
INTRODUCTION
The term autophagy identifies the basic self-degradative physiological process by which the cell removes worn-out or damaged components, such as protein aggregates, mitochondria, Endoplasmic Reticulum (ER), peroxisomes and intracellular pathogens [1-3]. Autophagy is evolutionarily conserved and involves the double-membrane sequestration and lysosomal breakdown of the cargo [1-3]. Although there is a basal autophagy in the cell, this process is further induced in response to stress, e.g. nutrient deprivation, hypoxia and pressure overload to catabolize cellular substrates and generate energy [4]. Defects in the autophagy machinery have been associated with the pathogenesis of the major human diseases including cancer [1,4]. Three major forms of autophagy have been described: micro-, macro- and chaperone-mediated autophagy, all involving the lysosomal breakdown of sequestered material [4,5]. The emerging role of macro-autophagy (from here on referred to as autophagy) has been the focus of recent cutting edge research and it is now accepted that it plays a critical role in both health and disease [1-3]. Autophagy is a multistep process involving its induction, the development of an isolation membrane, the completion and maturation of an ‘autophagosome’ containing cytosolic components for recycling and the ultimate fusion with a lysosome (forming the autophagolysosome) for degradation by lysosomal enzymes [1-3].
While oncogenic transformation and tumour development are associated with resistance to, or loss of, the apoptotic pathway, an increasingly number of experimental data suggest that autophagy imbalance plays an important role in tumorigenesis and is a relevant target for cancer therapy [1, 5-7]. Indeed, autophagy-deficient mice develop a large number of spontaneous tumours, supporting the idea that autophagy acts as tumor suppression mechanism [6-7]. Moreover, the heterozygous deletion in mice of essential autophagy genes such as Beclin1 and Ambra1 leads to high incidence of spontaneous tumours [8, 9]. The role played by Beclin 1 and Ambra1 as tumour suppressors is also evidenced by the identification of their binding partners, most of which are implicated in tumorigenesis, such as Bcl-2. The anti-apoptotic member of the bcl-2 family constitutively binds to Beclin1 and Ambra1 complex inhibiting autophagy induction [10]. Furthermore, overexpression of the positive regulator of Beclin1 UVRAG activates autophagy and suppresses tumour cell growth, whereas its down-regulation results in decreases autophagy levels and triggers uncontrolled cell proliferation [7, 8]. Finally, it has been recently published that Ambra1 regulates cell proliferation by facilitating the degradation of the proto-oncogene c-Myc by favouring its dephosphorylation by PP2A and thereby reducing the cell division rate [8].
Although the current view is that autophagy acts as a tumour suppressor process it is now also emerging that some established cancers require autophagy to survive, thus suggesting a pro-tumour autophagy activity. In keeping with this notion, the Ras-dependent tumorigenesis has been shown to be associated to autophagy induction [11, 12]. It is also now clear that the genotype of an individual tumor influences the autophagic function. For instance, in melanoma, BRAF has a direct effect on autophagy regulation. In fact, the overexpression of the wild type BRAF vs the mutated BRAFV600E in melanoma cells leads to increased basal levels of autophagy [13].
The paradox of autophagy acting as both a cell survival pathway and a tumour suppressor pathway is now being in some way reconciled; the current view is that while autophagy suppresses tumour growth at early stages of oncogenesis, it promotes growth in established tumors [6, 11, 12]. However, due to several controversial data the exact role of autophagy in the different steps of cancer development, especially in hepatocellular carcinoma (HCC), remains elusive [14-16].
HCC is the third cause of cancer-related deaths worldwide, and is characterized by poor prognosis and few treatment options [17]. The development of HCC is a multistep process. HCCs arise most frequently in the setting of chronic liver inflammation and fibrosis due to viral infection, metabolic injury, toxic insults, or autoimmune reactions. These tumors originate from premalignant lesions, ranging from dysplastic foci to dysplastic hepatocyte nodules that are often seen in damaged and cirrhotic livers, and are more proliferative than the surrounding parenchyma [18]. Since no effective treatment for HCC exists and, upon diagnosis, most patients with advanced disease have a remaining lifespan of only 4–6 months, it is critical to detect cellular and molecular changes taking place in preneoplastic lesions and identify biomarkers and molecular targets useful for an early diagnosis and for therapy.
Since the knowledge of molecular events occurring in early stages of HCC development is hampered by the difficulties in the histomorphologic distinction between non-malignant nodular lesions and early HCCs, animal models allowing the study of different stages of hepatocarcinogenesis, represent a very helpful tool to detect cellular and molecular alterations occurring in early preneoplastic stages. Therefore, in our present study, we employed the Resistant-Hepatocyte (R-H) model which allows dissecting the several steps of hepatocarcinogenesis and whose translational value has already been demonstrated [19-21], to investigate the role of autophagy in the early stages of HCC development.
RESULTS
Differential autophagy modulation takes place in early preneoplastic nodules
The R-H model allows dissecting the different steps of the carcinogenic process, as phenotypically distinct lesions can be identified at well-defined timings [19]. Therefore, it is possible to investigate changes in the autophagic machinery at very early times in HCC development. To this aim, we performed ultrastructural examination of preneoplastic nodules dissected from the surface of the liver exposed to the R-H model and developed 10 weeks after treatment with DENA. Electron microscopy of hepatocytes in the preneoplastic nodules revealed extensive morphological alterations of cytoplasmic organelles (Figure 1B) compared with control liver (Figure 1A). Most hepatocytes in the nodules displayed abnormal mitochondria (Figure 1C), which appeared swollen with loss of cristae and ruptures of the outer membrane. No sign of clearance by autophagy of the degenerating mitochondria was visible. Autophagic vacuoles containing only partially degraded materials (Figure 1C) and lipid droplets accumulation were frequently observed (Figure 1D). All these features are indicative of an impairment of the autophagic process, demonstrating that derangement of the autophagic machinery occurs at very early stages of HCC development.
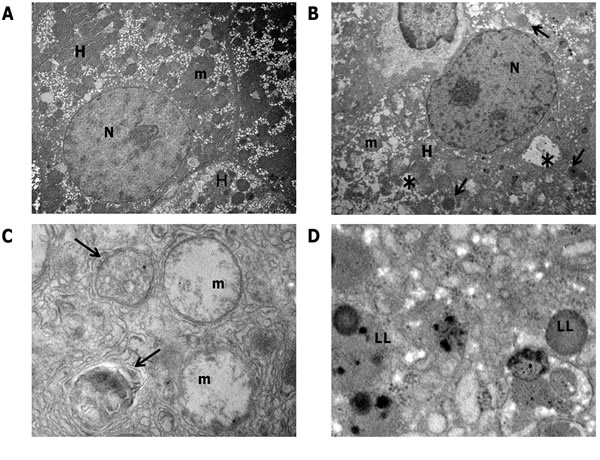
Figure 1: Transmission electron microscopy of preneoplastic nodules. A. Representative image of control liver. Hepatocytes (H) typically display a large, round nucleus (N); numerous mitochondria (m) with regular matrix and prominent cristae. B. Ultrastructure of a preneoplastic hepatocyte (H). Irregular nucleus (N), extensive vacuolation (asterisks), lipid droplets accumulation (see arrows) characterize the preneoplastic modifications. C. Higher magnification of nodule hepatocytes revealed that mitochondria undergo pathological modifications, consisting of swelling and progressive loss of cristae. Autophagosomes containing only partially digested material are also visible (arrows). D. Lipid inclusions (LL) accumulation in the cytoplasm of preneoplastic hepatocytes. Original magnifications: A, B= x3000; C, D= x30000.
Autophagy modulation in KRT-19+ negative vs positive preneoplastic nodules
We have previously shown in the R-H model [20,21] that although preneoplastic nodules positive for the putative progenitor cell marker cytokeratin-19 (KRT-19+) represent a minority of the total preneoplastic lesions, most HCCs are positive for this marker. This suggests that KRT-19+ preneoplastic nodules have an advantage in the progression to malignancy, while KRT-19- lesions undergo spontaneous remodeling during the carcinogenic process, a previously described phenomenon [22]. Therefore, we wished to investigate as to whether autophagy modulation could be a general phenomenon of preneoplastic stages or it is restricted to the most aggressive (KRT-19+) lesions. To this aim, immunohistochemistry analysis of typical autophagic markers, such as p62, Ambra1 and Beclin1, that are involved in the multistep process of autophagy, was performed on serial sections of livers from rats subjected to the R-H protocol and sacrificed 10 weeks after DENA. Interestingly, specific staining of KRT-19+ - but not of KRT-19- -preneoplastic lesions was observed by positivity of the liver sections for two other proteins, such as Ambra1 and Beclin1, involved in the early regulatory steps of the autophagic process and p62 which is mediating the recruitment of cargos in the autophagosomes (Figure 2A, 2B). Remarkably, 99%, 94% and 71% of GST-P+/KRT-19+ nodules were also positive for Ambra1, p62 and Beclin1, respectively (Table 1). Almost none of KRT-19- nodules – nor the surrounding parenchymal tissue- exhibited staining for these proteins. As shown in Figure 2A and 2B, IHC revealed cytoplasmic and nuclear accumulation of p62 only in a subset of hepatocytes (about 50% of the total) in the GST-P+/KRT-19+ nodules, but not in GST-P+/KRT-19- preneoplastic lesions, indicating that a marked differential modulation of the autophagic process occurs also inside the GST-P+/KRT-19+ nodules.
Table 1: Percentage and number of KRT-19+/Ambra1+, KRT-19+/p62+ and KRT-19+/Beclin1+ nodules with respect to the total amount of KRT-19+ nodules.
Mean N° of GST-P+ nodules |
Mean N° of KRT 19+ /GST-P+ nodules |
Mean N° of Ambra1+ /KRT-19+ nodules |
% of Ambra1+ /KRT-19+ nodules |
Mean N° of p62+ /KRT-19+ nodules |
% of p62+ /KRT-19+ nodules |
Mean N° of Beclin1+ /KRT-19+ nodules |
% of Beclin1+ /KRT-19+ nodules |
|
74.6 ± 13.3 |
18.4 ± 4.0 |
18.2 ± 4.0 |
99.0 |
17.3 ± 4.0 |
94.0 |
13.0 ± 2.2 |
70.6 |
The analysis of serial liver sections was performed on a total number of 373 nodules, obtained from 5 rats.

Figure 2: Autophagy impairment is restricted to a more aggressive subset of preneoplastic hepatic nodules. IHC on serial sections of liver from rats subjected to the R-H protocol and sacrificed 10 weeks after DENA. A. Presence of intense staining for Ambra1, Beclin1 and p62 in a nodule positive for GST-P and KRT-19 (magnification x4). A higher enlargement of the same nodule showing nuclear p62 staining (magnification x20). B. Absence of Ambra1, Beclin1 and p62 staining in a GST-P+/KRT-19- nodule (magnification x4).
Enhanced transcription is not the cause of the increased accumulation of p62, Ambra1 and Beclin1
To assess whether the accumulation of p62, Ambra1 and Beclin1 is the consequence of transcriptional or post-transcriptional modifications of genes/gene products involved in the autophagic process, we performed qRT-PCR analysis of the expression of these and other autophagy-associated genes, on laser micro-dissected GST-P+/KRT-19+ preneoplastic nodules. As shown in Figure 3A and 3B, no increased transcription of any of the examined genes involved in the autophagic process was observed. Rather, we found down-regulation of some of them, such as Ulk1, Ambra1, p62, LC3 and FoxO3. These results clearly demonstrate that the accumulation of these proteins in aggressive preneoplastic lesions is not due to an active transcription. However, the interpretation of these findings should take into consideration that, as highlighted by the IHC, the accumulation of the Ambra1, p62 and Beclin1 has been detected only in about 50% of the hepatocytes.
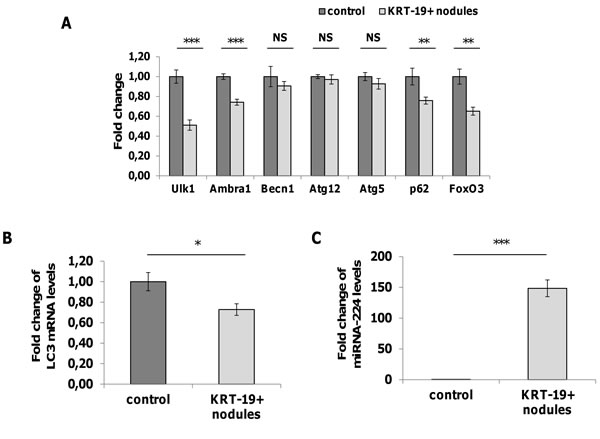
Figure 3: Analysis of mRNA levels of autophagy-associated genes and miRNA-224. A. qRT-PCR analysis of Ulk1, Ambra1, Beclin1, Atg12, Atg5, p62 and FoxO3 mRNA in KRT-19+ nodules generated 10 weeks after treatment with DENA. Gene expression is reported as fold-change relative to age-matched controls; ***P<0.001, **P<0.01, NS: not significant. B. LC3 expression in KRT-19+ preneoplastic nodules. Gene expression is reported as fold-change relative to age-matched controls; *P<0.05. C. qRT-PCR analysis of miR-224 expression in KRT-19+ nodules. miR-224 expression is reported as fold-change relative to age-matched controls; ***P<0.0001.
MiR-224 is highly expressed in KRT-19 positive nodules
Recently, it was reported that inhibition of autophagy in hepatitis B-associated human HCCs is responsible for the strong accumulation of miR-224 - one of the most up-regulated miRs in human cancer [23, 24] - in these tumors [25]. Therefore, we investigated the expression of miR-224 in KRT-19+ preneoplastic lesions. As shown in Figure 3C, miR-224 was strongly up-regulated in KRT-19 positive lesions (150-fold) compared to normal liver, supporting the notion that the increased miR-224 expression found in experimental and human HCC is most likely the consequence of its accumulation when autophagic machinery is deregulated.
The autophagic inducer, Amiodarone, stimulates the growth of KRT-19+ nodules
The results so far obtained demonstrate that autophagy deregulation is an early event in HCC development and characterizes about 50% of the parenchymal cells in the most aggressive lesions. Since previous works suggested that autophagy mainly contributes to tumor suppression during the early stage of tumorigenesis, we wished to further investigate whether impairment of the autophagic process favours cancer progression. To address this question, we administered Amiodarone, an autophagy inducer [26], or chloroquine (CQ), an autophagy inhibitor [27], to nodule-bearing rats developed 6 weeks after DENA. Interestingly, while Amiodarone did not significantly modify the number of GST-P+ preneoplastic lesions (Figure 4A), an impressive increase of the size (Figure 4B, 4E) and of the % hepatic area (Figure 4C, 4E) occupied by these lesions was observed compared to the control group. These enhancing effects were associated with a strong increase in nodule hepatocyte proliferation (Figure 5A, 5B). Interestingly, Amiodarone also led to an increase of the percentage of KRT-19+ nodules (Figure 4D, 4E). On the opposite, when compared to untreated animals, the autophagic inhibitor CQ caused a decrease of the size and of the % hepatic area occupied by these lesions (Figure 4A-4C, 4E). Moreover, the % of KRT-19+ nodules was found to be significantly lower than that of the Amiodarone-treated group. Remarkably, treatment with CQ strongly inhibited proliferation of nodule hepatocytes (Figure 5C) when compared to either control- or Amiodarone-treated group.
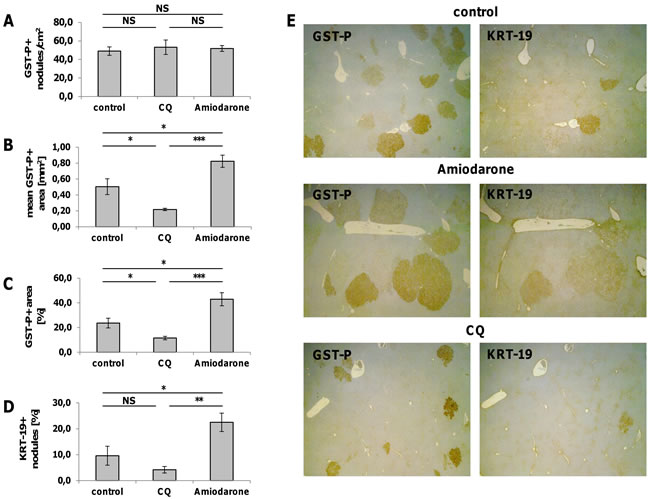
Figure 4: Effect of Amiodarone and Chloroquine on the number of GST-P+ nodules, the mean GST-P+ area, the percentage of the area occupied by GST-P+ hepatocytes and the number of KRT-19+ nodules. Rats exposed to the R-H protocol were injected intraperitoneally with Amiodarone (30 mg/kg, four doses) or Chloroquine (CQ, 50 mg/kg, 4 doses) starting 2 weeks after 2-AAF withdrawal (6 weeks after treatment with DENA). Animals were euthanized 7 days after the treatment. Effect of Amiodarone and Chloroquine administration on the number of GSTP-positive nodules A., the mean GST-P-positive area B., the percentage of the area occupied by GST-P-positive hepatocytes C. and the number of KRT-19+ nodules D. Values are expressed as mean ± SEM. ***P<0.001, **P<0.01, *P<0.05, NS: not significant. E. GST-P and KRT-19 immunohistochemistry of liver sections from rats treated with Amiodarone and CQ (magnification x1.25).
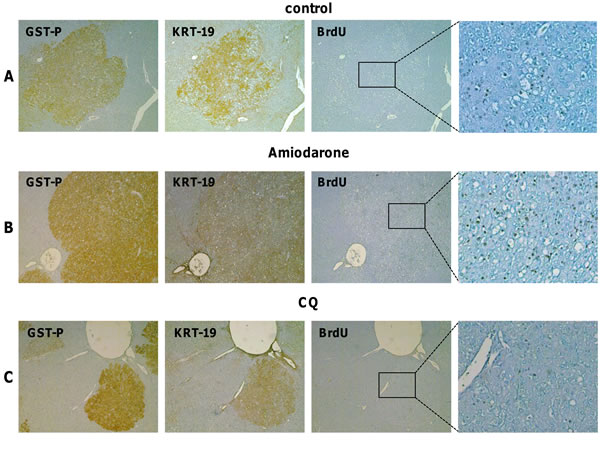
Figure 5: Effect of Amiodarone and Chloroquine administration on hepatocyte proliferation. A. GST-P, KRT-19 and BrdU IHC on serial sections of liver from rats subjected to the R-H protocol and sacrificed 7 weeks after DENA (magnification x4, inset x10). B. GST-P, KRT-19 and BrdU IHC on serial sections of liver from rats subjected to the R-H protocol, treated with four doses of Amiodarone and sacrificed 7 days after treatment (magnification x4, inset x10). C. GST-P, KRT-19 and BrdU IHC on serial sections of liver from rats subjected to the R-H protocol, treated with four doses of Chloroquine (CQ) and sacrificed 7 days after treatments (magnification x4, inset x10).
Previous report [25] has shown that Amiodarone-induced autophagy leads to a reduction of the tumor weight. Therefore, we wished to establish if the observed promoting effects on nodule growth induced by Amiodarone was associated with increased autophagy. To this aim, electron microscopy examination of nodules scooped out from the liver surface was performed. As shown in (Figure 6A), the fine structure of Amiodarone-treated hepatocytes revealed a less damaged morphology; lipid droplets scattered in the cytoplasm and dilated intercellular space were observed; however, after Amiodarone treatment cells in the nodules displayed intact rough endoplasmic reticulum and mitochondria which were similar to that of normal untreated hepatocytes (Figure 6C). Electron microscopy was also performed after CQ treatment; since the small size of CQ-treated rat nodules hampered the possibility of dissecting them from the liver, electron microscopy examination was performed in randomly selected areas of the liver. As shown in Figure 6B, EM analysis confirmed the inhibitory effect of CQ on autophagy; indeed, many vacuoles containing undigested cytoplasmic materials were observed. In addition, apoptotic and necrotic hepatocytes were often observed (data not shown). These findings indicate that while induction of autophagy stimulates the growth of preneoplastic lesions to HCC progression, inhibition of autophagy exerts the opposite effect.
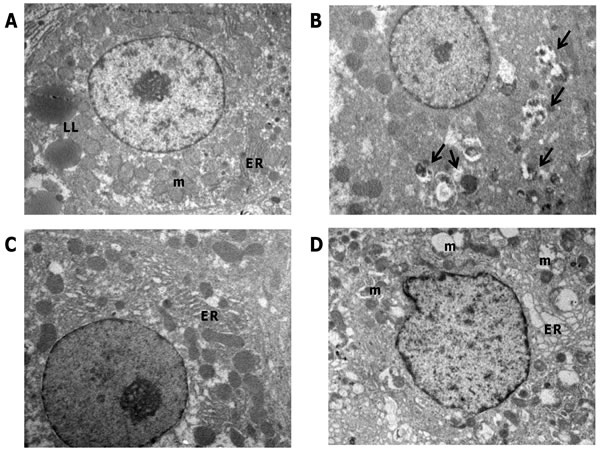
Figure 6: Ultrastructure of livers from nodule-bearing rats receiving amiodarone and chloroquine. A. TEM examination of liver tissues from rats receiving amiodarone show that endoplasmic reticulum (ER) appears well preserved and mitochondria (m) swelling and vacuolation are reduced compared to untreated preneoplastic liver D.; Few lipid inclusions (LL) are displayed by these hepatocytes compared to control liver C. B. Massive accumulation of autophagic vesicles is observed following treatment with chloroquine. Original magnifications: x3000.
DISCUSSION
Recent years have brought an enormous advance in the elucidation of the molecular machinery involved in mammalian autophagy [1-3]. Although the contribution of autophagy in tumour development is unquestioned, nonetheless, its role is not completely understood. Paradoxically, autophagy seems to play a dual role, acting as tumor promoter or tumor suppressor, depending on the tumor stage [28]. Numerous studies suggest that autophagy contributes to tumor suppression during the early stages of tumorigenesis [28-29]. This suppressive effect is likely due to some important functions of autophagy, such as defective organelle elimination in order to reduce oxidative stress and prevent DNA damage. During the late phases of tumorigenesis, autophagy is supposed to act as tumor promoter mechanism, enabling tumor cells to cope with high metabolic demand [6, 11].
As far as the role of autophagy in HCC development is concerned, it was reported that homozygous deficient mice for the autophagy genes ATG5 or ATG7 develop benign liver tumors that do not progress to metastatic cancer, which suggests that loss of autophagy may be sufficient for the initiation of tumorigenesis, but residual autophagy is required for the progression to cancer [6]. In line with these results, it was also reported that autophagy suppressed hepatocarcinogenesis at early stages by protecting normal cell stability and promoted hepatocarcinogenesis at late stages by supporting tumor cells growth [28, 29]. Finally, Ambra1 haplodeficiency in mice leads to the development of liver tumors, and, in humans, Beclin1 expression was decreased in HCC tissues compared with adjacent non-tumoral tissues and Ambra1 [4, 5].
In this study, we attempted to investigate the status of the autophagic process in the very early stages of hepatocarcinogenesis, taking advantage of the well-established R-H model in the rat. The main findings stemming from our work indicate that modulation of autophagy i) is a very early event in the multistage process of hepatocarcinogenesis, and, ii) takes place selectively in in KRT-19+ preneoplastic lesions, but not in KRT-19- lesions. We have previously shown that although KRT-19+ lesions represent a minority of the total preneoplastic lesions, most HCCs arising in this model are KRT-19+ [20, 21]. Therefore, our results are in line with the observation that autophagy defects favour cancer development and loss of autophagy may increase the propensity of cells toward oncogenic transformation [30, 31].
Another relevant finding of the present work is the association between high miR-224 expression and autophagy impairment. A recent study demonstrated that inhibition of autophagy in HBV-associated human HCCs is the mechanism responsible for the strong accumulation of the mature form miR-224 in these tumors [25]. These results were also confirmed in liver tumors of HBV X gene transgenic mice. Importantly, miRNA-224, involved in cell proliferation, migration and invasion [32], is one of the most up-regulated miRs in rodent and human HCC [21-23, 24]. Indeed, our data show a drastic upregulation of miRNA-224 in those subset of nodules which are more aggressive and develop HCC.
An additional and novel finding of the present study is that while the autophagic inducer, Amiodarone, promotes a striking increase in the proliferation of nodular hepatocytes and the size of preneoplastic lesions, the inhibitor of autophagy, CQ, induces the opposite, thus leading to a decrease of the size of preneoplastic nodules. Similarly, Sun et al. [29] demonstrated that autophagy inhibition by CQ in the tumor-forming stage of DENA-induced HCC, remarkably reduced tumor growth by decreasing cell survival and proliferation. Moreover, it has been also reported that the combination of sorafenib with chloroquine produced more pronounced tumor suppression in HCC, both in vivo and in vitro [33]. In further support of the hypothesis that inhibition of autophagy plays an anti-tumoral effect in the early stages of the carcinogenic process, Amiodarone increases the percentage of KRT-19+ nodules, endowed with a higher proliferative capacity. By contrast, inhibition of autophagy by CQ resulted in an almost complete loss of nodules positive for this marker. These results, in turn, suggest a link between autophagy and the accumulation of KRT-19. Indeed, we found that the most aggressive KRT-19-positive nodules express high level of Ambra1 that, through its interaction with the protein phosphatase PP2A, regulates the stability of the oncoprotein and pro-mitotic factor c-Myc [8]. Thus, the expression of Ambra1 can both potentiates autophagy through its interaction with Beclin1 and VPS34, as well as favours the proliferation of the most aggressive nodules by binding to PP2A.
In conclusion, the present work raised the interesting hypothesis that autophagy in the liver may act as an homeostatic mechanism limiting the progression of preneoplastic cells to a more malignant stage. The finding that the administration of chloroquine is able to rapidly reduce the size of the KRT-19+ preneoplastic lesions suggests that the pharmacological modulation of autophagy may represent a possible therapeutic approach to interfere with the development of HCC.
MATERIALS AND METHODS
Animals and treatment
Male Fischer rats were obtained from Charles River (Milano, Italy). Guidelines for Care and Use of Laboratory Animals were followed during the investigation. All animal procedures were approved by the Ethical Commission of the University of Cagliari and the Italian Ministry of Health. Animals were treated with a single dose of diethylnitrosamine (DENA, 150 mg/kg) and, two weeks later, were subjected to the R-H protocol, consisting of a 2 week-diet supplemented with 0.02% 2-acetylaminofluorene (2-AAF) and a two/third partial hepatectomy (PH) [19]. Rats were then switched to basal diet all throughout the experiment and sacrificed 10 weeks after DENA administration (See Supp. Figure 1A.)
Another group of rats exposed to R-H protocol was given four doses of Chloroquine (50 mg/kg, Sigma-Aldrich, C6628) or Amiodarone (30 mg/kg, Sigma-Aldrich, A8423) starting 2 weeks after 2-AAF withdrawal (6 weeks after treatment with DENA). Rats were sacrificed 7 days after the first dose (See Supp. Figure 1B and 1C). BrdU was given in drinking water (1mg/1ml) for 5 days before the sacrifice.
Histology and immunohistochemistry
Liver sections were fixed in 10% formalin and included in paraffin or quickly frozen by immersion in liquid nitrogen and processed for hematoxylin-eosin, or GST-P and KRT-19 immunohistochemistry, as described [21]. We considered as KRT-19 positive, all those lesions exhibiting a KRT-19 positive area of at least 5% of the total area of the preneoplastic lesion. The average area occupied by KRT-19 positive hepatocytes was at least 20% of the total area of the nodules microdissected for further analyses. Paraffin-embedded liver sections were used for p62, Ambra and Beclin 1 immunohistochemistry. Sections, were deparaffinized in xylene, incubated for 5 min each in 100%, 90%, 70%, and 50% ethanol for rehydration and immersed in 10 mM sodium citrate, pH 6.0, and microwaved for antigen retrieval. Endogenous peroxidase activity was blocked by 3% H2O2 for 5 min. After rinsing in phosphate-saline buffer (PBS) nonspecific antibody binding was reduced by incubating the sections with normal goat serum for 5 min. Sections were washed in PBS/1% BSA buffer and incubated with primary antibodies: rabbit anti-p62/SQSTM1 from MBL (Woburn, MA, USA) 1:400, rabbit anti AMBRA1 (ProSci) 1:100 and rabbit anti-BECN1 Antibody (Santa Cruz) 1:50 were used. Reactions were visualized using a streptavidin-biotin-immunoperoxidase system with DAB (Biogenex, San Ramon, CA) as chromogen substrates. Negative control staining was performed by omitting the primary antibody. Sections were counterstained in Mayer’s acid hemalum.
RNA extraction and qRT-PCR
Total RNA was extracted from preneoplastic lesions with the MirVana kit (Life Technologies) and stored at -80ºC until needed. While RNA quantity was measured by NanoDrop ND1000 (Thermo Scientific), RNA integrity was assessed by Agilent Bioanalyzer 2100. Only RNA samples with a RIN (RNA Integrity Number) ≥ 7 were included in the study. RNA was retrotranscribed with High Capacity cDNA Reverse Transcription Kit (Life Technologies) using random primers. The expression levels of the examined genes were evaluated by Real-Time PCR analysis with an ABI PRISM 7300HT thermocycler (Life Technologies) on 6 samples of GST-P-positive preneoplastic nodules. All samples were run in triplicate. For qRT-PCR, The Power SYBR Green PCR Master Mix (Life Technologies) was used. The complete list of primer sequences used is listed in Supp. Table 1. After checking the specificity of the PCR products with the melting curve, data were then normalized to GAPDH expression and the expression level of different targets was calculated by 2-∆∆Ct. Analysis of LC3 expression was performed using specific TaqMan probes (Life Technologies) and GAPDH as endogenous control.
Analysis of MicroRNA-224
cDNA was synthesized using the TaqMan MicroRNA Reverse Transcription Kit (Life Technologies) in accordance with the manufacturer’s instructions. qRT-PCR amplification was performed with the reverse transcription product, TaqMan 2X Universal PCR Master Mix, No AmpErase UNG, miRNA-224 primers and probe mix (Life Technologies). The endogenous control 4.5S RNA(H) was used to normalize miRNA expression levels.
Laser capture microdissection
GST-P+/KRT-19+ nodules were identified by immunohistochemical staining of 6µm-thick frozen liver sections. Nodules microdissection was done on 16µm serial sections with a Leica LMD6000, as previously described [21].
Electron microscopy
Tissue samples were fixed with 2.5% glutaraldehyde (Assing Spa, R1012) in 0.1 M cacodylate buffer for 1 h at 4°C (sodium cacodylate trihydrate, Sigma-Aldrich, C4945), and postfixed in 1% osmium tetroxide (Sigma-Aldrich, 75632) in 0.1 M cacodylate buffer for 1 h. Samples were then dehydrated in graded ethanol and embedded in Epon resin (AGAR 100, Agar Scientific R1045). Ultrathin sections were stained with 2% uranyl acetate (Sigma-Aldrich, 73943) and observed under a Zeiss EM900 transmission electron microscope. Images were captured digitally with a Mega View II digital camera (SIS; Zeiss).
Statistics
Data are expressed as mean ± standard deviation (SD) or mean ± standard error (SEM). Analysis of significance was done by t Student’s test using the GraphPad software (La Jolla, California).
Abbreviations
2-AAF, 2-acetylaminofluorene; Atg, autophagy-related genes; BrdU, 5-bromo-2’-deoxyuridine; CQ, chloroquine; DENA, diethylnitrosamine; GAPDH; glyceraldehyde 3-phosphate dehydrogenase; GST-P, placental glutathione S-transferase; HBV, hepatitis B virus; HCC, hepatocellular carcinoma; IHC, immunohistochemistry; KRT-19, cytokeratin-19; PH, partial hepatectomy; qRT-PCR, quantitative reverse transcriptase polymerase chain reaction; R-H model, Resistant-Hepatocyte model.
Acknowledgments
This work was supported by grants from the Italian Ministry of University and Research (FIRB Accordi di Programma 2011), the Italian Ministry of Health (Ricerca Finalizzata and Ricerca Corrente), and from Associazione Italiana Ricerca sul Cancro (AIRC, Grants to AC (IG-15279), MP), Ministero Università e Ricerca Scientifica (PRIN-2010LC747T to AC), R.A.S. 2012 to AC and Fondazione Banco di Sardegna to AC, GMLC and A Perra.
Authors’ contribution
L. Falasca: TEM analysis, study concept, critical revision of the manuscript; MA. Kowalik: study concept, design, immunohistochemical, qRT-PCR analyses, critical revision of the manuscript; A. Perra: laser microdissection, RNA extraction, immunohistochemistry; GM. Ledda-Columbano: pathological analysis of liver samples, acquisition and interpretation of data, critical revision of the manuscript; M. G Ippolito, M interpretation of data, critical revision; Piacentini, A. Columbano: study concept and design, analysis and interpretation of data, drafting of the manuscript.
conflicts of interest
No conflict of interest to declare.
REFERENCES
1. Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008; 132: 27-42.
2. Shaid S, Brandts CH, Serve H, Dikic I. Ubiquitination and selective autophagy. Cell Death Differ. 2013; 20: 21-30.
3. Ashrafi G, Schwarz TL. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013; 20: 31-42.
4. Garg AD, Martin S, Golab J, Agostinis P. Danger signalling during cancer cell death: origins, plasticity and regulation. Cell Death Differ. 2014; 21: 26-38.
5. Galluzzi L, Pietrocola F, Bravo-San Pedro JM, Amaravadi RK, Baehrecke EH, Cecconi F, Codogno P, Debnath J, Gewirtz DA, Karantza V, Kimmelman A, Kumar S, et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015; 34: 856-880.
6. Ko A, Kanehisa A, Martins I, Senovilla L, Chargari C, Dugue D, Mariño G, Kepp O, Michaud M, Perfettini JL, Kroemer G, Deutsch E. Autophagy inhibition radiosensitizes in vitro, yet reduces radioresponses in vivo due to deficient immunogenic signaling. Cell Death Differ. 2014; 21: 92-99.
7. Sui X, Chen R, Wang Z, Huang Z, Kong N, Zhang M, Han W, Lou F, Yang J, Zhang Q, Wang X, He C, Pan H. Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death Dis. 2013; 4: e838.
8. Cianfanelli V, Fuoco C, Lorente M, Salazar M, Quondamatteo F, Gherardini PF, De Zio D, Nazio F, Antonioli M, D’Orazio M, Skobo T, Bordi M, Rohde M et al. AMBRA1 links autophagy to cell proliferation and tumorigenesis by promoting c-Myc dephosphorylation and degradation. Nat Cell Biol. 2015; 17: 20-30.
9. Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, Cattoretti G, Levine B. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003; 112: 1809-1820.
10. Strappazzon F, Vietri-Rudan M, Campello S, Nazio F, Florenzano F, Fimia GM, Piacentini M, Levine B, Cecconi F. Mitochondrial BCL-2 inhibits AMBRA1-induced autophagy. EMBO J. 2011; 30: 1195-1208.
11. Schmukler E, Kloog Y, Pinkas-Kramarski R. Ras and autophagy in cancer development and therapy. Oncotarget. 2014; 5: 577-586. doi: 10.18632/oncotarget.1775.
12. Vessoni AT, Filippi-Chiela EC, Menck CF, Lenz G. Autophagy and genomic integrity. Cell Death Differ. 2013; 20: 1444-1454.
13. Corazzari M, Rapino F, Ciccosanti F, Giglio P, Antonioli M, Conti B, Fimia GM, Lovat PE, Piacentini M. Oncogenic BRAF induces chronic ER stress condition resulting in increased basal autophagy and apoptotic resistance of cutaneous melanoma. Cell Death Differ. 2015; 22: 946-958.
14. He Z, Simon HU. Autophagy protects from liver injury. Cell Death Differ. 2013; 20: 850-851.
15. Zhang W, Hou J, Wang X, Jiang R, Yin Y, Ji J, Deng L, Huang X, Wang K, Sun B. PTPRO-mediated autophagy prevents hepatosteatosis and tumorigenesis. Oncotarget. 2015; 6: 9420-9433. Doi: 10.18632/oncotarget.3353.
16. Tian Y, Kuo CF, Sir D, Wang L, Govindarajan S, Petrovic LM, Ou JH. Autophagy inhibits oxidative stress and tumor suppressors to exert its dual effect on hepatocarcinogenesis. Cell Death Differ. 2015; 22: 1025-1034.
17. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014; 64: 9-29.
18. Thorgeirsson SS, Lee JS, Grisham JW. Functional genomics of hepatocellular carcinoma. Hepatology. 2006; 43: S145-150.
19. Solt DB, Medline A, Farber E. Rapid emergence of carcinogen-induced hyperplastic lesions in a new model for the sequential analysis of liver carcinogenesis. Am J Pathol. 1977; 88: 595-618.
20. Andersen JB, Loi R, Perra A, Factor VM, Ledda-Columbano GM, Columbano A, Thorgeirsson SS. Progenitor-derived hepatocellular carcinoma model in the rat. Hepatology. 2010; 51: 1401-1409.
21. Petrelli A, Perra A, Cora D, Sulas P, Menegon S, Manca C, Migliore C, Kowalik MA, Ledda-Columbano GM, Giordano S, Columbano A. MicroRNA/gene profiling unveils early molecular changes and nuclear factor erythroid related factor 2 (NRF2) activation in a rat model recapitulating human hepatocellular carcinoma (HCC). Hepatology 2014; 59: 228-241
22. Enomoto K, Farber E. Kinetics of phenotypic maturation of remodeling of hyperplastic nodules during liver carcinogenesis. Cancer Res. 1982; 42: 2330-2335.
23. Wang Y, Lee AT, Ma JZ, Wang J, Ren J, Yang Y, Tantoso E, Li KB, Ooi LL, Tan P, Lee CG. Profiling microRNA expression in hepatocellular carcinoma reveals microRNA-224 up-regulation and apoptosis inhibitor-5 as a microRNA-224-specific target. J Biol Chem. 2008; 283: 13205-13215.
24. Ladeiro Y, Couchy G, Balabaud C, Bioulac-Sage P, Pelletier L, Rebouissou S, Zucman-Rossi J. MicroRNA profiling in hepatocellular tumors is associated with clinical features and oncogene/tumor suppressor gene mutations. Hepatology. 2008; 47: 1955-1963.
25. Lan SH, Wu SY, Zuchini R, Lin XZ, Su IJ, Tsai TF, Lin YJ, Wu CT, Liu HS. Autophagy suppresses tumorigenesis of hepatitis B virus-associated hepatocellular carcinoma through degradation of microRNA-224. Hepatology. 2014; 59: 505-517.
26. Lin CW, Chen YS, Lin CC, Chen YJ, Lo GH, Lee PH, Kuo PL, Dai CY, Huang JF, Chung WL, Yu ML. Amiodarone as an autophagy promoter reduces liver injury and enhances liver regeneration and survival in mice after partial hepatectomy. Sci Rep. 2015; 5: 15807.
27. Chen P, Hu T, Liang Y, Jiang Y, Pan Y, Li C, Zhang P, Wei D, Li P, Jeong LS, Chu Y, Qi H, Yang M et al. Synergistic inhibition of autophagy and neddylation pathways as a novel therapeutic approach for targeting liver cancer. Oncotarget. 2015; 6: 9002-9017. doi: 10.18632/oncotarget.3282.
28. Sun K, Deng W, Zhang S, Cai N, Jiao S, Song J, Wei L. Paradoxical roles of autophagy in different stages of tumorigenesis: protector for normal or cancer cells. Cell Biosci. 2013; 3: 35.
29. Sun K, Guo XL, Zhao QD, Jing YY, Kou XR, Xie XQ, Zhou Y, Cai N, Gao L, Zhao X, Zhang SS, Song JR, Li D et al. Paradoxical role of autophagy in the dysplastic and tumor-forming stages of hepatocarcinoma development in rats. Cell Death Dis. 2013; 4: e501.
30. Mathew R, White E. Autophagy in tumorigenesis and energy metabolism: friend by day, foe by night. Curr Opin Genet Dev. 2011; 21: 113-119.
31. Kimmelman AC. The dynamic nature of autophagy in cancer. Genes Dev. 2011; 25: 1999-2010.
32. Li Q, Wang G, Shan JL, Yang ZX, Wang HZ, Feng J, Zhen JJ, Chen C, Zhang ZM, Xu W, Luo XZ, Wang D. MicroRNA-224 is upregulated in HepG2 cells and involved in cellular migration and invasion. J Gastroenterol Hepatol. 2010; 25: 164-171.
33. Shi YH, Ding ZB, Zhou J, Hui B, Shi GM, Ke AW, Wang XY, Dai Z, Peng YF, Gu CY, Qiu SJ, Fan J. Targeting autophagy enhances sorafenib lethality for hepatocellular carcinoma via ER stress-related apoptosis. Autophagy. 2011; 7: 1159-1172.