HSC-CM decreased CD11c, MHC II, CD86 and CD80 expression, suggesting less BM cell differentiation into macrophages and immature DCs. Meanwhile, Gr-1 increased significantly and a slight increase in B7-H1.
Next, the expression of DCs, MDSCs, and MDSC subsets were detected after BM cells were cultured with HSC-CM. The gating strategy of these cells is shown in Figure 1B. As shown in the Figure 1C, the number of CD11b+CD11C+ DCs decreased by approximately half (decreased from 55.9 ± 2.1% to 33.7 ± 1.9%, P < 0.01. Figure 1C, upper panel). CD11b/Gr-1 co-staining confirmed the presence of MDSCs, which doubled (25 ± 2.9% in control vs. 54.9 ± 2.4% in HSC-CM, Figure 1C, middle panel). MDSCs can be divided phenotypically into granulocytic (MDSCs, CD11b+/Ly6G+/Ly6Cint/low) and monocytic (Mo-MDSCs, CD11b+/Ly6G-/Ly6Chigh) subgroups, which have been shown to be immunosuppressive via different pathways. Figure 1C (bottom panel), depicts G-MDSCs and Mo-MDSCs induction in HSC-CM culture and that upregulation of Mo-MDSC was most prominent and these findings agree with those of Qian’s group [16]. To verify the specific HSC-CM effect on MDSC expansion, we used CM from MEF cells as controls and noted that MEF-CM had no effect on MDSC expansion, and the influence of HSC-CM on BM cells was concentration-dependent (Figure 1D).
To study immunosuppression of MDSCs, Gr-1+ cells were isolated using MACS, and more than 90% of the Gr-1+ cells were CD11b+Gr-1+. MDSCs were cultured with T cells (1:1). As shown in Figure 1E, MDSCs inhibited T-cell proliferation. It has been reported that elevated expression of Arg-1, iNOS, and IL-4Rα accounts for suppression of T-cell function by MDSCs [11, 25]. For this reason, the mRNA expression of each protein was detected, and a 2-fold increase in iNOS mRNA, a 4.5-fold increase in Arg-1 mRNA, and a 4-fold increase in IL-4Rα expression were detected (Figure 1F). In this way, HSC-CM inhibited DC development and promoted MDSC accumulation in vitro.
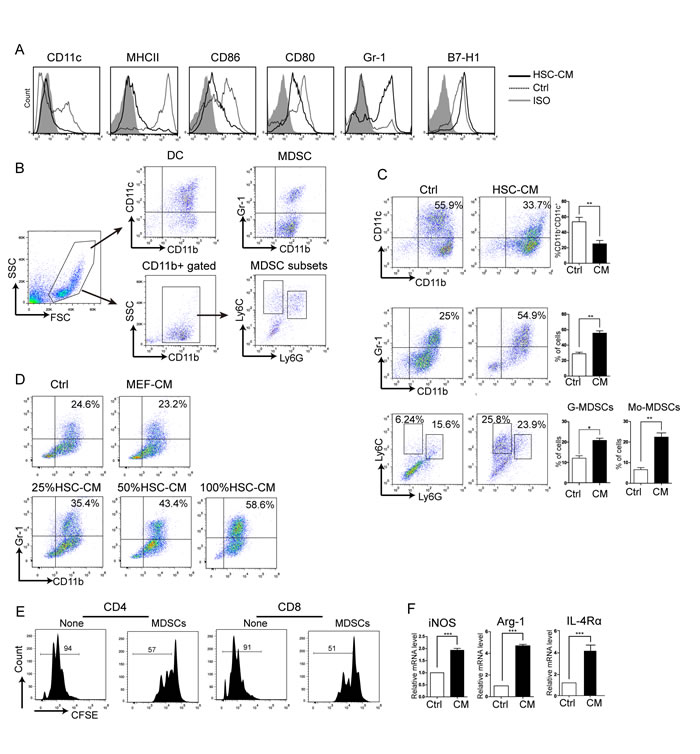
Figure 1: Effects of HSC-CM on BM-derived DC differentiation in vitro. A. Cell surface marker expression in myeloid cells after HSC-CM treatment measured by flow cytometry. Filled areas are isotype controls; dotted lines are RPMI 1640 medium controls; full lines are HSC-CM. B. Gating strategy of DCs, total MDSCs and subsets. C. The effect of HSC-CM on DCs, MDSCs and MDSC subgroups. Number is percent of the cell population represented by immature DCs (top panel), MDSCs (middle panel) or Mo-MDSCs, G-MDSCs (bottom panel). Percent G-MDSCs was calculated as follows: corrected G-MDSC percent = 100% × CD11b+ percent × Ly6G+Ly6Clow percent. Corrected Mo-MDSC percent = 100% × CD11b+ percent × Ly6G-Ly6Chigh percent. D. HSC-CM induced MDSCs in a concentration-dependent manner. MEF-CM was a control CM. E. Gr-1+ cells inhibited T-cell proliferation. F. iNOS, Arg-1, and IL-4Rα expression in Gr-1+ cells according to RT-PCR. Data represent 3-5 independent experiments and are expressed as means ± SD; *P < 0.05, **P < 0.01, ***P < 0.001.
Induction of MDSCs from BM cells by HSC-conditioned media is mediated by PGE2 production
Factors such as cytokines (IL-6) [26, 27], stem-cell factor (SCF) [28], growth factors VEGF, TGF-β, GM-CSF, G-CSF, and macrophage colony-stimulating factor (M-CSF), COX-2, and PGE2 [21, 29, 30] have been reported to induce MDSC expansion. IL-6 is abundantly produced by activated HSCs according to previous work from our laboratory and that of others [9], so we speculated that HSCs may induce MDSC accumulation via IL-6. To test this hypothesis, IL-6 neutralizing antibody was added to HSC-CM cultures of BM cells and MDSCs did not decrease compared to the HSC-CM group (Figure 2A), so IL-6 does not apparently mediate the effect of HSC-CM on MDSC induction.
PGE2 influences MDSC expansion and is secreted by activated HSCs [23, 24], so to assess whether HSC-CM induced MDSC expansion was correlated with PGE2 production, HSCs were pretreated with SC-236, a COX-2 inhibitor. Figure 2B depicts no significant cytotoxicity of SC-236 against HSCs (5-20 μM), but PGE2 production was inhibited by SC-236 (Figure 2C).
BM cells were cultured with the HSC-CM or conditioned media from 20 μM SC-236-pretreated HSCs (SC-236-CM) for 3 days. Flow cytometry indicated a significant increase of MDSCs in the HSC-CM group compared to controls (Figure 2E). SC-236-CM significantly reduced MDSCs induced by HSC-CM (Figure 2D), suggesting the importance of PGE2 in inducing MDSCs by HSC-CM.
We next investigated the effect of SC-236 on MDSC subgroups. HSC-CM upregulated expression of both CD11b+Ly6G+Ly6Cint/low G-MDSCs and CD11b+Ly6G-Ly6Chigh Mo-MDSCs, especially Mo-MDSCs (Figure 2E). SC-236-CM abolished G-MDSCs promotion by HSCs, and they decreased in the HSC-CM and SC-236-CM groups. However, the effect of HSCs on Mo-MDSCs induction was not altered significantly by SC-236 treatment, suggesting that induction of Mo-MDSCs was independent of PGE2 production in HSCs.
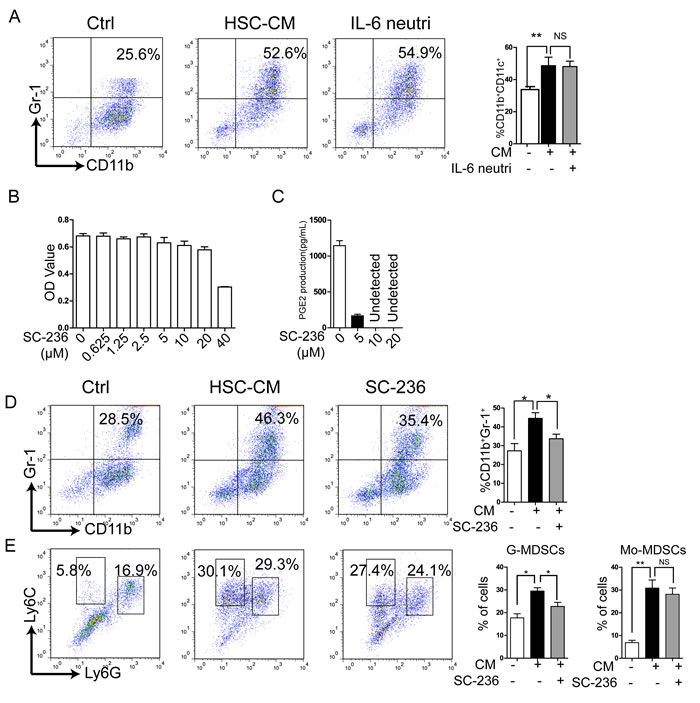
Figure 2: Inhibition of PGE2 production by HSCs reversed the effect of HSC-CM on MDSC induction in vitro. A. Effect of IL-6 neutralization on MDSC expression. Number represents percent of MDSCs in entire cell population (right panel). B. Cell counts according to CCK8 assay. C. PGE2 secretion measured by ELISA. D.-E. MDSC, G-MDSC, and Mo-MDSC populations assessed with flow cytometry. Number represents percent of entire cell population (right panels), calculated as described in Figure 1. Number is percent of the cell population represented by Mo-MDSCs and G-MDSCs. Data are means of 3-5 independent experiments ± SD; *P < 0.05, **P < 0.01, ***P < 0.001.
EP4 antagonists block MDSC accumulation
MDSCs from 4T1 tumor-bearing mice are known to express four E-prostanoid receptors (EP) for PGE2 [30]. To validate that HSCs induces MDSC accumulation via the PGE2/EP signaling way, BM cells were cultured with IL-4 and GM-CSF with/without CM, or with/without EP antagonists. Figure 3A shows that HSC-CM upregulated expression of MDSCs. When BM cells were cultured with HSC-CM plus EP antagonists, only the EP4 antagonist inhibited induction of MDSCs by HSC-CM. Data show that accumulation of MDSCs induced by HSCs was mediated through the PGE2/EP4 signaling.
Because the stimulatory effect of HSCs on Mo-MDSCs only slightly decreased with SC-236 treatment. Next, experiments were conducted to determine whether the G-MDSC and Mo-MDSC subsets induced by HSCs were regulated by the PGE2/EP4 signaling. As shown in Figure 3B, AH23848 (EP4 antagonist) inhibited the G-MDSC but not Mo-MDSC expansion by HSC-CM. Taken together, these in vitro data indicated that HSC-derived PGE2 induced the expansion and differentiation of MDSCs, especially the G-MDSC subset, through the EP4 receptor.
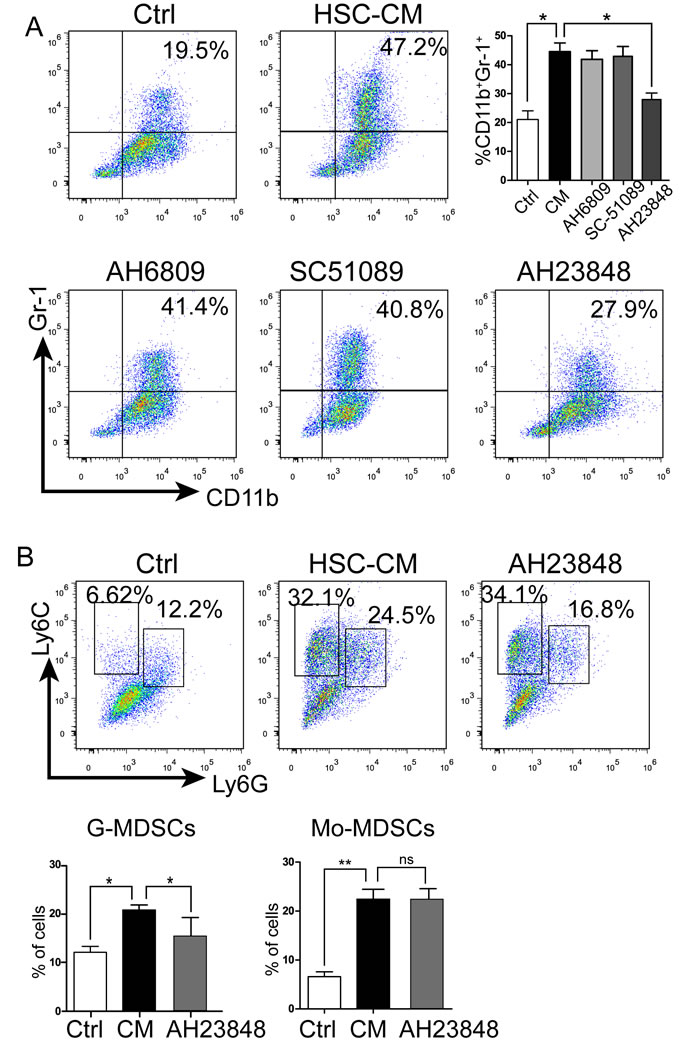
Figure 3: HSCs promoted MDSC accumulation via the EP4 receptor. A. Effect of EP antagonists on MDSC expression. Number is percent of the cell population represented by MDSCs (EP1 antagonist SC-51089; EP1 and EP2 antagonist AH6809; EP4 antagonist AH23848). B. Expression of G-MDSCs and Mo-MDSCs in BM cells cultured with/without EP4 receptor antagonist AH23848; number is percent of the cell population represented by Mo-MDSCs and G-MDSCs as calculated in Figure 1. Data are means of 3-5 independent experiments ± SD; *P < 0.05, **P < 0.01, ***P < 0.001.
Inhibition of COX-2 activity in HSCs impairs MDSC induction in vivo
To understand how HSCs-produced PGE2 affects accumulation of MDSC cells in vivo, a murine orthotopic HCC model was established. The liver capsule was injected with H22 cells alone (1 × 106 cells/mouse, control group), or H22 plus HSCs (2 × 105 cells/mouse, HSC co-transplanted group), or H22 and SC-236-pretreated HSCs (2 × 105 cells/mouse, SC-236 pretreated group) (n = 8/group). Figure 4A shows that tumors from the HSC co-transplanted group were significantly larger than those of controls, a finding that agreed with previous observations [10]. In addition, tumors were smaller in the SC-236 pretreated group compared to the HSC co-transplanted group, suggesting that HSC pretreated with SC-236 lost tumor-promoting activity.
Because the primary tumor burden by itself does not dictate the level of MDSCs [30], we then further investigated the accumulation of MDSCs in the spleen and inside the tumor among the 3 groups via flow cytometric analysis (Figure 4B). Significant differences in the percent of MDSCs in the spleen between control group and HSC co-transplanted group with or without SC-236 pretreatment were observed (8.4% vs. 16.7% for control vs. HSC co-transplanted group, P < 0.01; 16.7% vs. 12.3% for HSC co-transplanted group vs. SC-236 pretreated group, P < 0.05). Further analysis of MDSCs population in the tumors revealed a similar pattern. As shown in Figure 4B (bottom panel), there was a significant increase in the percent of MDSCs in the tumor in the HSC co-transplanted group relative to the H22 alone group (8.9% vs. 6.1%; P < 0.05). HSCs pretreated with SC-236 resulted in a 25% decrease in the MDSC population as compared to that observed in the HSC group (P < 0.05).
Next, we sought to evaluate the subsets of MDSCs in tumors co-transplanted with HSCs or SC-236-pretreated HSCs. As Figure 4C shows, co-transplanted HSCs significantly increased both the percent of G-MDSCs and Mo-MDSCs in spleen and tumor, whereas HSCs pretreated with SC-236 presented a reduction in G-MDSCs and Mo-MDSCs expression both in the spleen and tumors, respectively. To evaluate MDSC immunosuppressive activity, MDSCs were isolated from the control group, HSC co-transplanted group, and SC-236 pretreated group. As shown in Figure 4D, MDSC immunosuppressive activity showed no obvious difference among three groups. The data from the subcutaneous model (Supplementary Figure 2) were consistent with the orthotopic HCC model. To confirm whether the increase in MDSCs in the HSC group was due to the higher tumor burden of these mice, the population of MDSCs was assessed in the spleens of mice at 1 week, 2 weeks, and 3 weeks after tumor injection. The data show that the MDSCs in the HSCs were higher than those in the control group and SC-236 group since at the first week when tumors did not different in size among groups (Supplementary Figure 3). These findings suggest that COX-2 antagonist SC-236-pretreated HSCs could reduce the tumor size and the population of MDSCs in tumor-bearing mice.
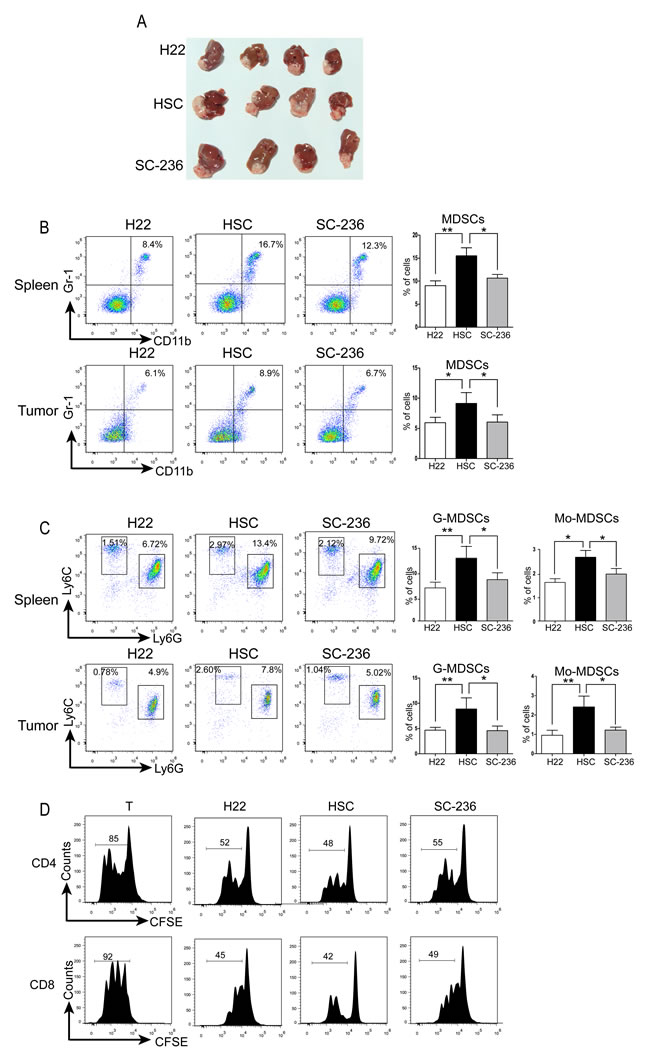
Figure 4: HSCs promoted HCC growth by inducing MDSCs via PGE2 signaling. A. Representative tumors. B. MDSC accumulation in splenocytes and tumors were counted; number is percent of the cell population represented by MDSCs (right panels). C. G-MDSCs and Mo-MDSCs were measured with flow cytometry. Number is percent of the cell population represented by Mo-MDSC and G-MDSC, calculated as described in Figure 1. D. CD4+ and CD8+ T cells measured by CFSE dilution assay and flow cytometry. Data are means ± SD; *P < 0.05, **P < 0.01, ***P < 0.001.
Discussion
Researchers across the world have made great strides in understanding the role of the tumor environment in tumor progression, but cross-talk between stromal and immune cells in cancer progression and metastasis is entirely unexplored. Our previous work indicates that activated HSCs can create an immunosuppressive microenvironment in an orthotopic liver tumor mouse model by inducing expansion of Tregs and MDSCs [10], but how this occurs is unclear. Qian’s group reported that MDSCs could be induced by HSC derived C3 to protect islet allografts, but HSCs deficient in C3 did not completely lose their capacity to induce MDSCs [16]. Here, we investigated other factors involved in induction of MDSCs and measured their effects on specific subsets of MDSCs and noted that HSCs promoted MDSC accumulation, particularly Mo-MDSCs. Furthermore, data showed that HSCs promoted G-MDSC expansion via COX2-PGE2-EP4 signaling, and inhibiting HSC-derived PEG2 with a COX-2 inhibitor reduced tumor grow and MDSCs accumulation, which indicated that the PGE2/EP4 pathway was involved in induction of MDSCs by HSCs.
MDSCs are critical mediators of tumor-induced immune dysfunction and cancer progression [32]. Previous work indicates that MDSC expansion is regulated by factors such as COX2, PGE2, SCF, M-CSF, IL-6, GM-CSF, and VEGF [11, 28, 30, 33], most of which are produced by activated HSCs. IL-6, VEGF, GM-CSF, and G-CSF have been implicated in MDSC development [6, 34-37]. Reports suggest that induction of MDSCs by HSCs is unlikely to be mediated by VEGF, GM-CSF, or G-CSF [7, 16]. IL-6 is sufficient to drive MDSC expansion, compared to other factors produced by various tumors and it causes a rapid generation of MDSCs from mouse and human BM [38-40]. In BM cell culture, however, IL-6 neutralizing antibody had little or no effect on generation of CD11b+Gr-1+ MDSCs (Figure 2A). PGE2 is known to be a regulator of MDSC accumulation, MDSC-mediated T cell inhibition, and expression of Arg1 and iNOS [41-43]. Evidence is mounting that activation of COX-2 signaling results in an alteration of HSCs [44, 45]. In addition, COX-2 and COX-2-dependent PGE2 expression was persistently upregulated in fully activated HSCs and participated in the chemokines regulation, VEGF production, and HSC proliferation and migration [46-48]. PGE2 also induces MDSC expansion [21, 49-54], so activated HSCs that produce abundant PGE2 were evaluated next.
The expression and expansion of MDSCs were reduced after treatment of HSCs with COX-2 inhibitor SC-236, which indicated that MDSC induction by HSCs was mediated by the COX-2/PGE2 pathway. IFN-γ has been reported to be critical for HSC-mediated MDSC generation [7] and HSCs deficiency in IFN-γ R1 largely lose the ability to induce MDSCs. PGE2 signaling is thought to mobilize the cAMP-PKA pathway via EP2/EP4 receptors and induce CREB- and its co-activator CRTC2-mediated transcription of IL-12Rβ2 and IFN-γR1 [55]. Therefore, inhibiting PGE2 signaling may affect transcription of IFN-γ R1, thereby decreasing MDSCs.
We assessed the effect of COX2 inhibitor SC-236 on MDSC subset generation and noted that only G-MDSC expansion decreased with SC-236 CM but not Mo-MDSCs. PGE2 exerts its functions by interacting with PGE2 receptors, of which there are four subtypes (EP1-4) [56]. PGE2 is reported to induce accumulation of Gr-1+CD11b+ MDSCs from BM cells via EP1, EP2, and/or EP4 receptors [30]. In our study, only an EP4 receptor antagonist inhibited MDSC expansion and G-MDSC expansion appeared to be the only cell type affected, although both G-MDSCs and Mo-MDSCs expressed the EP4 receptor (Supplementary Figure 1). Thus, expansion of G-MDSCs induced by HSCs was through the COX2-PGE2-EP4 signaling pathway and different factors might be involved in Mo-MDSC expansion.
Qian’s group reported that BM cells cultured with HSCs deficient in C3 expressed less Ly6C compared to wild type HSCs, suggesting that C3 may be crucial for inducing Mo-MDSC (CD11b+ Ly6G-Ly6Chigh cell) expression [16]. Also shRNA-mediated reductions in C3 expression in HSCs impaired Mo-MDSC expression (data not shown). Possibly, C3 has greater priority over PGE2 for inhibiting Mo-MDSC expression. SC-236 had no obvious effect on immunosuppressive activity of MDSCs isolated from tumors, perhaps because of the abundant inflammatory factors secreted by tumor cells that stimulate immunosuppressive activity of MDSCs. TGF-β1-induced miR-494 expression in MDSCs is essential for accumulation and function of tumor-expanded MDSCs [57].
In our animal model, reduction of HSC-derived PGE2 with the COX2 inhibitor reduced Gr-1+ CD11b+ cells accumulation and decreased growth of implanted tumors. Interestingly, HSCs pretreated with COX2 inhibitor could better inhibit Mo-MDSC accumulation in vivo, although the COX2 inhibitor and the EP receptor antagonist had less effect on Mo-MDSC accumulation in co-culture. This may be explained by differences in the tumor environment. Fewer Mo-MDSCs in vivo may result from differentiation of Mo-MDSCs into other types of immune cells. Ly6C expression in Mo-MDSC sorted from the spleen of EL-4 TB mice was reported to be down-regulated expression by ~30% after 3 days of culture with GM-CSF [58].
Overproduction of COX2 and PGE2 in the tumor microenvironment may also explain the local accumulation of MDSCs observed in different cancers [32, 59]. HSC co-transplantation induced more PGE2 expression than control group, while co-transplantation with COX-2 inhibitor-pretreated HSCs produced a PGE2-reduced microenvironment than in the HSC co-transplantation group, which reduced Mo-MDSC accumulation (Supplemental Figure 4), However, HSC-secreted PGE2 influenced MDSC accumulation but not their immunosuppressive activity. Different chemokine/chemokine receptor signal axes play different roles in mediating the mobilization of MDSC subsets. The CXCR4-CXCL12 axis is necessary for mediating the attraction of monocytic MDSCs into the tumor environment and PGE2 is important to regulation of CXCL12 production in cancer-associated fibroblasts and the cancer environment, and to enhancement of CXCR4 expression in Mo-MDSCs [29]. CCL2 has been reported to foster PMN-MDSC accumulation in tumors [60]. The expression of Cxcl12 and Ccl2 was also measured in the tumor microenvironment, and data show the expression of Cxcl12 and Ccl2 to be increased in the HSC co-transplantation group. Relative to the HSC co-transplantation group, Cxcl12 and Ccl2 mRNA expression was down-regulated in the SC-236 group.
Our data indicate a relationship between activated HSCs and MDSC subsets in the HCC tumor microenvironment. HSCs promote G-MDSC accumulation via COX2-PGE2-EP4 signaling and COX2 inhibition blocks HSC-derived PGE2 and HSC-mediated induction of MDSCs, decreasing HSC tumor-promoting ability (Figure 5). We offer a novel mechanistic explanation for the link between HSCs and HCC progression, targeting PGE2 as an innovative strategy for future cancer immunotherapy development.
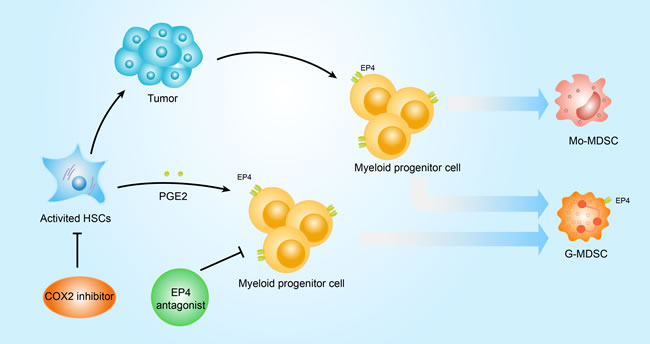
Figure 5: Scheme of MDSC induction signaling pathway by HSCs. In vitro, activated HSC-derived PGE2 inhibits myeloid progenitor cell maturity and induction of G-MDSC accumulation via the EP4 receptor, which can be reversed with a selective COX2 inhibitor or EP4 antagonist. Inhibition of activated HSC-derived PGE2 reduces HCC growth and HSC-induced MDSC accumulation.
Materials and Methods
Animals and cell lines
All experimental protocols were reviewed and approved by our Institutional Review Board and animal experimental protocols were performed in compliance with the Guidelines for the Institutional Animal Care and Use Committee of Xiamen University.
Adult male BALB/c mice (H-2d, haplotype, 8-12 weeks-of-age) mice were purchased from the National Rodent Laboratory Animal Resources, Shanghai, China. The mouse H22 hepatoma cell line was purchased from Shanghai Cell Bank, Chinese Academy of Sciences, and maintained in RPMI 1640 medium (HyClone, Logan, UT), supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 µg/mL streptomycin, as previously described [9].
HSCs isolation and culture
HSCs were isolated from BALB/c mouse livers as described previously [9]. Livers were perfused with phosphate buffered saline (PBS) and type IV collagenase (Sigma, city, state), and soaked in collagenase for further digestion. Isolated HSCs were cultured in RPMI 1640 medium (HyClone, Logan, UT, U.S.) supplemented with 10% heat-inactivated FBS (Gibco, BRL Co. Ltd.), 100 U/mL penicillin, and 100 mg/mL streptomycin in 5% CO2/95% air at 37ºC. Cells at passage 4-10 were used for experiments. Cell activation was measured via a-SMA staining [9, 31].
Culture of DCs
DCs were prepared according to published methods [31]. After lysing RBCs with lysis buffer (Beyotime, Jiangsu, China), 1 × 106 BM cells/well from tibias and femurs of BALB/c mice were cultured in RPMI 1640 medium containing 10% FBS and mouse recombinant GM-CSF (10 ng/mL, R&D, Minneapolis, MN, U.S.; IL-4 10 ng/mL, PeproTech, Rocky Hill, NJ, U.S.) for 6 days. To measure the effect of HSCs on BM, (HSC-CM) or SC-236, CM was used to replace RPMI 1640 medium at DC culture initiation. Floating cells were harvested, washed, and resuspended in RPMI 1640 medium.
Isolation of MDSCs
Tumors or spleen were cut into small pieces and dissociated with a MACS dissociator (Miltenyi Biotec, Bergisch Gladbach, Germany) according to the manufacturer’s instructions. After dissociation, dead cells were excluded by Dead Cell Removal Kit (Miltenyi Biotec, Bergisch Gladbach, Germany). Then, the remaining cells were used to isolate MDSCs by bition-Gr-1 antibody (BD, San Diego, CA, U.S.) and Streptavidin MicroBeads (Miltenyi Biotec).
Mixed leukocyte reaction
A mixed leukocyte reaction (MLR) culture was performed in 24-well plates (Corning, Corning, NY). Nylon wool-eluted splenic T cells (1 × 106/well) from BALB/c mice were used as responders. T-cell proliferation was elicited with anti-CD3 mAb (1 μg/mL) and CD28 (1 μg/mL). Cultures were maintained in RPMI 1640 complete medium for 3 days under 5% CO2. T-cell proliferation was measured with a CFSE dilution assay and flow cytometry.
Tumor inoculation
Mice were injected intra-hepatically with 0.1 mL cell suspension containing either 1 × 106 H22 cells or a mixture of 1 × 106 H22 cells and 2 × 105 activated HSCs (N = 8/group). To understand the role of HSC-derived PGE2, HSCs were pretreated with the COX2 antagonist SC-236 (20 µM) 3 days before inoculation. HSCs treated with DMSO were controls. At the end of the experiment, mice were euthanized, and spleens and tumors were collected and stored for subsequent analysis.
CCK8 and PGE2 assay
Cells were treated with different concentration of SC-236 for 24 h, and 10 µl CCK8 reagent was added. After 4 h, absorbance was read (λ 450 nm). A PGE2 ELISA kit was obtained from R&D Systems and assays were conducted according to kit instructions.
Quantitative real-time PCR assay
Total RNA was extracted with TRIzol Reagent (Invitrogen, city, state). Complementary DNA (cDNA) was synthesized with SuperScript II reverse transcriptase (Invitrogen). Quantitative PCR (qPCR) primers were as follows: Arginase-1: Forward: 5’-CACGGCAGTGGCTTTAACCT-3’, Reverse: 5′-TGGCGCATTCACAGTCACTT-3′; iNOS: Forward: 5′-GGAATCTTGGAGCGAGTTGT-3′, Reverse: 5′- CCTCTTGTCTTTGACCCAGTAG-3′. IL-4Rα: Forward: 5′-CCTACACTACAGGCTGATGTTC-3′, Reverse: 5′-TGGACCGGCCTATTCATTTC-3′. mRNA was measured with a 7500 Fast PCR system (Applied Biosystems, Foster City, CA, U.S.) in duplicate and were normalized to GAPDH mRNA.
Flow cytometry
Splenocytes were harvested and disaggregated in 10 mL of RPMI 1640 complete medium. RBCs were lysed with RBC Lysis Buffer (Beyotime, Nanjing, China). Tumors were cut into small pieces and dissociated with a MACS dissociator (Miltenyi Biotec, Bergisch Gladbach, Germany) according to the manufacturer’s instructions. After dissociation, dead cells were exclude with DAPI (0.2 μg/mL), and leukocytes were gated on FSC and SSC for analysis. Monoclonal antibodies against CD11b, CD11C, Ly6C, Ly6G, and Gr-1 were purchased from BD PharMingen (San Diego, CA, U.S.), and antibodies against CD40, CD86, CD80, MHCI, MHCII, and B7-H1 were purchased from eBioscience (San Diego, CA, U.S.). Fluorescently-labeled cells were analyzed with flow cytometry (Beckman Gallios). Analysis and graphical output were performed using with FlowJo software and appropriate isotype control antibodies were used.
Statistical analysis
Data were analyzed using SPSS software (version 13.0) and are expressed as means ± SD. Statistical analyses were performed with a Student’s t test (P < 0.05).
Acknowledgments
We thank Ms. Huiqin Zhuo and Ms. Qin Yao for their help with flow cytometry. This work was supported by grants from the National Natural Science Foundation of China (Grant No. 81572335), the National Key Sci-Tech Special Project of China (Grant No. 2016ZX10002016-001-004), the Natural Science Foundation of Fujian Province, China (Grant No. 2014D012).
Grant support
This work was supported by grants from the National Natural Science Foundation of China (Grant No. 81572335), the National Key Sci-Tech Special Project of China (Grant No. 2016ZX10002016-001-004), the Natural Science Foundation of Fujian Province, China (Grant No. 2014D012).
Conflicts of interest
The authors disclose no potential conflicts of interest.
References
1. Thun MJ, DeLancey JO, Center MM, Jemal A and Ward EM. The global burden of cancer: priorities for prevention. Carcinogenesis. 2010; 31:100-110.
2. Ferlay J, Shin HR, Bray F, Forman D, Mathers C and Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010; 127:2893-2917.
3. Yoshihara K, Shahmoradgoli M, Martinez E, Vegesna R, Kim H, Torres-Garcia W, Trevino V, Shen H, Laird PW, Levine DA, Carter SL, Getz G, Stemke-Hale K, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun. 2013; 4:2612.
4. Puche JE, Saiman Y and Friedman SL. Hepatic stellate cells and liver fibrosis. Compr Physiol. 2013; 3:1473-1492.
5. Lee YS and Jeong WI. Retinoic acids and hepatic stellate cells in liver disease. J Gastroenterol Hepatol. 2012; 27 Suppl 2:75-79.
6. Yu MC, Chen CH, Liang X, Wang L, Gandhi CR, Fung JJ, Lu L and Qian S. Inhibition of T-cell responses by hepatic stellate cells via B7-H1-mediated T-cell apoptosis in mice. Hepatology. 2004; 40:1312-1321.
7. Chou H-S, Hsieh C-C, Yang H-R, Wang L, Arakawa Y, Brown K, Wu Q, Lin F, Peters M, Fung JJ, Lu L and Qian S. Hepatic stellate cells regulate immune response by way of induction of myeloid suppressor cells in mice. Hepatology. 2011; 53:1007-1019.
8. Sun Y, Xi D, Ding W, Wang F, Zhou H and Ning Q. Soluble FGL2, a novel effector molecule of activated hepatic stellate cells, regulates T-cell function in cirrhotic patients with hepatocellular carcinoma. Hepatol Int. 2014; 8:567-575.
9. Zhao W, Zhang L, Yin Z, Su W, Ren G, Zhou C, You J, Fan J and Wang X. Activated hepatic stellate cells promote hepatocellular carcinoma development in immunocompetent mice. Int J Cancer. 2011; 129:2651-2661.
10. Zhao W, Zhang L, Xu Y, Zhang Z, Ren G, Tang K, Kuang P, Zhao B, Yin Z and Wang X. Hepatic stellate cells promote tumor progression by enhancement of immunosuppressive cells in an orthotopic liver tumor mouse model. Lab Invest. 2014; 94:182-191.
11. Gabrilovich DI and Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009; 9:162-174.
12. Kohanbash G and Okada H. Myeloid-derived suppressor cells (MDSCs) in gliomas and glioma-development. Immunol Invest. 2012; 41:658-679.
13. Youn JI, Nagaraj S, Collazo M and Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol. 2008; 181:5791-5802.
14. Ioannou M, Alissafi T, Lazaridis I, Deraos G, Matsoukas J, Gravanis A, Mastorodemos V, Plaitakis A, Sharpe A, Boumpas D and Verginis P. Crucial role of granulocytic myeloid-derived suppressor cells in the regulation of central nervous system autoimmune disease. J Immunol. 2012; 188:1136-1146.
15. Hochst B, Schildberg FA, Sauerborn P, Gabel YA, Gevensleben H, Goltz D, Heukamp LC, Turler A, Ballmaier M, Gieseke F, Muller I, Kalff J, Kurts C, et al. Activated human hepatic stellate cells induce myeloid derived suppressor cells from peripheral blood monocytes in a CD44-dependent fashion. J Hepatol. 2013; 59:528-535.
16. Hsieh CC, Chou HS, Yang HR, Lin F, Bhatt S, Qin J, Wang L, Fung JJ, Qian S and Lu L. The role of complement component 3 (C3) in differentiation of myeloid-derived suppressor cells. Blood. 2013; 121:1760-1768.
17. Chen CH, Kuo LM, Chang Y, Wu W, Goldbach C, Ross MA, Stolz DB, Chen L, Fung JJ, Lu L and Qian S. In vivo immune modulatory activity of hepatic stellate cells in mice. Hepatology. 2006; 44:1171-1181.
18. Sugimoto Y and Narumiya S. Prostaglandin E receptors. J Biol Chem. 2007; 282:11613-11617.
19. Kalinski P. Regulation of immune responses by prostaglandin E2. J Immunol. 2012; 188:21-28.
20. Obermajer N, Muthuswamy R, Lesnock J, Edwards RP and Kalinski P. Positive feedback between PGE2 and COX2 redirects the differentiation of human dendritic cells toward stable myeloid-derived suppressor cells. Blood. 2011; 118:5498-5505.
21. Obermajer N and Kalinski P. Generation of myeloid-derived suppressor cells using prostaglandin E2. Transplant Res. 2012; 1:15.
22. Cardoso CC, Paviani ER, Cruz LA, Guma FC, Borojevic R and Guaragna RM. Effect of pentoxifylline on arachidonic acid metabolism, neutral lipid synthesis and accumulation during induction of the lipocyte phenotype by retinol in murine hepatic stellate cell. Mol Cell Biochem. 2003; 254:37-46.
23. Olaso E, Salado C, Egilegor E, Gutierrez V, Santisteban A, Sancho-Bru P, Friedman SL and Vidal-Vanaclocha F. Proangiogenic role of tumor-activated hepatic stellate cells in experimental melanoma metastasis. Hepatology. 2003; 37:674-685.
24. Hui AY, Dannenberg AJ, Sung JJ, Subbaramaiah K, Du B, Olinga P and Friedman SL. Prostaglandin E2 inhibits transforming growth factor beta 1-mediated induction of collagen alpha 1(I) in hepatic stellate cells. J Hepatol. 2004; 41:251-258.
25. Condamine T and Gabrilovich DI. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. 2011; 32:19-25.
26. Sumida K, Wakita D, Narita Y, Masuko K, Terada S, Watanabe K, Satoh T, Kitamura H and Nishimura T. Anti-IL-6 receptor mAb eliminates myeloid-derived suppressor cells and inhibits tumor growth by enhancing T-cell responses. Eur J Immunol. 2012; 42:2060-2072.
27. Wu CT, Hsieh CC, Lin CC, Chen WC, Hong JH and Chen MF. Significance of IL-6 in the transition of hormone-resistant prostate cancer and the induction of myeloid-derived suppressor cells. J Mol Med (Berl). 2012; 90:1343-1355.
28. Pan PY, Wang GX, Yin B, Ozao J, Ku T, Divino CM and Chen SH. Reversion of immune tolerance in advanced malignancy: modulation of myeloid-derived suppressor cell development by blockade of stem-cell factor function. Blood. 2008; 111:219-228.
29. Obermajer N, Muthuswamy R, Odunsi K, Edwards RP and Kalinski P. PGE(2)-induced CXCL12 production and CXCR4 expression controls the accumulation of human MDSCs in ovarian cancer environment. Cancer Res. 2011; 71:7463-7470.
30. Sinha P, Clements VK, Fulton AM and Ostrand-Rosenberg S. Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res. 2007; 67:4507-4513.
31. Zhao W, Su W, Kuang P, Zhang L, Liu J, Yin Z, Wang X. The role of hepatic stellate cells in the regulation of T-cell function and the promotion of hepatocellular carcinoma. Int J Oncol. 2012; 41:457-64.
32. Rabinovich GA, Gabrilovich D and Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol. 2007; 25:267-296.
33. Bunt SK, Yang L, Sinha P, Clements VK, Leips J and Ostrand-Rosenberg S. Reduced inflammation in the tumor microenvironment delays the accumulation of myeloid-derived suppressor cells and limits tumor progression. Cancer Res. 2007; 67:10019-10026.
34. Maher JJ. Interactions between hepatic stellate cells and the immune system. Semin Liver Dis. 2001; 21:417-426.
35. Serafini P, Carbley R, Noonan KA, Tan G, Bronte V and Borrello I. High-dose granulocyte-macrophage colony-stimulating factor-producing vaccines impair the immune response through the recruitment of myeloid suppressor cells. Cancer Res. 2004; 64:6337-6343.
36. Gabrilovich D, Ishida T, Oyama T, Ran S, Kravtsov V, Nadaf S and Carbone DP. Vascular endothelial growth factor inhibits the development of dendritic cells and dramatically affects the differentiation of multiple hematopoietic lineages in vivo. Blood. 1998; 92:4150-4166.
37. Nefedova Y, Nagaraj S, Rosenbauer A, Muro-Cacho C, Sebti SM and Gabrilovich DI. Regulation of dendritic cell differentiation and antitumor immune response in cancer by pharmacologic-selective inhibition of the janus-activated kinase 2/signal transducers and activators of transcription 3 pathway. Cancer Res. 2005; 65:9525-9535.
38. Sander LE, Sackett SD, Dierssen U, Beraza N, Linke RP, Muller M, Blander JM, Tacke F and Trautwein C. Hepatic acute-phase proteins control innate immune responses during infection by promoting myeloid-derived suppressor cell function. J Exp Med. 2010; 207:1453-1464.
39. Marigo I, Bosio E, Solito S, Mesa C, Fernandez A, Dolcetti L, Ugel S, Sonda N, Bicciato S, Falisi E, Calabrese F, Basso G, Zanovello P, Cozzi E, Mandruzzato S and Bronte V. Tumor-induced tolerance and immune suppression depend on the C/EBPbeta transcription factor. Immunity. 2010; 32:790-802.
40. Lechner MG, Liebertz DJ and Epstein AL. Characterization of cytokine-induced myeloid-derived suppressor cells from normal human peripheral blood mononuclear cells. J Immunol. 2010; 185:2273-2284.
41. Rodriguez PC, Hernandez CP, Quiceno D, Dubinett SM, Zabaleta J, Ochoa JB, Gilbert J and Ochoa AC. Arginase I in myeloid suppressor cells is induced by COX-2 in lung carcinoma. J Exp Med. 2005; 202:931-939.
42. Xiang X, Poliakov A, Liu C, Liu Y, Deng ZB, Wang J, Cheng Z, Shah SV, Wang GJ, Zhang L, Grizzle WE, Mobley J and Zhang HG. Induction of myeloid-derived suppressor cells by tumor exosomes. Int J Cancer. 2009; 124:2621-2633.
43. Donkor MK, Lahue E, Hoke TA, Shafer LR, Coskun U, Solheim JC, Gulen D, Bishay J and Talmadge JE. Mammary tumor heterogeneity in the expansion of myeloid-derived suppressor cells. Int Immunopharmacol. 2009; 9:937-948.
44. Cheng J, Imanishi H, Liu W, Iwasaki A, Ueki N, Nakamura H and Hada T. Inhibition of the expression of alpha-smooth muscle actin in human hepatic stellate cell line, LI90, by a selective cyclooxygenase 2 inhibitor, NS-398. Biochem Biophys Res Commun. 2002; 297:1128-1134.
45. Koga H, Sakisaka S, Ohishi M, Kawaguchi T, Taniguchi E, Sasatomi K, Harada M, Kusaba T, Tanaka M, Kimura R, Nakashima Y, Nakashima O, Kojiro M, et al. Expression of cyclooxygenase-2 in human hepatocellular carcinoma: relevance to tumor dedifferentiation. Hepatology. 1999; 29:688-696.
46. Mallat A, Gallois C, Tao J, Habib A, Maclouf J, Mavier P, Preaux AM and Lotersztajn S. Platelet-derived growth factor-BB and thrombin generate positive and negative signals for human hepatic stellate cell proliferation. Role of a prostaglandin/cyclic AMP pathway and cross-talk with endothelin receptors. J Biol Chem. 1998; 273:27300-27305.
47. Failli P, De FR, Caligiuri A, Gentilini A, Romanelli RG, Marra F, Batignani G, Guerra CT, Laffi G, Gentilini P and Pinzani M. Nitrovasodilators inhibit platelet-derived growth factor-induced proliferation and migration of activated human hepatic stellate cells. Gastroenterology. 2000; 119:479-492.
48. Efsen E, Bonacchi A, Pastacaldi S, Valente AJ, Wenzel UO, Tosti-Guerra C, Pinzani M, Laffi G, Abboud HE, Gentilini P and Marra F. Agonist-specific regulation of monocyte chemoattractant protein-1 expression by cyclooxygenase metabolites in hepatic stellate cells. Hepatology. 2001; 33:713-721.
49. Mao Y, Sarhan D, Steven A, Seliger B, Kiessling R and Lundqvist A. Inhibition of tumor-derived prostaglandin-e2 blocks the induction of myeloid-derived suppressor cells and recovers natural killer cell activity. Clin Cancer Res. 2014; 20:4096-4106.
50. Fujita M, Kohanbash G, Fellows-Mayle W, Hamilton RL, Komohara Y, Decker SA, Ohlfest JR and Okada H. COX-2 blockade suppresses gliomagenesis by inhibiting myeloid-derived suppressor cells. Cancer Res. 2011; 71:2664-2674.
51. Serafini P. Editorial: PGE2-producing MDSC: a role in tumor progression? J Leukoc Biol. 2010; 88:827-829.
52. Veltman JD, Lambers ME, van Nimwegen M, Hendriks RW, Hoogsteden HC, Aerts JG and Hegmans JP. COX-2 inhibition improves immunotherapy and is associated with decreased numbers of myeloid-derived suppressor cells in mesothelioma. Celecoxib influences MDSC function. BMC Cancer. 2010; 10:464.
53. Obermajer N, Wong JL, Edwards RP, Odunsi K, Moysich K and Kalinski P. PGE(2)-driven induction and maintenance of cancer-associated myeloid-derived suppressor cells. Immunol Invest. 2012; 41:635-657.
54. Zhang Y, Liu Q, Zhang M, Yu Y, Liu X and Cao X. Fas signal promotes lung cancer growth by recruiting myeloid-derived suppressor cells via cancer cell-derived PGE2. J Immunol. 2009; 182:3801-3808.
55. Yao C, Hirata T, Soontrapa K, Ma X, Takemori H and Narumiya S. Prostaglandin E(2) promotes Th1 differentiation via synergistic amplification of IL-12 signalling by cAMP and PI3-kinase. Nat Commun. 2013; 4:1685.
56. Candelario-Jalil E, Slawik H, Ridelis I, Waschbisch A, Akundi RS, Hull M and Fiebich BL. Regional distribution of the prostaglandin E2 receptor EP1 in the rat brain: accumulation in Purkinje cells of the cerebellum. J Mol Neurosci. 2005; 27:303-310.
57. Liu Y, Lai L, Chen Q, Song Y, Xu S, Ma F, Wang X, Wang J, Yu H, Cao X and Wang Q. MicroRNA-494 is required for the accumulation and functions of tumor-expanded myeloid-derived suppressor cells via targeting of PTEN. J Immunol. 2012; 188:5500-5510.
58. Youn J-I, Kumar V, Collazo M, Nefedova Y, Condamine T, Cheng P, Villagra A, Antonia S, McCaffrey JC, Fishman M, Sarnaik A, Horna P, Sotomayor E et al. Epigenetic silencing of retinoblastoma gene regulates pathologic differentiation of myeloid cells in cancer. Nat Immunol. 2013; 14:211-220.
59. Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer. 2005; 5:263-274.
60. Chun E, Lavoie S, Michaud M, Gallini CA, Kim J, Soucy G, Odze R, Glickman JN and Garrett WS. CCL2 Promotes Colorectal Carcinogenesis by Enhancing Polymorphonuclear Myeloid-Derived Suppressor Cell Population and Function. Cell Rep. 2015; 12:244-257.