
Introduction
Cancer is one of the major causes of mortality worldwide. More than half of all cancer patients receive radiation therapy as part of a curative or palliative treatment often in combination with surgery or chemotherapy. While most tumors initially respond to treatment, they often acquire resistance to therapy and eventually recur. The varied clinical responses observed between and within patients are in part the result of tumor heterogeneity and both acquired and intrinsic treatment resistance often caused by deregulation of signaling pathways that control normal cell renewal in adult tissues. The Notch signaling pathway is one of these frequently altered pathways in many tumors.
The involvement of ionizing radiation in the majority of cancer treatments, the pivotal role of Notch signaling in many fundamental processes such as self-renewal and differentiation, together with the fact that Notch signaling is often deregulated in cancer, suggest that targeting the Notch pathway may be beneficial for many cancer patients. Here, we review the opportunities and challenges of targeting Notch signaling to improve treatment response to radiation therapy.
Radiation resistance of cancer cells
Resistance to radiation is a common phenomenon and a major obstacle in cancer therapy [1]. Intrinsic determinants of radiation resistance include pathways regulating survival and apoptosis, cell cycle status as well as DNA repair capability. Extrinsic factors including extracellular matrix molecules, cytokines, hypoxia and angiogenesis also influence radiosensitivity.
Additionally, biological heterogeneity within the tumor population leads to differential radiation response. Pre-clinical models as well as clinical observations have demonstrated substantial genotypic and phenotypic heterogeneity between (inter-tumor) and within (intra-tumor) tumors [2]. This tumor heterogeneity poses a challenge to both cancer diagnostics and therapy. First because small tumor biopsies are unlikely to capture the complete genomic landscape of a patient’s tumor and thereby fail to identify (all) treatment-resistant cancer genotypes [3]. Second, while the sensitive tumor population will shrink, treatment-resistant tumor cells invariably will take over and tumors recur. Understanding factors underlying this heterogeneous treatment response will help to overcome treatment resistance.
Heterogenic treatment response may arise from influences of the microenvironment and genetic instability generating (epi)genetic changes. Malignant cell populations may alternatively (or complementary) consist of a developmentally defined hierarchy of heterogeneous phenotypes derived from a small subset of so-called cancer stem cells (CSC) [4]. Such cells have been best characterized in hematological [5], breast [6-7] and CNS malignancies [8]. Mounting evidence indicates that CSC populations in solid tumors are resistant to radiation. Mechanisms to explain this intrinsic resistance include efficient repair of DNA damage, lower numbers of DNA breaks, redistribution of cells in the cell cycle, repopulation, and reoxygenation of hypoxic tumor areas. These studies have been extensively reviewed elsewhere [9-10]. If these CSC rely on specific pathways for their survival after treatment, identification of such pathways would provide opportunities for the targeted and selective killing of treatment resistant cancer cells [11]. One of the promising candidates in this context is Notch signaling, a pathway active in many developmental as well as adult stem cell pathways and frequently altered in human cancers [12-13]
Here, we will discuss the role of Notch signaling in the radiation response of human tumors and highlight the opportunities to exploit inactivation of Notch using pharmacological inhibitors in conjunction with radiation therapy.
Notch signaling and radiation resistance
Notch proteins are short-range cell-cell signaling receptors that have key roles during development and in adult tissue self-renewal, proliferation and differentiation. In mammals, the Notch pathway consists of 4 Notch receptors (Notch1-4) that are present in signal receiving cells and 5 ligands on adjacent signal sending cells. In the canonical pathway, Notch receptor activation via ligand interaction leads to a consecutive series of proteolytic cleavages finally resulting in the release of the Notch intracellular domain (NICD) that translocates to the nucleus to act as transcription regulator. The list of target genes regulated by Notch is cell type dependent and includes genes involved in cell cycle regulation [14], cellular differentiation [15] and stem cell maintenance [16].
Consistent with its fundamental role in many aspects of vertebrate development, deregulation of the Notch pathway is implicated in various developmental syndromes. In adult tissues, deregulation or mutation of NOTCH proteins is observed in many cancer types and has been shown to contribute to carcinogenesis and treatment resistance [13]. Notch inhibitors have been under pre-clinical investigation for over a decade and shown strong responses in many cancer models. Several clinical trials of Notch pathway inhibitors in patients with leukemia have been reported and several are ongoing in solid cancers [17]. Here, we focus specifically on the role of Notch in resistance to radiotherapy and the different intrinsic and extrinsic mechanisms involved.
Intrinsic resistance
a) Targeting DNA repair
It has recently been demonstrated that Notch has a direct role in DNA damage response (DDR). The activity of Notch1 and ataxia-telengiectasia mutated kinase (ATM, the primary DNA sensor kinase in DDR) were shown to be inversely correlated in C.elegans and in human cell lines. ATM is activated specifically upon double strand (ds) DNA breaks induced by ionizing radiation. Notch1 directly binds to ATM thereby inactivating its kinase activity. Importantly, inactivation of ATM via Notch activation contribute to the survival of Notch driven human leukemia (T-ALL). Blocking Notch using a γ-secretase inhibitor (GSI) in the presence of DNA damage leads to increased radiation sensitivity in an ATM-dependent manner [18]. Activated Notch1 and pATM levels were also significantly inversely correlated in human primary breast cancer, validated by immunohistochemistry and in expression microarray datasets [18]. This result suggests that cancer cells treated with DNA-damaging agents such as radiation may undergo more robust cell death if treated with a Notch inhibitor. Another very recent and interesting observation came from a study by Deng et al. [19] in which they show that inactivation of homologous recombination in human Notch-driven cancer results in significant radiosensitization. This provides a basis for Notch-directed cancer therapy via blocking of homology-directed dsDNA break repair.
b) Targeting cancer stem cells (CSC)
There is increasing evidence supporting the role of Notch in maintenance and self-renewal of CSC in T-ALL [20-22], brain [23-24], breast [11, 25], lung [26] and colon tumors [27]. In glioma, it has been reported that blocking Notch using GSI depleted CD133+ glioma CSCs, attenuated neurosphere formation and lowered tumorigenicity [28]. In line with this, Notch inhibition selectively impaired clonogenic survival of the glioma CD133+ CSCs sub-population thereby enhancing its radiation sensitivity. The intrinsic radioresistance may be caused by alteration of DNA damage checkpoints [8] or through up-regulation of the pro-survival factors Akt and Mcl-1 in CSCs [29]. Hovinga et al. reported that Notch inhibition enhances the response to radiation by reducing proliferation and self-renewal of CSCs in tumor explants only when endothelial cells were present, suggesting a critical role for Notch not only in tumor cells but within the entire microenvironment [28]. Consistently, the use of Notch inhibitors in an orthotopic glioma model slowed tumor growth and prolonged survival by decreasing the number of the CD133+ CSCs [30-31].
Also in breast cancer, inhibition of Notch decreased breast CSCs (ESA+/CD44+/CD24low) activity and reduced tumor initiation in vivo [32]. Notch inhibition after radiotherapy prevented up-regulation of radiation-induced expression of Notch2, Notch3, Dll1, Dll3, Jag1 and was associated with a reduction in breast CSCs [33]. Radiation resistance in breast CSC has also been associated with lower levels of DNA damaging reactive oxygen species (ROS) due to increased production of free radical scavengers such as of glutathione [34]. Although a role for Notch signaling in regulating ROS in CSCs has not yet been reported, Notch inhibition in endothelial cells has been shown to increase ROS generation, proliferation, migration and adhesion, suggesting that increased ROS production upon Notch inhibition after radiotherapy could also reduce the number of breast CSCs in a non-cell autonomous manner. Also, in airway basal stem cells ROS regulates self-renewal in a Notch dependent manner [35], yet a direct relationship with the response to radiation and Notch has not been established.
In non-small cell lung cancer (NSCLC), cells with stem cell properties have also been shown to be dependent on Notch activity. These cells are more treatment resistant and tumorigenic in vivo, whereas GSI-treated xenografts failed to regenerate tumors upon re-implantation in suitable hosts [26]. A similar role for Notch in the maintenance and renewal of colon cancer initiating cells has been described [27]. Although in these studies, the direct role of Notch in response to radiation has not been investigated, the data suggest that Notch inhibition in these malignancies may result in improved tumor radiation sensitivity by inhibiting the viability of the cancer initiating cells.
c) Cross-talk with other signaling pathways
Cancers are driven by the interaction of multiple signaling pathways. Inhibiting an individual pathway will thus almost never be sufficient to cure cancer. Even in tumors that are “oncogene addicted” (referring to the dependency of some cancers on one or few genes for proliferation and survival) [36], targeting the specific genes that are critical for maintenance of the malignant phenotype will eventually result in tumor recurrence due to emergence of therapy-resistance [37]. The effect of Notch signaling on radiation response most likely also occurs through cross-talk with other signaling pathways. In glioma, Notch activity increased the radiation resistance of glioma CSCs by activating the Akt pathway. Notch blockade prior to irradiation was shown to inhibit Akt activation, an effect that was rescued by ectopic expression of the active form of Notch1 and Notch2 [29]. Likewise, in glioma spheres, Notch inhibition significantly decreased Akt and STAT3 phosphorylation and reduced survival of glioma CSCs [23]. These studies suggest that Notch can promote radiation resistance by activating Akt signaling.
In NSCLC, one of the most important overexpressed cellular targets is epidermal growth factor receptor (EGFR). Increased EGFR expression has been associated with radiation resistance [38] and combination of radiation with EGFR inhibition have yielded in relatively small but statistically significant radiosensitizing effects [39]. Others have shown that pharmacological inhibition of EGFR using erlotinib increased the stem like-cells (ALDH+) in EGFR-mutated NSCLC cell lines and that Notch transcriptional activity was increased in these cells. Strikingly, Notch inhibition eliminated the ALDH+ population, an effect attributed specifically to Notch3-dependent signaling [40]. As stem-like cells have been shown to contribute to radioresistance [41], combined EGFR/Notch targeting in lung cancer cells bearing activating mutations in EGFR could offer a very powerful approach to reduce the radiation resistant populations.
K-RAS is one of the most commonly mutated oncogenes in human cancer. Activated RAS oncogene was shown to increase radiation resistance in human cells [45]. Notch has been demonstrated to cooperate with the RAS pathway to promote carcinogenesis in various tumor types. For example, Notch1 activity was shown to be upregulated in RAS-transformed cells. Genetic or pharmacological down-regulation of Notch signaling was sufficient to abolish the RAS-induced neoplastic phenotype including proliferation and anchorage-independent growth in vitro and in vivo [46]. Taking into account the role of oncogenic K-RAS in radiation resistance, these data support a rational for targeting both pathways. However, caution should be taken and application of such an approach not generalized. Indeed, in a K-RAS driven NSCLC mouse model opposing tumorigenic functions of Notch1 and Notch2 were reported. In these mice, Notch1 ablation resulted in decreased levels of the Notch target gene expression and of pERK1/2, resulting in reduced tumor formation, while Notch2 ablation showed an increase in HES1 expression and resulted in increased carcinogenesis [47].
Several reports describe a direct effect of Notch signaling on the cell cycle. This may be exploited in the context of fractionated radiation, as it is well established that cells in different phases of the cell cycle exhibit different radiation sensitivity. Notch can directly induce cyclin D1 and cyclin-dependent kinase2 activity [48-49] and in breast epithelial cells Notch promotes transformation by inducing cyclin D1 [50]. c-Myc, an oncogene and potent driver of cell cycle entry, is a direct target of Notch and essential for cell cycle progression in T-ALL [51-52] and mouse mammary tumors [53].
d) Regulating EMT
There is mounting evidence showing that Notch signaling contributes to the acquisition of the EMT phenotype, for instance by up-regulating Snail and Slug, both transcriptional repressors of E-cadherin [54-55]. During EMT, epithelial cells undergo a morphological change resulting in increased motility, invasion and stemness [56] a process associated with chemo- and radiation therapy resistance [57-60]. For example, radioresistant NSCLC have been shown to share many phenotypical properties with cells that have undergone EMT [61]. Notch1 signaling was shown to enhance the EMT process in EGFR inhibitor resistant lung cancer cells [62]. In lung adenocarcinoma, a population of metastasis-prone cells with significantly enriched expression of Notch receptors and ligands that drive EMT were identified [63]. While the majority of the metastatic lung cancer cells were shown to be radioresistant [64], Notch targeting suggests a role in increasing radiation sensitivity by inhibiting genes involved in the EMT process.
In NSCLC, c-MET amplification is shown to direct invasion and metastasis [65]. Others have shown that co-expression of c-MET and Notch1 induces EMT in NSCLC patients and promotes invasion [66], likely due to their interaction by cross-talk. Given the role of Notch and c-MET expression in poor radiation response [67] as well as direct interaction between c-MET and Notch [68], Notch blocking in combination with c-MET targeted therapy could be critical to inhibit the aggressive behavior of NSCLCs and increase the radiation sensitivity.
The association between EMT and radioresistance and the prominent role of Notch signaling as driving force in the EMT process, suggest that Notch inhibition will result in radiosensitization of tumors that underwent EMT.
Cross-talk with microRNAs
Increasing evidence implicates microRNAs (miRNA) in the regulation of drug and radiation resistance [69-73]. The most extensively studied miRNA in the context of Notch signaling and cancer is the tumor suppressive miR-34. In glioma, miR-34 was shown to inhibit tumor growth in vivo by down-regulating Notch1 and Notch2 expression [73]. Similarly, in colorectal cancer, high miR-34a levels inhibited colon CSCs self-renewal in vitro as well as xenograft tumor formation by suppressing Notch signaling whereas low miR-34a levels up-regulated Notch and promoted a CSC phenotype [74]. Also in NSCLC the expression of miR-34 was reported to be low [75] and its ectopic expression enhanced the radiation sensitivity of lung cancer cells [76]. Kang et al. showed that this effect is Notch1-dependent and demonstrated that miR-34 induced Notch1 downregulation thereby promoting apoptosis resulting in a radiosensitizing effect [77]. Similarly, in P53-deficient gastric and pancreatic cancer cells, restoration of miR-34 reduced the expression of Notch pathway members and was associated with reduced in vivo tumor formation as well as increased treatment sensitization [78-79]. Overall, these studies provide insight on the role of miR-34 in radiation resistance, partly mediated via regulation of Notch.
Extrinsic resistance
a) Angiogenesis
Notch signaling is important in pro-angiogenic role in tumor vasculature. Endothelial cells (ECs) express several Notch receptors (Notch1, 4) and ligands (Delta-like 1, 4 and Jagged1). VEGF acts as a proliferative driver of angiogenesis, while Dll4/Notch signaling helps to control vessel sprouting and branching [80-81]. Tumor vasculature is abnormal, and the endothelial cells of tumor blood vessels are different from those of normal vasculature [82]. Consequently, increased tumor angiogenesis, as indicated by increased microvessel density or by increased VEGF expression, does not necessarily correlate with increased blood flow and oxygen availability. This situation, together with the existence of heterogeneous hypoxic regions within tumors results into reduced response to radiation therapy. Based on the crucial role of Dll4/Notch signaling in the vascular sprouting and tumor angiogenesis, pharmacological targeting of the Dll4/Notch has been shown to be effective as a novel anti-angiogenic therapy by blocking non-productive vessel growth and tumor collapse [83-85]. Adding chemotherapeutic agents to the Dll4 cocktail inhibited the induction of anti-apoptotic genes and resulted in further additive anti-tumor activity by decreasing the CSC population. Also when combined with ionizing radiation, Dll4 blockade impaired the tumor growth by promoting non-functional tumor angiogenesis [86]. As it is the case for all anti-angiogenic treatment, the temporal relationship with radiation is crucial [87] and therefore the optimal order and timing for administration of the radiation/anti-Dll4 combination in the context of vascular normalization requires careful attention (see below).
b) Hypoxia
Hypoxia is a common feature of human tumors and is associated with increased malignancy and resistance to chemo- and radiotherapy [88]. Hypoxic cells are 2-3 fold less sensitive to the effects of radiation because they lack the oxygen radicals that contribute to irreversible DNA damage [89]. Pre-treatment oxygenation of tumors is prognostic and predictive for radiotherapy response in head and neck squamous carcinoma and many other tumors [90-91]. Previously, we have shown that NSCLC xenografts expressing a constitutively active Notch1 were more resistant to single dose radiation therapy. Tumors with high constitutive Notch activity proliferated faster and consistently had a higher hypoxic fraction [92]. This was accompanied by increased vessel density while the total number of perfused vessels remained similar to control tumor cells pointing towards an increase in non-functional vasculature (unpublished data). These data suggest that Notch signaling may increase the survival of hypoxic cells and thereby influence the response to radiotherapy.
The link between hypoxia and Notch signaling has been described in various studies. Notch has been reported to activate the hypoxia response pathway through HES1-induced STAT3 phosphorylation resulting in transcriptional up-regulation of HIF1-α and its target genes [93]. Interestingly, hypoxia in turn also elevates Notch activity [94-95] both via HIF1-α dependent [94] and HIF1-α independent pathways [96]. Especially the latter is intriguing, as it was shown that hypoxia-induced Notch signaling contributed to increased proliferation, maintenance of stem cell properties and suppression of senescence via a metabolic shift to glycolysis [96]. Glycolysis would not only provide a growth advantage for the fast proliferating cells but is also involved in cellular immortalization via reduction of intrinsic ROS [97-99]. As such, the metabolic effects of Notch on glycolysis may be indirectly responsible for increasing survival and radiation resistance by suppressing ROS.
Although many of these oncogenic pathways are found to cross-talk with Notch at some level, these interactions are cell type and context dependent. Thus, the direct effect of Notch blockade on cancer cells may vary.
Strategies to target Notch and its potential risks
In most cases, deregulation of Notch has oncogenic effects. The first evidence for the involvement of Notch in cancer was the detection of a rearrangement between the intracellular part of Notch1 (NICD1) and the T-cell receptor beta (TRB) leading to high-level expression of truncated and constitutively active Notch1 in T-ALL [100-101]. Mutations and chromosomal rearrangements have also been reported in splenic marginal zone B-cell lymphoma [102-103] and in triple-negative breast cancer cells [104]. Notch has also been shown to act as oncogene in lung [26, 92], colon [105], melanoma [106], pancreatic [107], glioma [31, 108], head and neck [109] and many other cancer types.
In other cases, Notch functions as a tumor suppressor as shown in skin tumors [110-111], bladder cancer [112], squamous cell lung carcinoma [113], squamous cell carcinoma in cutaneous and head-and-neck tumors [114-117] and potentially also in small cell lung carcinoma (SCLC), which is a neuroendocrine subtype of lung cancer [118].
This indicates that the outcome for aberrant Notch activity is highly context-dependent. Notch inhibitors are being investigated in clinical studies to treat these malignancies where Notch acts as an oncogene whereas methodologies activating the Notch pathway may have therapeutic potential in those cancers where Notch suppresses tumor growth, although any experimental evidence for the latter is still lacking.
We will first provide an overview of the different possibilities to inhibit the Notch pathway (Figure 1) and discuss how these may interact with radiation in a subsequent section. In canonical Notch signaling, Notch receptors are subjected to a series of sequential proteolytic cleavages. The site1 (S1) cleavage is controlled by a Furin convertase and is responsible for the receptor maturation. Therefore, it is possible to interfere with Notch maturation in Golgi by using Furin inhibitors [119-120]. After receptor maturation, the receptor is transported to the cell’s surface, a process that can be blocked by using an inhibitor of calcium transporter Atp2a3/SERCA [121]. Following surface expression, receptor activation is mediated by binding to ligand on adjacent cells. Notch-ligand interactions can be blocked using soluble versions of the receptor that function as decoy [122-123] or by blocking the ligand-induced conformational changes in the Notch receptor [124-126]. Upon receptor-ligand interaction, mammalian Notch receptors are cleaved by the disintegrin metalloprotease ADAM10 at site 2 (S2) [127-128] leading to shedding of the large Notch ectodomain (NECD). This cleavage is followed by a S3-cleavage caused by a γ-secretase complex and results in the release of the cytoplasmic NICD, which subsequently translocates to the nucleus where it binds to the DNA binding protein CSL (CBF1/Suppressor of Hairless/Lag-1; also known as Rbp-j) and the co-activator Mastermind-like (MAML) to induce expression of target genes [128-129]. The S2 cleavage can be inhibited by blocking ADAM proteases [128, 130-132] and S3 cleavage by γ-secretase inhibitors (GSI).
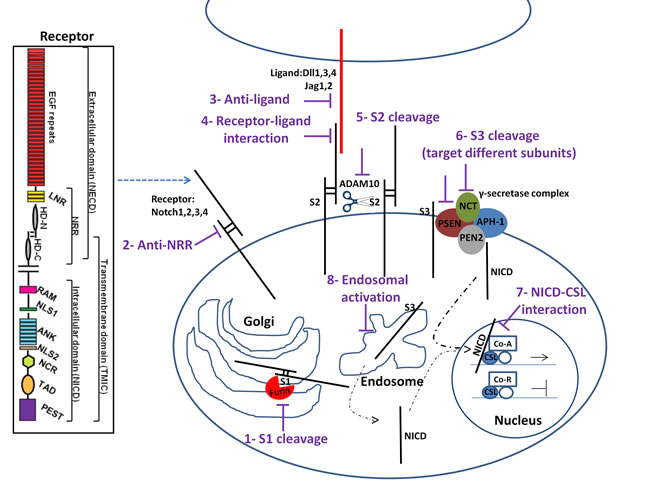
Figure 1: Notch signaling pathway and potential drug intervention sites (see text for details). 1) Furin cleavage at S1 site can be inhibited. 2) Notch antibodies targeting Notch receptors 3) or ligands would target individual receptor pathway 4) Targeting the interaction of Notch receptor with ligand after receptor maturation abrogates the pathway activity 5) Cleavage by a disintegrin and metalloproteinase Adam10 at S2 site and 6) γ-secretase complex at S3 site can be inhibited to limit Notch signaling. 7) Interfering with NICD/CSL interaction using small peptides disrupts the canonical Notch pathway signaling. 8) Inhibition of endosomal Notch trafficking could potentially reduce Notch signaling activity regardless of ligand activity. The Notch receptor is comprised of a Notch extracellular domain (NECD) and Notch intracellular domain (NICD). EGFR: epidermal growth factor repeats; HD: heterodimerization domain; NRR: negative regulatory region; LNR: cysteine-rich LNR repeats; RAM: RAM domain; NLS: nuclear localization signals; ANK: ankyrin repeat domain; NCR: cysteine response region; TAD: transactivation domain; PEST: region rich in proline (P), glutamine (E), serine (S) and threonine (T) residues.
A detailed overview of ways to intervene with Notch signaling in various disease has been described elsewhere [17]. Here, we focus on different strategies to target Notch specifically in cancer.
1- GSIs: GSIs are pan-Notch inhibitors used in both pre-clinical and clinical settings. They target γ-secretase cleavage of NOTCH by presenilin, a rate-limiting step in the Notch activation cascade. Use of GSI is however hampered by the dose-limiting toxicity in the gut as Notch inhibitors promote goblet cell metaplasia leading to severe diarrhea in animals and humans [133-135]. Intermittent dosing schedules [136-138], glucocorticoid administration or anti-estrogen therapy [139] have been shown to mitigate the adverse effects while maintaining GSI’s anti-tumor efficacy [140-141]. Obviously, most drugs used in oncology, including targeted agents and immunotherapeutics have significant acute and chronic toxicities, and in every situation the ratio between risks and benefits must be carefully weighted. Cyclical (intermittent) dosing, dose adjustments and patient stratification are therefore important to minimize the toxicities of chemo- and radiation therapy [139, 142-146].
While GSI’s are potent inhibitors of the Notch signaling pathway, they are not designed to be receptor-specific and they target all four Notch isoforms that within the same cell type can have either tumor promoting or tumor suppressive roles. Thus, there is a need for receptor specific antagonists.
2- Notch receptor specific targeting: Monoclonal antibodies have been developed that target the negative regulatory region (NRR) of Notch1, Notch2 [125] and Notch2/3 [126, 147] and act by keeping the receptor in an unresponsive “closed” confirmation or by blocking receptor-ligand interactions through hindering EGF repeats required for binding [148]. These antibodies are able to target cancers by inhibiting simultaneously cancer cell growth and by disrupting tumor angiogenesis that depends on DLL4/Notch1 signaling.
3- Notch ligand targeting: To interfere with tumor angiogenesis, Notch ligands are important targets. Along this line, Dll4 blocking antibodies have been used to suppress tumor vascularization and tumor growth [83, 149]. Data from a Phase1 clinical trial showed that Demsizumab (anti-Dll4) suppressed tumor vascularization, was well tolerated and resulted in reduced tumor size [150-151]. In animals, the long-term use of these antibodies, however caused marked histopathological changes in liver endothelial cells and induced vascular tumors [152]. While Dll4 mainly has a function in the vasculature, Jagged1 is important in immunosuppressive T regulatory cells and promotes the maintenance or expansion of hematopoietic precursor cells [153] as well as tumor/stem cells [154-155]. Targeting Jagged1 in stroma and tumor cells can thus result in synergistic effects as demonstrated in ovarian cancer [54, 156].
Alternatively, activating Notch signaling could be a way to inhibit angiogenesis. While overexpression of Dll4 was shown to promote tumor growth, overexpression of a soluble DSL domain of Dll1 resulted in reduced tumor growth by attenuating vascularization [157]. Therefore, it is important to further explore the differential activities of the Notch ligands on both stroma and tumor cells.
Recent work has shown that the Notch pathway can also be used non-canonically [158] and that Notch proteins can become activated in a DSL-independent manner as shown in various cancers including melanoma [159] and T-ALL [160]. Breast cancer stem cell expansion has also been shown to be dependent on a ligand-independent Notch activation mechanism [161]. Taking into account the radioresistant phenotype of breast CSC, targeting Notch ligands in such case may be suboptimal, and other strategies to target Notch cleavage or downstream events may be more effective.
4- Alternatives: Cleavage of Notch proteins by ADAM metalloproteases is a rate-limiting step preceding γ-secretase cleavage. Specifically, metalloproteases ADAM 10 and 17 have been implicated in ligand dependent and independent signaling, respectively [127, 162-163]. ADAM metalloproteases bind many signaling molecules and receptors, including TNFα and EGF receptors and thus unless specifically targeted to tumors are likely to yield dose-limiting toxicities in normal tissues. Moreover, we reported Notch cleavage and transcriptional activity of oncogenic Notch1 signaling in cells treated with both broad-spectrum metalloprotease inhibitors as well as specific ADAM17/10 hydroxamate inhibitors [128], suggesting the involvement of unknown proteases engaged in the activation of oncogenic Notch1. While this hypothesis requires further validation, it may open the possibility of targeting disease-specific Notch proteases while leaving normal Notch signaling intact.
The main components of the γ-secretase complex are presenilin, APH-1, Pen-2, and Nicastrin. Currently used GSIs inhibit the catalytic activity of presenilin and lead to off-target effects on the wide range of γ-secretase complex substrates. [164]. Targeting Nicastrin as the key component of the γ-secretase complex using neutralizing antibodies reduced proliferation of cancer cells [165] and resulted in anti-tumor and anti-metastatic effects in a model of triple negative breast cancer [166].
γ-secretase cleavage and activity not only occurs at the cell surface but also in the acidic environment of the endosomes and lysosomes [167-168] where the activity of vacuolar ATPase (V-ATPase) regulates acidification of endocytic compartments necessary for Notch signaling activation [23, 169]. Endocytic trafficking is also essential for Notch ligand internalization and promoting Notch activation [170-171]. Pharmacologic inhibition of V-ATPase decreases Notch signaling activity [172] and pretreatment with V-ATPase inhibitors can sensitize solid human tumors to chemotherapy drugs and might also a be a good strategy for radiosensitization [173].
Finally, hydrocarbon-stapled peptides that mimic a dominant negative fragment of Notch-CSL-mastermind-like (dnMAML) and that prevent binding of full-length MAML to NICD/CSL have been developed. Unlike GSIs, these peptides have shown a reduced gastrointestinal toxicity in treated animals [174].
Treatment scheduling and personalized treatment
While numerous clinical trials using various Notch inhibitors were ongoing, several of these trials have stopped due to dose-limiting toxicity and lack of efficacy. Combining Notch inhibition with (chemo) radiation can only be successful if these hurdles can be overcome. Appropriate treatment scheduling and patient selection will be key to achieve this goal.
a) Treatment scheduling
One of the most important aspects that have been understudied is the scheduling for Notch inhibitors in conjunction with other treatments. For example, it has been reported that Notch inhibition caused hypersprouting of non-functional vasculature resulting in decreased tumor growth [175-176]. Impaired angiogenesis has also in other studies been shown to reduce tumor growth, yet at the same time, these tumors became strongly hypoxic [177]. Thus, administration of a Notch inhibitor in patients before radiotherapy may induce hypoxia and contribute to a more malignant phenotype and radio- and chemotherapy resistance [88].
It has also been shown that irradiation can induce Notch expression and activity and promote stem cell like characteristics [7, 33, 178-180]. Therefore, it may be critically important to continue Notch inhibition after radiotherapy. Significantly enhanced radiation-mediated tumor cytotoxicity has indeed been demonstrated upon treatment with GSI following irradiation in lung xenografts [178]. A similar study in glioma determined that Notch inhibition before temozolomide administration diminished the efficacy of chemotherapy while Notch inhibition after chemotherapy strongly inhibited tumor formation [181]. The reason for this could likely be due to the induced Notch activity after chemotherapy treatment [47]. More data are clearly needed to determine the most appropriate treatment schedule and such results will be invaluable for translation into the clinic with the aim to improve outcome. In colorectal cancer patients, sequence of the drug treatment was shown to be more important and effective than the drug exposure itself likely due to enhancing the subsequent treatment [182].
It will also be of utmost importance to determine the interaction between Notch inhibition, chemotherapy and fractionated radiation. Recovery from radiation injury and tumor cell repopulation between fractions reduces tumor control, while reoxygenation of hypoxic cells and redistribution of cells into a more radiosensitive phase increases tumor control [183-184]. In addition, the effect on the therapeutic ratio when Notch inhibition will be combined with alternative fractionation schedules such as accelerated (decreasing the overall treatment time) or hypofractionated (lower number of fractions with a higher dose per fraction) treatment needs careful attention as such alternative fractionation schemes have been shown to improve tumor control [185-186].
Alternatively, increased radiotherapy effectiveness can potentially also be achieved by properly scheduling in combination with angiostatic drugs such as anti-Dll4 antibodies through “vascular normalization”. This concept proposes that in order to have an effect when using anti-angiogenesis drugs, an equilibrium between pro- and anti-angiogenic factors in the tumor microenvironment rather than complete angiogenesis inhibition is needed. Dysfunctional vasculatures become then more normal, hence tumor oxygenation and perfusion will be improved thereby increasing the efficacy of administered drugs and/or radiation [187]. Defining the optimal time point during radiotherapy at which anti-angiogenesis drugs such as Notch inhibitors should be administered will be crucial [188].
Table 1: Possible sites to intervene in Notch pathway
Intervening Notch at various sites |
Mechanism of action |
Pre-clinical studies |
Clinical studies |
S1 cleavage |
- Inhibition of Furin and block receptor maturation |
- Not reported (experimental studies available) |
- Not Reported |
Receptor -Anti Notch1,Notch2, Notch3 -Anti-Notch4 |
- Unresponsive receptor to ligand binding by targeting NRR in case of Notch1,2 and 3 - Blocking receptor–ligand interactions by hindering EGF repeats required for binding |
- Anti-Notch1, Notch2 [125, 202] - Anti-Notch3 [147] - Anti-Notch4 [203] |
- Anti-Notch2, Notch3 [138] - Anti-Notch1 [204] |
Ligand -Anti-Dll4 -Anti-Jagged1 |
- Non-functional vasculature - Abrogation of angiogenesis, targeting CSCs, targeting EMT and inhibiting the immunosuppressive T- regulatory cells |
- [83] |
- [151] - Not reported |
Receptor-Ligand Interaction -Notch1 receptor decoy -Dll1 and Jag1 ligand decoy |
- Ligand dependent Notch antagonist |
- [122] |
- Not reported |
S2 cleavage (Adam metalloproteases) |
- Targeting both Adam metalloprotease 10/17 and block ectodomain shedding |
- [131] |
|
S3 cleavage (γ-secretase complex) |
- Inhibition of different subunits of γ-secretase complex and block NICD release |
- [208] |
- [209] |
NICD-CSL interaction |
- Suppressing transcriptional activation by preventing binding of MAML1 to the ICN–CSL complex |
- Not reported |
|
Endosomal activation |
- Disrupting γ-secretase cleavage in acidic endosome - Inhibition of V-ATPase |
- [211] - [173] |
- Not Reported |
b) Genetic profile of cancer types and signatures
The different classes of gene expression profiles, reflecting the consequences of different sets of oncogenic mutations, correlate with different prognoses and different responses to therapy. Therefore, cancer cells vary widely in their response to radiation therapy [189] as well as Notch targeted therapies reflecting their particular genetic profile. For example, estrogen receptor negative (ERα-) breast cancer cells have higher Notch activity and respond better to Notch inhibition. In ERα+ cells when estrogen is deprived or upon anti-estrogen-treatment, breast cancer stem cells are selectively enriched and Notch-4 activity increased [190-191]. Combination of a Notch inhibitor with an anti-estrogen could therefore be a promising therapeutic strategy in ERα+ breast cancer cells. In both ERα- and ERα+ tumors radiosensitivity is expected to increase, as Notch inhibition will specifically target the more radioresistant stem cell compartment. Yet, there are currently no valid predictive factors that reliably identify patients who would greatly benefit from radiation treatment. In triple-negative breast cancer (TNBC) patients, approximately 19.5% carry BRCA mutations [192]. These mutation carriers are defective in DNA repair; therefore, it would be expected that these tumors might exhibit sensitivity rather than insensitivity to radiation therapy. One possible explanation for this response is that these tumors might possess compensatory DNA repair mechanisms that are more effective at dealing with radiation-induced DNA damage. In this regard, identification of a marker in TNBC cells would be invaluable in identifying potential radiosensitizing agents. Gene expression profiling analysis performed on these tumors revealed that oncogenic PEST domain mutations in Notch1, 2 and 3 receptors occur in ~13% of TNBC cells conferring GSI sensitivity [104] and provides a strong rationale for a Notch-driven personalized medicine strategy. Notch4, but not Notch1-3 was shown to contribute to the induction of proliferation, tumorigenesis and invasiveness in TNBC cells and its inhibition was shown to suppress tumorigenicity and tumor volume [193-194]. Furthermore, mutations of Notch receptors resulting in an active Notch pathway are frequent in TNBC conferring GSI sensitivity [104]. Notch targeting can thus be a potential therapeutic target for the radiosensitization of TNBC cells.
In skin squamous cell carcinomas (SCCs), EGFR signaling plays a significant role in suppressing differentiation through negative regulation of Notch1 gene expression and activity [195]. Especially for large skin SCC and at sites where surgery is not an option, radiation is often used as first-line treatment. Notch blockade counteracts the differentiation-inducing effects of EGFR inhibitors, while at the same time, synergizes with these compounds in induction of apoptosis. This indicates an attractive combination therapy that may enhance the potency of EGFR inhibitory agent. This study provides a mechanistic explanation for the Notch loss-of-function mutations found in squamous skin carcinomas [113]. Squamous tumors without such mutations may thus be sensitive to Notch inhibitors, and treatment efficacy enhanced especially for indications involving radiotherapy.
In NSCLC patients, Dll4/Notch1 signaling was reported to negatively influence NSCLC growth via PTEN up-regulation [97]. This indicates that the therapeutic application of a Notch inhibitor could be adversely affected in different categories of lung cancer [196]. Notch inhibition could be specifically beneficial in lung cancers with inactive PTEN [197]. In contrast, in glioma, loss of PTEN has been reported as a critical event that leads to Notch inhibitor resistance by transferring the “oncogene addiction” from the Notch to the PI3K/AKT pathway [198], supporting the regulatory link between Notch and the PTEN/PI3K/AKT pathway. Therefore, attenuation of cell growth using a combination of Notch inhibition and PI3K inhibitors in PTEN mutant glioma CSCs may lead to increased treatment efficacy [199]. As glioma stem cells promote radioresistance by preferential activation of the DNA damage response [8] and Notch has been shown to enhance radiation resitance in glioma [29], the combination of Notch inhibitors with radiation can be expected to yield beneficial outcomes in these patients.
These data and similar other data arising from genomic, transcriptional and proteomic analysis in glioma [31] or breast cancer [200] exemplify how understanding the molecular signatures that could predict the therapeutic response allow identification of a subset of patients who are likely to benefit from the Notch inhibition/radiotherapy combination therapies.
Patient selection could also be performed based on determination of activated (i.e. cleaved) Notch proteins levels or target genes as shown in TNBC [201], indicating their potential as prognostic biomarker to identify TNBC patients who are most likely to respond to anti-Notch based therapeutics. Likewise, adenoid cystic carcinoma (ACC) tumor xenografts with activating Notch1 mutations responded to Notch inhibition, whereas the tumors without Notch1 mutation and low levels of NICD1 were resistant [201]. Therefore, establishing an association between the drug responses and molecular subclasses of the specific cancer type may help to identify potential cohorts of patients for targeted therapy and to be treated in combination with radiotherapy.
Taken together, while Notch deregulation is frequent in cancers, the failure of clinical trials using Notch inhibitors may be explained by our incomplete understanding of the unique and redundant functions of the Notch receptors and our inability to select the correct patients and lack of knowledge on the correct timing of intervention. However, it appears that Notch signaling plays a key role in tumor initiation, progression and treatment response and that combining Notch therapeutics with radiotherapy may lead to synergistic improvements. More basic and translational research is needed to address these issues prior to conducting clinical trials. Only then can we expect to see therapeutic profit from Notch inhibitors on cancer response.
Acknowledgments
Supported by the EU, ERC-Consolidator Grant (617060).
Conflicts of interest
None.
References
1. Milas L and Hittelman WN. Cancer stem cells and tumor response to therapy: current problems and future prospects. Semin Radiat Oncol. 2009; 19(2):96-105.
2. Marusyk A and Polyak K. Tumor heterogeneity: causes and consequences. Biochim Biophys Acta. 2010; 1805(1):105-117.
3. Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, McDonald NQ, Butler A, Jones D, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012; 366(10):883-892.
4. Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, Visvader J, Weissman IL and Wahl GM. Cancer stem cells—perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006; 66(19):9339-9344.
5. Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, Minden M, Paterson B, Caligiuri MA and Dick JE. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994; 367(6464):645-648.
6. Woodward WA, Chen MS, Behbod F, Alfaro MP, Buchholz TA and Rosen JM. WNT/beta-catenin mediates radiation resistance of mouse mammary progenitor cells. Proc Natl Acad Sci U S A. 2007; 104(2):618-623.
7. Phillips TM, McBride WH and Pajonk F. The response of CD24(-/low)/CD44+ breast cancer-initiating cells to radiation. J Natl Cancer Inst. 2006; 98(24):1777-1785.
8. Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD and Rich JN. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006; 444(7120):756-760.
9. Baumann M, Krause M and Hill R. Exploring the role of cancer stem cells in radioresistance. Nat Rev Cancer. 2008; 8(7):545-554.
10. Pajonk F, Vlashi E and McBride WH. Radiation resistance of cancer stem cells: the 4 R’s of radiobiology revisited. Stem Cells. 2010; 28(4):639-648.
11. Korkaya H and Wicha MS. Selective targeting of cancer stem cells: a new concept in cancer therapeutics. BioDrugs. 2007; 21(5):299-310.
12. Koch U, Lehal R and Radtke F. Stem cells living with a Notch. Development. 2013; 140(4):689-704.
13. Ranganathan P, Weaver KL and Capobianco AJ. Notch signalling in solid tumours: a little bit of everything but not all the time. Nat Rev Cancer. 2011; 11(5):338-351.
14. Joshi I, Minter LM, Telfer J, Demarest RM, Capobianco AJ, Aster JC, Sicinski P, Fauq A, Golde TE and Osborne BA. Notch signaling mediates G1/S cell-cycle progression in T cells via cyclin D3 and its dependent kinases. Blood. 2009; 113(8):1689-1698.
15. Ross DA, Rao PK and Kadesch T. Dual roles for the Notch target gene Hes-1 in the differentiation of 3T3-L1 preadipocytes. Mol Cell Biol. 2004; 24(8):3505-3513.
16. VanDussen KL, Carulli AJ, Keeley TM, Patel SR, Puthoff BJ, Magness ST, Tran IT, Maillard I, Siebel C, Kolterud A, Grosse AS, Gumucio DL, Ernst SA, Tsai YH, Dempsey PJ and Samuelson LC. Notch signaling modulates proliferation and differentiation of intestinal crypt base columnar stem cells. Development. 2012; 139(3):488-497.
17. Andersson ER and Lendahl U. Therapeutic modulation of Notch signalling—are we there yet? Nat Rev Drug Discov. 2014; 13(5):357-378.
18. Vermezovic J, Adamowicz M, Santarpia L, Rustighi A, Forcato M, Lucano C, Massimiliano L, Costanzo V, Bicciato S, Del Sal G and d’Adda di Fagagna F. Notch is a direct negative regulator of the DNA-damage response. Nat Struct Mol Biol. 2015; 22(5):417-424.
19. Deng X, Michaelson D, Tchieu J, Cheng J, Rothenstein D, Feldman R, Lee SG, Fuller J, Haimovitz-Friedman A, Studer L, Powell S, Fuks Z, Hubbard EJ and Kolesnick R. Targeting Homologous Recombination in Notch-Driven C. elegans Stem Cell and Human Tumors. PLoS One. 2015; 10(6):e0127862.
20. Giambra V, Jenkins CR, Wang H, Lam SH, Shevchuk OO, Nemirovsky O, Wai C, Gusscott S, Chiang MY, Aster JC, Humphries RK, Eaves C and Weng AP. NOTCH1 promotes T cell leukemia-initiating activity by RUNX-mediated regulation of PKC-theta and reactive oxygen species. Nat Med. 2012; 18(11):1693-1698.
21. Tatarek J, Cullion K, Ashworth T, Gerstein R, Aster JC and Kelliher MA. Notch1 inhibition targets the leukemia-initiating cells in a Tal1/Lmo2 mouse model of T-ALL. Blood. 2011; 118(6):1579-1590.
22. Armstrong F, Brunet de la Grange P, Gerby B, Rouyez MC, Calvo J, Fontenay M, Boissel N, Dombret H, Baruchel A, Landman-Parker J, Romeo PH, Ballerini P and Pflumio F. NOTCH is a key regulator of human T-cell acute leukemia initiating cell activity. Blood. 2009; 113(8):1730-1740.
23. Fan X, Khaki L, Zhu TS, Soules ME, Talsma CE, Gul N, Koh C, Zhang J, Li YM, Maciaczyk J, Nikkhah G, Dimeco F, Piccirillo S, Vescovi AL and Eberhart CG. NOTCH pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells. 2010; 28(1):5-16.
24. Fan X, Matsui W, Khaki L, Stearns D, Chun J, Li YM and Eberhart CG. Notch pathway inhibition depletes stem-like cells and blocks engraftment in embryonal brain tumors. Cancer Res. 2006; 66(15):7445-7452.
25. Ercan C, van Diest PJ and Vooijs M. Mammary development and breast cancer: the role of stem cells. Curr Mol Med. 2011; 11(4):270-285.
26. Hassan KA, Wang L, Korkaya H, Chen G, Maillard I, Beer DG, Kalemkerian GP and Wicha MS. Notch pathway activity identifies cells with cancer stem cell-like properties and correlates with worse survival in lung adenocarcinoma. Clin Cancer Res. 2013; 19(8):1972-1980.
27. Sikandar SS, Pate KT, Anderson S, Dizon D, Edwards RA, Waterman ML and Lipkin SM. NOTCH signaling is required for formation and self-renewal of tumor-initiating cells and for repression of secretory cell differentiation in colon cancer. Cancer Res. 2010; 70(4):1469-1478.
28. Hovinga KE, Shimizu F, Wang R, Panagiotakos G, Van Der Heijden M, Moayedpardazi H, Correia AS, Soulet D, Major T, Menon J and Tabar V. Inhibition of notch signaling in glioblastoma targets cancer stem cells via an endothelial cell intermediate. Stem Cells. 2010; 28(6):1019-1029.
29. Wang J, Wakeman TP, Lathia JD, Hjelmeland AB, Wang XF, White RR, Rich JN and Sullenger BA. Notch promotes radioresistance of glioma stem cells. Stem Cells. 2010; 28(1):17-28.
30. Chu Q, Orr BA, Semenkow S, Bar EE and Eberhart CG. Prolonged inhibition of glioblastoma xenograft initiation and clonogenic growth following in vivo Notch blockade. Clin Cancer Res. 2013; 19(12):3224-3233.
31. Saito N, Fu J, Zheng S, Yao J, Wang S, Liu DD, Yuan Y, Sulman EP, Lang FF, Colman H, Verhaak RG, Yung WK and Koul D. A high Notch pathway activation predicts response to gamma secretase inhibitors in proneural subtype of glioma tumor-initiating cells. Stem Cells. 2014; 32(1):301-312.
32. Harrison H, Farnie G, Howell SJ, Rock RE, Stylianou S, Brennan KR, Bundred NJ and Clarke RB. Regulation of breast cancer stem cell activity by signaling through the Notch4 receptor. Cancer research. 2010; 70(2):709-718.
33. Lagadec C, Vlashi E, Alhiyari Y, Phillips TM, Bochkur Dratver M and Pajonk F. Radiation-induced Notch signaling in breast cancer stem cells. Int J Radiat Oncol Biol Phys. 2013; 87(3):609-618.
34. Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, Qian D, Lam JS, Ailles LE, Wong M, Joshua B, Kaplan MJ, Wapnir I, Dirbas FM, Somlo G, Garberoglio C, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009; 458(7239):780-783.
35. Paul MK, Bisht B, Darmawan DO, Chiou R, Ha VL, Wallace WD, Chon AT, Hegab AE, Grogan T, Elashoff DA, Alva-Ornelas JA and Gomperts BN. Dynamic changes in intracellular ROS levels regulate airway basal stem cell homeostasis through Nrf2-dependent Notch signaling. Cell Stem Cell. 2014; 15(2):199-214.
36. Weinstein IB and Joe AK. Mechanisms of disease: Oncogene addiction—a rationale for molecular targeting in cancer therapy. Nat Clin Pract Oncol. 2006; 3(8):448-457.
37. Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG and Halmos B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005; 352(8):786-792.
38. Akimoto T, Hunter NR, Buchmiller L, Mason K, Ang KK and Milas L. Inverse relationship between epidermal growth factor receptor expression and radiocurability of murine carcinomas. Clin Cancer Res. 1999; 5(10):2884-2890.
39. Schutze C, Dorfler A, Eicheler W, Zips D, Hering S, Solca F, Baumann M and Krause M. Combination of EGFR/HER2 tyrosine kinase inhibition by BIBW 2992 and BIBW 2669 with irradiation in FaDu human squamous cell carcinoma. Strahlenther Onkol. 2007; 183(5):256-264.
40. Arasada RR, Amann JM, Rahman MA, Huppert SS and Carbone DP. EGFR Blockade Enriches for Lung Cancer Stem-like Cells through Notch3-Dependent Signaling. Cancer Res. 2014; 74(19):5572-5584.
41. Lundholm L, Haag P, Zong D, Juntti T, Mork B, Lewensohn R and Viktorsson K. Resistance to DNA-damaging treatment in non-small cell lung cancer tumor-initiating cells involves reduced DNA-PK/ATM activation and diminished cell cycle arrest. Cell Death Dis. 2013; 4:e478.
42. Chiang MY, Xu L, Shestova O, Histen G, L’Heureux S, Romany C, Childs ME, Gimotty PA, Aster JC and Pear WS. Leukemia-associated NOTCH1 alleles are weak tumor initiators but accelerate K-ras-initiated leukemia. J Clin Invest. 2008; 118(9):3181-3194.
43. Izrailit J, Berman HK, Datti A, Wrana JL and Reedijk M. High throughput kinase inhibitor screens reveal TRB3 and MAPK-ERK/TGFbeta pathways as fundamental Notch regulators in breast cancer. Proc Natl Acad Sci U S A. 2013; 110(5):1714-1719.
44. Liu ZJ, Xiao M, Balint K, Smalley KS, Brafford P, Qiu R, Pinnix CC, Li X and Herlyn M. Notch1 signaling promotes primary melanoma progression by activating mitogen-activated protein kinase/phosphatidylinositol 3-kinase-Akt pathways and up-regulating N-cadherin expression. Cancer Res. 2006; 66(8):4182-4190.
45. Bernhard EJ, Stanbridge EJ, Gupta S, Gupta AK, Soto D, Bakanauskas VJ, Cerniglia GJ, Muschel RJ and McKenna WG. Direct evidence for the contribution of activated N-ras and K-ras oncogenes to increased intrinsic radiation resistance in human tumor cell lines. Cancer Res. 2000; 60(23):6597-6600.
46. Weijzen S, Rizzo P, Braid M, Vaishnav R, Jonkheer SM, Zlobin A, Osborne BA, Gottipati S, Aster JC, Hahn WC, Rudolf M, Siziopikou K, Kast WM and Miele L. Activation of Notch-1 signaling maintains the neoplastic phenotype in human Ras-transformed cells. Nat Med. 2002; 8(9):979-986.
47. Baumgart A, Mazur PK, Anton M, Rudelius M, Schwamborn K, Feuchtinger A, Behnke K, Walch A, Braren R, Peschel C, Duyster J, Siveke JT and Dechow T. Opposing role of Notch1 and Notch2 in a Kras-driven murine non-small cell lung cancer model. Oncogene. 2014.
48. Ronchini C and Capobianco AJ. Induction of cyclin D1 transcription and CDK2 activity by Notch(ic): implication for cell cycle disruption in transformation by Notch(ic). Mol Cell Biol. 2001; 21(17):5925-5934.
49. Capobianco AJ, Zagouras P, Blaumueller CM, Artavanis-Tsakonas S and Bishop JM. Neoplastic transformation by truncated alleles of human NOTCH1/TAN1 and NOTCH2. Mol Cell Biol. 1997; 17(11):6265-6273.
50. Ling H, Sylvestre JR and Jolicoeur P. Notch1-induced mammary tumor development is cyclin D1-dependent and correlates with expansion of pre-malignant multipotent duct-limited progenitors. Oncogene. 2010; 29(32):4543-4554.
51. Weng AP, Millholland JM, Yashiro-Ohtani Y, Arcangeli ML, Lau A, Wai C, Del Bianco C, Rodriguez CG, Sai H, Tobias J, Li Y, Wolfe MS, Shachaf C, Felsher D, Blacklow SC, Pear WS, et al. c-Myc is an important direct target of Notch1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes Dev. 2006; 20(15):2096-2109.
52. Loosveld M, Castellano R, Gon S, Goubard A, Crouzet T, Pouyet L, Prebet T, Vey N, Nadel B, Collette Y and Payet-Bornet D. Therapeutic Targeting of c-Myc in T-Cell Acute Lymphoblastic Leukemia, T-ALL. Oncotarget. 2014; 5(10):3168-3172. doi: 10.18632/oncotarget.1873.
53. Sharma VM, Calvo JA, Draheim KM, Cunningham LA, Hermance N, Beverly L, Krishnamoorthy V, Bhasin M, Capobianco AJ and Kelliher MA. Notch1 contributes to mouse T-cell leukemia by directly inducing the expression of c-myc. Mol Cell Biol. 2006; 26(21):8022-8031.
54. Wang Z, Li Y, Kong D, Banerjee S, Ahmad A, Azmi AS, Ali S, Abbruzzese JL, Gallick GE and Sarkar FH. Acquisition of epithelial-mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of the notch signaling pathway. Cancer Res. 2009; 69(6):2400-2407.
55. Leong KG, Niessen K, Kulic I, Raouf A, Eaves C, Pollet I and Karsan A. Jagged1-mediated Notch activation induces epithelial-to-mesenchymal transition through Slug-induced repression of E-cadherin. J Exp Med. 2007; 204(12):2935-2948.
56. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J and Weinberg RA. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008; 133(4):704-715.
57. Singh A and Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010; 29(34):4741-4751.
58. Bhangu A, Wood G, Brown G, Darzi A, Tekkis P and Goldin R. The role of epithelial mesenchymal transition and resistance to neoadjuvant therapy in locally advanced rectal cancer. Colorectal Dis. 2014; 16(4):O133-143.
59. Yauch RL, Januario T, Eberhard DA, Cavet G, Zhu W, Fu L, Pham TQ, Soriano R, Stinson J, Seshagiri S, Modrusan Z, Lin CY, O’Neill V and Amler LC. Epithelial versus mesenchymal phenotype determines in vitro sensitivity and predicts clinical activity of erlotinib in lung cancer patients. Clin Cancer Res. 2005; 11(24 Pt 1):8686-8698.
60. Gort EH, Groot AJ, van der Wall E, van Diest PJ and Vooijs MA. Hypoxic regulation of metastasis via hypoxia-inducible factors. Curr Mol Med. 2008; 8(1):60-67.
61. Gomez-Casal R, Bhattacharya C, Ganesh N, Bailey L, Basse P, Gibson M, Epperly M and Levina V. Non-small cell lung cancer cells survived ionizing radiation treatment display cancer stem cell and epithelial-mesenchymal transition phenotypes. Mol Cancer. 2013; 12(1):94.
62. Xie M, Zhang L, He CS, Xu F, Liu JL, Hu ZH, Zhao LP and Tian Y. Activation of Notch-1 enhances epithelial-mesenchymal transition in gefitinib-acquired resistant lung cancer cells. J Cell Biochem. 2012; 113(5):1501-1513.
63. Yang Y, Ahn YH, Gibbons DL, Zang Y, Lin W, Thilaganathan N, Alvarez CA, Moreira DC, Creighton CJ, Gregory PA, Goodall GJ and Kurie JM. The Notch ligand Jagged2 promotes lung adenocarcinoma metastasis through a miR-200-dependent pathway in mice. J Clin Invest. 2011; 121(4):1373-1385.
64. Choi SH, Yang H, Lee SH, Ki JH, Nam DH and Yoo HY. TopBP1 and Claspin contribute to the radioresistance of lung cancer brain metastases. Mol Cancer. 2014; 13:211.
65. Breindel JL, Haskins JW, Cowell EP, Zhao M, Nguyen DX and Stern DF. EGF receptor activates MET through MAPK to enhance non-small cell lung carcinoma invasion and brain metastasis. Cancer Res. 2013; 73(16):5053-5065.
66. Wang X, Song N, Zhang Y, Cai Y, Liu Y, Qu X, Li Z, Li D, Hou K, Kang J and Hu X. Coexpression of c-Met and Notch-1 correlates with poor prognosis in resected non-small-cell lung cancer. Tumour Biol. 2015; 36(9):7053-7059.
67. Akervall J, Nandalur S, Zhang J, Qian CN, Goldstein N, Gyllerup P, Gardinger Y, Alm J, Lorenc K, Nilsson K, Resau J, Wilson G and Teh B. A novel panel of biomarkers predicts radioresistance in patients with squamous cell carcinoma of the head and neck. Eur J Cancer. 2014; 50(3):570-581.
68. Apostolou P, Toloudi M, Ioannou E, Kourtidou E, Chatziioannou M, Kopic A, Komiotis D, Kiritsis C, Manta S and Papasotiriou I. Study of the interaction among Notch pathway receptors, correlation with stemness, as well as their interaction with CD44, dipeptidyl peptidase-IV, hepatocyte growth factor receptor and the SETMAR transferase, in colon cancer stem cells. J Recept Sig Transd. 2013; 33(6):353-358.
69. Sarkar FH, Li Y, Wang Z, Kong D and Ali S. Implication of microRNAs in drug resistance for designing novel cancer therapy. Drug Resist Updat. 2010; 13(3):57-66.
70. Shiiba M, Shinozuka K, Saito K, Fushimi K, Kasamatsu A, Ogawara K, Uzawa K, Ito H, Takiguchi Y and Tanzawa H. MicroRNA-125b regulates proliferation and radioresistance of oral squamous cell carcinoma. Br J Cancer. 2013; 108(9):1817-1821.
71. Zhang B, Chen J, Ren Z, Chen Y, Li J, Miao X, Song Y, Zhao T, Li Y, Shi Y, Ren D and Liu J. A specific miRNA signature promotes radioresistance of human cervical cancer cells. Cancer Cell Int. 2013; 13(1):118.
72. Chun-Zhi Z, Lei H, An-Ling Z, Yan-Chao F, Xiao Y, Guang-Xiu W, Zhi-Fan J, Pei-Yu P, Qing-Yu Z and Chun-Sheng K. MicroRNA-221 and microRNA-222 regulate gastric carcinoma cell proliferation and radioresistance by targeting PTEN. BMC Cancer. 2010; 10:367.
73. Li Y, Guessous F, Zhang Y, Dipierro C, Kefas B, Johnson E, Marcinkiewicz L, Jiang J, Yang Y, Schmittgen TD, Lopes B, Schiff D, Purow B and Abounader R. MicroRNA-34a inhibits glioblastoma growth by targeting multiple oncogenes. Cancer Res. 2009; 69(19):7569-7576.
74. Bu P, Chen KY, Chen JH, Wang L, Walters J, Shin YJ, Goerger JP, Sun J, Witherspoon M, Rakhilin N, Li J, Yang H, Milsom J, Lee S, Zipfel W, Jin MM, et al. A microRNA miR-34a-regulated bimodal switch targets Notch in colon cancer stem cells. Cell Stem Cell. 2013; 12(5):602-615.
75. Bommer GT, Gerin I, Feng Y, Kaczorowski AJ, Kuick R, Love RE, Zhai Y, Giordano TJ, Qin ZS, Moore BB, MacDougald OA, Cho KR and Fearon ER. p53-mediated activation of miRNA34 candidate tumor-suppressor genes. Curr Biol. 2007; 17(15):1298-1307.
76. Duan W, Xu Y, Dong Y, Cao L, Tong J and Zhou X. Ectopic expression of miR-34a enhances radiosensitivity of non-small cell lung cancer cells, partly by suppressing the LyGDI signaling pathway. J Radiat Res. 2013; 54(4):611-619.
77. Kang J, Kim E, Kim W, Seong KM, Youn H, Kim JW, Kim J and Youn B. Rhamnetin and cirsiliol induce radiosensitization and inhibition of epithelial-mesenchymal transition (EMT) by miR-34a-mediated suppression of Notch-1 expression in non-small cell lung cancer cell lines. J Biol Chem. 2013; 288(38):27343-27357.
78. Ji Q, Hao X, Meng Y, Zhang M, Desano J, Fan D and Xu L. Restoration of tumor suppressor miR-34 inhibits human p53-mutant gastric cancer tumorspheres. BMC Cancer. 2008; 8:266.
79. Ji Q, Hao X, Zhang M, Tang W, Yang M, Li L, Xiang D, Desano JT, Bommer GT, Fan D, Fearon ER, Lawrence TS and Xu L. MicroRNA miR-34 inhibits human pancreatic cancer tumor-initiating cells. PLoS One. 2009; 4(8):e6816.
80. Hellstrom M, Phng LK, Hofmann JJ, Wallgard E, Coultas L, Lindblom P, Alva J, Nilsson AK, Karlsson L, Gaiano N, Yoon K, Rossant J, Iruela-Arispe ML, Kalen M, Gerhardt H and Betsholtz C. Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature. 2007; 445(7129):776-780.
81. Diez H, Fischer A, Winkler A, Hu CJ, Hatzopoulos AK, Breier G and Gessler M. Hypoxia-mediated activation of Dll4-Notch-Hey2 signaling in endothelial progenitor cells and adoption of arterial cell fate. Exp Cell Res. 2007; 313(1):1-9.
82. Eberhard A, Kahlert S, Goede V, Hemmerlein B, Plate KH and Augustin HG. Heterogeneity of angiogenesis and blood vessel maturation in human tumors: implications for antiangiogenic tumor therapies. Cancer Res. 2000; 60(5):1388-1393.
83. Noguera-Troise I, Daly C, Papadopoulos NJ, Coetzee S, Boland P, Gale NW, Lin HC, Yancopoulos GD and Thurston G. Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature. 2006; 444(7122):1032-1037.
84. Ridgway J, Zhang G, Wu Y, Stawicki S, Liang WC, Chanthery Y, Kowalski J, Watts RJ, Callahan C, Kasman I, Singh M, Chien M, Tan C, Hongo JA, de Sauvage F, Plowman G, et al. Inhibition of Dll4 signalling inhibits tumour growth by deregulating angiogenesis. Nature. 2006; 444(7122):1083-1087.
85. Hoey T, Yen WC, Axelrod F, Basi J, Donigian L, Dylla S, Fitch-Bruhns M, Lazetic S, Park IK, Sato A, Satyal S, Wang X, Clarke MF, Lewicki J and Gurney A. DLL4 blockade inhibits tumor growth and reduces tumor-initiating cell frequency. Cell Stem Cell. 2009; 5(2):168-177.
86. Liu SK, Bham SA, Fokas E, Beech J, Im J, Cho S, Harris AL and Muschel RJ. Delta-like ligand 4-notch blockade and tumor radiation response. J Natl Cancer Inst. 2011; 103(23):1778-1798.
87. Dings RP, Loren M, Heun H, McNiel E, Griffioen AW, Mayo KH and Griffin RJ. Scheduling of radiation with angiogenesis inhibitors anginex and Avastin improves therapeutic outcome via vessel normalization. Clin Cancer Res. 2007; 13(11):3395-3402.
88. Wilson WR and Hay MP. Targeting hypoxia in cancer therapy. Nat Rev Cancer. 2011; 11(6):393-410.
89. Edward C. Halperin CAP, Luther W. Brady. (2008). Perez and Brady’s Principles and Practice of Radiation Oncology. (Philadelphia: Lippincott Williams & Wilkins).
90. Nordsmark M, Bentzen SM, Rudat V, Brizel D, Lartigau E, Stadler P, Becker A, Adam M, Molls M, Dunst J, Terris DJ and Overgaard J. Prognostic value of tumor oxygenation in 397 head and neck tumors after primary radiation therapy. An international multi-center study. Radiother Oncol. 2005; 77(1):18-24.
91. Nordsmark M, Overgaard M and Overgaard J. Pretreatment oxygenation predicts radiation response in advanced squamous cell carcinoma of the head and neck. Radiother Oncol. 1996; 41(1):31-39.
92. Theys J, Yahyanejad S, Habets R, Span P, Dubois L, Paesmans K, Kattenbeld B, Cleutjens J, Groot AJ, Schuurbiers OC, Lambin P, Bussink J and Vooijs M. High NOTCH activity induces radiation resistance in non small cell lung cancer. Radiother Oncol. 2013; 108(3):440-445.
93. Lee JH, Suk J, Park J, Kim SB, Kwak SS, Kim JW, Lee CH, Byun B, Ahn JK and Joe CO. Notch signal activates hypoxia pathway through HES1-dependent SRC/signal transducers and activators of transcription 3 pathway. Mol Cancer Res. 2009; 7(10):1663-1671.
94. Gustafsson MV, Zheng X, Pereira T, Gradin K, Jin S, Lundkvist J, Ruas JL, Poellinger L, Lendahl U and Bondesson M. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev Cell. 2005; 9(5):617-628.
95. Bedogni B, Warneke JA, Nickoloff BJ, Giaccia AJ and Powell MB. Notch1 is an effector of Akt and hypoxia in melanoma development. J Clin Invest. 2008; 118(11):3660-3670.
96. Moriyama H, Moriyama M, Isshi H, Ishihara S, Okura H, Ichinose A, Ozawa T, Matsuyama A and Hayakawa T. Role of Notch signaling in the maintenance of human mesenchymal stem cells under hypoxic conditions. Stem Cells Dev. 2014.
97. Ding XY, Ding J, Wu K, Wen W, Liu C, Yan HX, Chen C, Wang S, Tang H, Gao CK, Guo LN, Cao D, Li Z, Feng GS, Wang HY and Xu ZF. Cross-talk between endothelial cells and tumor via delta-like ligand 4/Notch/PTEN signaling inhibits lung cancer growth. Oncogene. 2012; 31(23):2899-2906.
98. Kondoh H, Lleonart ME, Nakashima Y, Yokode M, Tanaka M, Bernard D, Gil J and Beach D. A high glycolytic flux supports the proliferative potential of murine embryonic stem cells. Antioxidants & Redox Signaling. 2007; 9(3):293-299.
99. Kondoh H, Lleonart ME, Gil J, Wang J, Degan P, Peters G, Martinez D, Carnero A and Beach D. Glycolytic enzymes can modulate cellular life span. Cancer Research. 2005; 65(1):177-185.
100. Weng AP, Ferrando AA, Lee W, Morris JPt, Silverman LB, Sanchez-Irizarry C, Blacklow SC, Look AT and Aster JC. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004; 306(5694):269-271.
101. Ellisen LW, Bird J, West DC, Soreng AL, Reynolds TC, Smith SD and Sklar J. TAN-1, the human homolog of the Drosophila notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell. 1991; 66(4):649-661.
102. Rossi D, Trifonov V, Fangazio M, Bruscaggin A, Rasi S, Spina V, Monti S, Vaisitti T, Arruga F, Fama R, Ciardullo C, Greco M, Cresta S, Piranda D, Holmes A, Fabbri G, et al. The coding genome of splenic marginal zone lymphoma: activation of NOTCH2 and other pathways regulating marginal zone development. J Exp Med. 2012; 209(9):1537-1551.
103. Kiel MJ, Velusamy T, Betz BL, Zhao L, Weigelin HG, Chiang MY, Huebner-Chan DR, Bailey NG, Yang DT, Bhagat G, Miranda RN, Bahler DW, Medeiros LJ, Lim MS and Elenitoba-Johnson KS. Whole-genome sequencing identifies recurrent somatic NOTCH2 mutations in splenic marginal zone lymphoma. J Exp Med. 2012; 209(9):1553-1565.
104. Wang K, Zhang Q, Li DN, Ching K, Zhang C, Zheng XX, Ozeck M, Shi S, Li XR, Wang H, Rejto P, Christensen J and Olson P. PEST Domain Mutations in Notch Receptors Comprise an Oncogenic Driver Segment in Triple-Negative Breast Cancer Sensitive to a gamma-Secretase Inhibitor. Clinical Cancer Research. 2015; 21(6):1487-1496.
105. Timme CR, Gruidl M and Yeatman TJ. Gamma-secretase inhibition attenuates oxaliplatin-induced apoptosis through increased Mcl-1 and/or Bcl-xL in human colon cancer cells. Apoptosis. 2013; 18(10):1163-1174.
106. Pinnix CC and Herlyn M. The many faces of Notch signaling in skin-derived cells. Pigment Cell Res. 2007; 20(6):458-465.
107. Abel EV, Kim EJ, Wu J, Hynes M, Bednar F, Proctor E, Wang L, Dziubinski ML and Simeone DM. The Notch pathway is important in maintaining the cancer stem cell population in pancreatic cancer. PLoS One. 2014; 9(3):e91983.
108. Jiang L, Wu J, Chen Q, Hu X, Li W and Hu G. Notch1 expression is upregulated in glioma and is associated with tumor progression. J Clin Neurosci. 2011; 18(3):387-390.
109. Sun W, Gaykalova DA, Ochs MF, Mambo E, Arnaoutakis D, Liu Y, Loyo M, Agrawal N, Howard J, Li R, Ahn S, Fertig E, Sidransky D, Houghton J, Buddavarapu K, Sanford T, et al. Activation of the NOTCH pathway in head and neck cancer. Cancer Res. 2014; 74(4):1091-1104.
110. Nicolas M, Wolfer A, Raj K, Kummer JA, Mill P, van Noort M, Hui CC, Clevers H, Dotto GP and Radtke F. Notch1 functions as a tumor suppressor in mouse skin. Nat Genet. 2003; 33(3):416-421.
111. Demehri S, Turkoz A and Kopan R. Epidermal Notch1 loss promotes skin tumorigenesis by impacting the stromal microenvironment. Cancer Cell. 2009; 16(1):55-66.
112. Rampias T, Vgenopoulou P, Avgeris M, Polyzos A, Stravodimos K, Valavanis C, Scorilas A and Klinakis A. A new tumor suppressor role for the Notch pathway in bladder cancer. Nat Med. 2014; 20(10):1199-1205.
113. Wang NJ, Sanborn Z, Arnett KL, Bayston LJ, Liao W, Proby CM, Leigh IM, Collisson EA, Gordon PB, Jakkula L, Pennypacker S, Zou Y, Sharma M, North JP, Vemula SS, Mauro TM, et al. Loss-of-function mutations in Notch receptors in cutaneous and lung squamous cell carcinoma. Proc Natl Acad Sci U S A. 2011; 108(43):17761-17766.
114. Pickering CR, Zhang J, Yoo SY, Bengtsson L, Moorthy S, Neskey DM, Zhao M, Ortega Alves MV, Chang K, Drummond J, Cortez E, Xie TX, Zhang D, Chung W, Issa JP, Zweidler-McKay PA, et al. Integrative genomic characterization of oral squamous cell carcinoma identifies frequent somatic drivers. Cancer Discov. 2013; 3(7):770-781.
115. Agrawal N, Jiao Y, Bettegowda C, Hutfless SM, Wang Y, David S, Cheng Y, Twaddell WS, Latt NL, Shin EJ, Wang LD, Wang L, Yang W, Velculescu VE, Vogelstein B, Papadopoulos N, et al. Comparative genomic analysis of esophageal adenocarcinoma and squamous cell carcinoma. Cancer Discov. 2012; 2(10):899-905.
116. Proweller A, Tu L, Lepore JJ, Cheng L, Lu MM, Seykora J, Millar SE, Pear WS and Parmacek MS. Impaired notch signaling promotes de novo squamous cell carcinoma formation. Cancer Res. 2006; 66(15):7438-7444.
117. Rothenberg SM and Ellisen LW. The molecular pathogenesis of head and neck squamous cell carcinoma. J Clin Invest. 2012; 122(6):1951-1957.
118. Vilimas T, Mascarenhas J, Palomero T, Mandal M, Buonamici S, Meng F, Thompson B, Spaulding C, Macaroun S, Alegre ML, Kee BL, Ferrando A, Miele L and Aifantis I. Targeting the NF-kappaB signaling pathway in Notch1-induced T-cell leukemia. Nat Med. 2007; 13(1):70-77.
119. Coppola JM, Bhojani MS, Ross BD and Rehemtulla A. A small-molecule furin inhibitor inhibits cancer cell motility and invasiveness. Neoplasia. 2008; 10(4):363-370.
120. Logeat F, Bessia C, Brou C, LeBail O, Jarriault S, Seidah NG and Israel A. The Notch1 receptor is cleaved constitutively by a furin-like convertase. Proc Natl Acad Sci U S A. 1998; 95(14):8108-8112.
121. Roti G, Carlton A, Ross KN, Markstein M, Pajcini K, Su AH, Perrimon N, Pear WS, Kung AL, Blacklow SC, Aster JC and Stegmaier K. Complementary Genomic Screens Identify SERCA as a Therapeutic Target in NOTCH1 Mutated Cancer. Cancer Cell. 2013; 23(3):390-405.
122. Funahashi Y, Hernandez SL, Das I, Ahn A, Huang J, Vorontchikhina M, Sharma A, Kanamaru E, Borisenko V, Desilva DM, Suzuki A, Wang X, Shawber CJ, Kandel JJ, Yamashiro DJ and Kitajewski J. A notch1 ectodomain construct inhibits endothelial notch signaling, tumor growth, and angiogenesis. Cancer Res. 2008; 68(12):4727-4735.
123. Kofler NM, Shawber CJ, Kangsamaksin T, Reed HO, Galatioto J and Kitajewski J. Notch signaling in developmental and tumor angiogenesis. Genes Cancer. 2011; 2(12):1106-16. doi: 10.1177/1947601911423030.
124. Gordon WR, Vardar-Ulu D, Histen G, Sanchez-Irizarry C, Aster JC and Blacklow SC. Structural basis for autoinhibition of Notch. Nat Struct Mol Biol. 2007; 14(4):295-300.
125. Wu Y, Cain-Hom C, Choy L, Hagenbeek TJ, de Leon GP, Chen Y, Finkle D, Venook R, Wu X, Ridgway J, Schahin-Reed D, Dow GJ, Shelton A, Stawicki S, Watts RJ, Zhang J, et al. Therapeutic antibody targeting of individual Notch receptors. Nature. 2010; 464(7291):1052-1057.
126. Tiyanont K, Wales TE, Siebel CW, Engen JR and Blacklow SC. Insights into Notch3 activation and inhibition mediated by antibodies directed against its negative regulatory region. J Mol Biol. 2013; 425(17):3192-3204.
127. Groot AJ, Habets R, Yahyanejad S, Hodin CM, Reiss K, Saftig P, Theys J and Vooijs M. Regulated proteolysis of NOTCH2 and NOTCH3 receptors by ADAM10 and presenilins. Mol Cell Biol. 2014; 34(15):2822-2832.
128. van Tetering G, van Diest P, Verlaan I, van der Wall E, Kopan R and Vooijs M. Metalloprotease ADAM10 is required for Notch1 site 2 cleavage. J Biol Chem. 2009; 284(45):31018-31027.
129. Kopan R and Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009; 137(2):216-233.
130. Moss ML, White JM, Lambert MH and Andrews RC. TACE and other ADAM proteases as targets for drug discovery. Drug Discov Today. 2001; 6(8):417-426.
131. Zhou BB, Peyton M, He B, Liu C, Girard L, Caudler E, Lo Y, Baribaud F, Mikami I, Reguart N, Yang G, Li Y, Yao W, Vaddi K, Gazdar AF, Friedman SM, et al. Targeting ADAM-mediated ligand cleavage to inhibit HER3 and EGFR pathways in non-small cell lung cancer. Cancer Cell. 2006; 10(1):39-50.
132. McGowan PM, Mullooly M, Caiazza F, Sukor S, Madden SF, Maguire AA, Pierce A, McDermott EW, Crown J, O’Donovan N and Duffy MJ. ADAM-17: a novel therapeutic target for triple negative breast cancer. Ann Oncol. 2013; 24(2):362-369.
133. van Es JH, van Gijn ME, Riccio O, van den Born M, Vooijs M, Begthel H, Cozijnsen M, Robine S, Winton DJ, Radtke F and Clevers H. Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature. 2005; 435(7044):959-963.
134. Vooijs M, Liu Z and Kopan R. Notch: architect, landscaper, and guardian of the intestine. Gastroenterology. 2011; 141(2):448-459.
135. Milano J, McKay J, Dagenais C, Foster-Brown L, Pognan F, Gadient R, Jacobs RT, Zacco A, Greenberg B and Ciaccio PJ. Modulation of notch processing by gamma-secretase inhibitors causes intestinal goblet cell metaplasia and induction of genes known to specify gut secretory lineage differentiation. Toxicol Sci. 2004; 82(1):341-358.
136. Krop I, Demuth T, Guthrie T, Wen PY, Mason WP, Chinnaiyan P, Butowski N, Groves MD, Kesari S, Freedman SJ, Blackman S, Watters J, Loboda A, Podtelezhnikov A, Lunceford J, Chen C, et al. Phase I pharmacologic and pharmacodynamic study of the gamma secretase (Notch) inhibitor MK-0752 in adult patients with advanced solid tumors. J Clin Oncol. 2012; 30(19):2307-2313.
137. Cullion K, Draheim KM, Hermance N, Tammam J, Sharma VM, Ware C, Nikov G, Krishnamoorthy V, Majumder PK and Kelliher MA. Targeting the Notch1 and mTOR pathways in a mouse T-ALL model. Blood. 2009; 113(24):6172-6181.
138. Smith C, Patnaik, Papadopoulos, Chambers, Thorpe, Xu, Kapoun, Dupont, Tolcher. (2012). A first-in-human phase I study to evaluate the fully human monoclonal antibody OMP-59R5 (anti-Notch2/3) administered intravenously to patients with advanced solid tumors. pp. 3025.
139. Takebe N, Miele L, Harris PJ, Jeong W, Bando H, Kahn M, Yang SX and Ivy SP. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nat Rev Clin Oncol. 2015; 12(8):445-464.
140. Samon JB, Castillo-Martin M, Hadler M, Ambesi-Impiobato A, Paietta E, Racevskis J, Wiernik PH, Rowe JM, Jakubczak J, Randolph S, Cordon-Cardo C and Ferrando AA. Preclinical analysis of the gamma-secretase inhibitor PF-03084014 in combination with glucocorticoids in T-cell acute lymphoblastic leukemia. Mol Cancer Ther. 2012; 11(7):1565-1575.
141. Real PJ, Tosello V, Palomero T, Castillo M, Hernando E, de Stanchina E, Sulis ML, Barnes K, Sawai C, Homminga I, Meijerink J, Aifantis I, Basso G, Cordon-Cardo C, Ai W and Ferrando A. Gamma-secretase inhibitors reverse glucocorticoid resistance in T cell acute lymphoblastic leukemia. Nat Med. 2009; 15(1):50-58.
142. Inaba H and Pui CH. Glucocorticoid use in acute lymphoblastic leukaemia. Lancet Oncol. 2010; 11(11):1096-1106.
143. Liu Z, Turkoz A, Jackson EN, Corbo JC, Engelbach JA, Garbow JR, Piwnica-Worms DR and Kopan R. Notch1 loss of heterozygosity causes vascular tumors and lethal hemorrhage in mice. J Clin Invest. 2011; 121(2):800-808.
144. Wang Y, Chan SL, Miele L, Yao PJ, Mackes J, Ingram DK, Mattson MP and Furukawa K. Involvement of Notch signaling in hippocampal synaptic plasticity. Proc Natl Acad Sci U S A. 2004; 101(25):9458-9462.
145. Qyang Y, Chambers SM, Wang P, Xia X, Chen X, Goodell MA and Zheng H. Myeloproliferative disease in mice with reduced presenilin gene dosage: effect of gamma-secretase blockage. Biochemistry-Us. 2004; 43(18):5352-5359.
146. Hadland BK, Manley NR, Su D, Longmore GD, Moore CL, Wolfe MS, Schroeter EH and Kopan R. Gamma -secretase inhibitors repress thymocyte development. Proc Natl Acad Sci U S A. 2001; 98(13):7487-7491.
147. Li K, Li Y, Wu W, Gordon WR, Chang DW, Lu M, Scoggin S, Fu T, Vien L, Histen G, Zheng J, Martin-Hollister R, Duensing T, Singh S, Blacklow SC, Yao Z, et al. Modulation of Notch signaling by antibodies specific for the extracellular negative regulatory region of NOTCH3. J Biol Chem. 2008; 283(12):8046-8054.
148. Aste-Amezaga M, Zhang NY, Lineberger JE, Arnold BA, Toner TJ, Gu MC, Huang LY, Vitelli S, Vo KT, Haytko P, Zhao JZ, Baleydier F, L’Heureux S, Wang HF, Gordon WR, Thoryk E, et al. Characterization of Notch1 Antibodies That Inhibit Signaling of Both Normal and Mutated Notch1 Receptors. Plos One. 2010; 5(2).
149. Kuramoto T, Goto H, Mitsuhashi A, Tabata S, Ogawa H, Uehara H, Saijo A, Kakiuchi S, Maekawa Y, Yasutomo K, Hanibuchi M, Akiyama S, Sone S and Nishioka Y. Dll4-Fc, an inhibitor of Dll4-notch signaling, suppresses liver metastasis of small cell lung cancer cells through the downregulation of the NF-kappaB activity. Mol Cancer Ther. 2012; 11(12):2578-2587.
150. Jimeno A LP, Strother RM, Diamond JR, Plato L, Younger A, et al. Phase Istudy of REGN421 (R)/SAR153192, a fully-human delta-like ligand 4 (Dll4) monoclonal antibody(mAb), in patients with advanced solid tumors. J Clin I Oncol. 2013; 31.
151. Smith DC, Eisenberg PD, Manikhas GM, Chugh R, Gubens MA, Stagg RJ, Kapoun AM, Xu L, Dupont J and Sikic BI. A Phase 1 Dose Escalation and Expansion Study of the Anti-Cancer Stem Cell Agent Demcizumab (Anti-DLL4) in Patients with Previously Treated Solid Tumors. Clin Cancer Res. 2014.
152. Yan M, Callahan CA, Beyer JC, Allamneni KP, Zhang G, Ridgway JB, Niessen K and Plowman GD. Chronic DLL4 blockade induces vascular neoplasms. Nature. 2010; 463(7282):E6-7.
153. Varnum-Finney B, Purton LE, Yu M, Brashem-Stein C, Flowers D, Staats S, Moore KA, Le Roux I, Mann R, Gray G, Artavanis-Tsakonas S and Bernstein ID. The Notch ligand, Jagged-1, influences the development of primitive hematopoietic precursor cells. Blood. 1998; 91(11):4084-4091.
154. Zhu TS, Costello MA, Talsma CE, Flack CG, Crowley JG, Hamm LL, He X, Hervey-Jumper SL, Heth JA, Muraszko KM, DiMeco F, Vescovi AL and Fan X. Endothelial cells create a stem cell niche in glioblastoma by providing NOTCH ligands that nurture self-renewal of cancer stem-like cells. Cancer Res. 2011; 71(18):6061-6072.
155. Gopisetty A, Bhattacharya P, Haddad C, Bruno JC, Jr., Vasu C, Miele L and Prabhakar BS. OX40L/Jagged1 cosignaling by GM-CSF-induced bone marrow-derived dendritic cells is required for the expansion of functional regulatory T cells. Journal of immunology. 2013; 190(11):5516-5525.
156. Steg AD, Katre AA, Goodman B, Han HD, Nick AM, Stone RL, Coleman RL, Alvarez RD, Lopez-Berestein G, Sood AK and Landen CN. Targeting the Notch Ligand Jagged1 in Both Tumor Cells and Stroma in Ovarian Cancer. Clinical Cancer Research. 2011; 17(17):5674-5685.
157. Zhao XC, Dou GR, Wang L, Liang L, Tian DM, Cao XL, Qin HY, Wang CM, Zhang P and Han H. Inhibition of tumor angiogenesis and tumor growth by the DSL domain of human Delta-like 1 targeted to vascular endothelial cells. Neoplasia. 2013; 15(7):815-825.
158. Palmer WH and Deng WM. Ligand-Independent Mechanisms of Notch Activity. Trends Cell Biol. 2015; 25(11):697-707.
159. Ma J, Tang X, Wong P, Jacobs B, Borden EC and Bedogni B. Noncanonical activation of Notch1 protein by membrane type 1 matrix metalloproteinase (MT1-MMP) controls melanoma cell proliferation. J Biol Chem. 2014; 289(12):8442-8449.
160. Malecki MJ, Sanchez-Irizarry C, Mitchell JL, Histen G, Xu ML, Aster JC and Blacklow SC. Leukemia-associated mutations within the NOTCH1 heterodimerization domain fall into at least two distinct mechanistic classes. Mol Cell Biol. 2006; 26(12):4642-4651.
161. Hirata N, Yamada S, Shoda T, Kurihara M, Sekino Y and Kanda Y. Sphingosine-1-phosphate promotes expansion of cancer stem cells via S1PR3 by a ligand-independent Notch activation. Nat Commun. 2014; 5:4806.
162. Sulis ML, Saftig P and Ferrando AA. Redundancy and specificity of the metalloprotease system mediating oncogenic NOTCH1 activation in T-ALL. Leukemia. 2011; 25(10):1564-1569.
163. Bozkulak EC and Weinmaster G. Selective use of ADAM10 and ADAM17 in activation of Notch1 signaling. Mol Cell Biol. 2009; 29(21):5679-5695.
164. Axelson H, Fredlund E, Ovenberger M, Landberg G and Pahlman S. Hypoxia-induced dedifferentiation of tumor cells—a mechanism behind heterogeneity and aggressiveness of solid tumors. Semin Cell Dev Biol. 2005; 16(4-5):554-563.
165. Hayashi I, Takatori S, Urano Y, Miyake Y, Takagi J, Sakata-Yanagimoto M, Iwanari H, Osawa S, Morohashi Y, Li T, Wong PC, Chiba S, Kodama T, Hamakubo T, Tomita T and Iwatsubo T. Neutralization of the gamma-secretase activity by monoclonal antibody against extracellular domain of nicastrin. Oncogene. 2012; 31(6):787-798.
166. Filipovic A, Lombardo Y, Fronato M, Abrahams J, Aboagye E, Nguyen QD, d’Aqua BB, Ridley A, Green A, Rahka E, Ellis I, Recchi C, Przulj N, Sarajlic A, Alattia JR, Fraering P, et al. Anti-nicastrin monoclonal antibodies elicit pleiotropic anti-tumour pharmacological effects in invasive breast cancer cells. Breast Cancer Res Treat. 2014.
167. Pasternak SH, Bagshaw RD, Guiral M, Zhang S, Ackerley CA, Pak BJ, Callahan JW and Mahuran DJ. Presenilin-1, nicastrin, amyloid precursor protein, and gamma-secretase activity are co-localized in the lysosomal membrane. J Biol Chem. 2003; 278(29):26687-26694.
168. Kaether C, Haass C and Steiner H. Assembly, trafficking and function of gamma-secretase. Neurodegener Dis. 2006; 3(4-5):275-283.
169. Vaccari T, Duchi S, Cortese K, Tacchetti C and Bilder D. The vacuolar ATPase is required for physiological as well as pathological activation of the Notch receptor. Development. 2010; 137(11):1825-1832.
170. Nichols JT, Miyamoto A and Weinmaster G. Notch signaling—constantly on the move. Traffic. 2007; 8(8):959-969.
171. Parks AL, Klueg KM, Stout JR and Muskavitch MA. Ligand endocytosis drives receptor dissociation and activation in the Notch pathway. Development. 2000; 127(7):1373-1385.
172. Kobia F, Duchi S, Deflorian G and Vaccari T. Pharmacologic inhibition of vacuolar H plus ATPase reduces physiologic and oncogenic Notch signaling. Molecular Oncology. 2014; 8(2):207-220.
173. De Milito A and Fais S. Tumor acidity, chemoresistance and proton pump inhibitors. Future Oncol. 2005; 1(6):779-786.
174. Moellering RE, Cornejo M, Davis TN, Del Bianco C, Aster JC, Blacklow SC, Kung AL, Gilliland DG, Verdine GL and Bradner JE. Direct inhibition of the NOTCH transcription factor complex. Nature. 2009; 462(7270):182-188.
175. Thurston G, Noguera-Troise I and Yancopoulos GD. The Delta paradox: DLL4 blockade leads to more tumour vessels but less tumour growth. Nat Rev Cancer. 2007; 7(5):327-331.
176. Dufraine J, Funahashi Y and Kitajewski J. Notch signaling regulates tumor angiogenesis by diverse mechanisms. Oncogene. 2008; 27(38):5132-5137.
177. Paez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Vinals F, Inoue M, Bergers G, Hanahan D and Casanovas O. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009; 15(3):220-231.
178. Mizugaki H, Sakakibara-Konishi J, Ikezawa Y, Kikuchi J, Kikuchi E, Oizumi S, Dang TP and Nishimura M. gamma-Secretase inhibitor enhances antitumour effect of radiation in Notch-expressing lung cancer. Br J Cancer. 2012; 106(12):1953-1959.
179. Charles N, Ozawa T, Squatrito M, Bleau AM, Brennan CW, Hambardzumyan D and Holland EC. Perivascular nitric oxide activates notch signaling and promotes stem-like character in PDGF-induced glioma cells. Cell Stem Cell. 2010; 6(2):141-152.
180. Masuda S. Re: Delta-like ligand 4-Notch blockade and tumor radiation response. J Natl Cancer Inst. 2012; 104(5):421; author reply 421-422.
181. Gilbert CA, Daou MC, Moser RP and Ross AH. Gamma-secretase inhibitors enhance temozolomide treatment of human gliomas by inhibiting neurosphere repopulation and xenograft recurrence. Cancer Res. 2010; 70(17):6870-6879.
182. Colorectal cancer: Sequence of drugs-more important than drug exposure? Nat Rev Clin Oncol. 2015; 12(10):562.
183. Yang J, Yue JB, Liu J and Yu JM. Repopulation of tumor cells during fractionated radiotherapy and detection methods (Review). Oncol Lett. 2014; 7(6):1755-1760.
184. De Ruysscher D, Pijls-Johannesma M, Bentzen SM, Minken A, Wanders R, Lutgens L, Hochstenbag M, Boersma L, Wouters B, Lammering G, Vansteenkiste J and Lambin P. Time between the first day of chemotherapy and the last day of chest radiation is the most important predictor of survival in limited-disease small-cell lung cancer. Journal of Clinical Oncology. 2006; 24(7):1057-1063.
185. Fu KK, Pajak TF, Trotti A, Jones CU, Spencer SA, Phillips TL, Garden AS, Ridge JA, Cooper JS and Ang KK. A Radiation Therapy Oncology Group (RTOG) phase III randomized study to compare hyperfractionation and two variants of accelerated fractionation to standard fractionation radiotherapy for head and neck squamous cell carcinomas: first report of RTOG 9003. Int J Radiat Oncol Biol Phys. 2000; 48(1):7-16.
186. Saunders M, Dische S, Barrett A, Harvey A, Gibson D and Parmar M. Continuous hyperfractionated accelerated radiotherapy (CHART) versus conventional radiotherapy in non-small-cell lung cancer: a randomised multicentre trial. CHART Steering Committee. Lancet. 1997; 350(9072):161-165.
187. Jain RK. Normalizing tumor vasculature with anti-angiogenic therapy: a new paradigm for combination therapy. Nat Med. 2001; 7(9):987-989.
188. Kleibeuker EA, Griffioen AW, Verheul HM, Slotman BJ and Thijssen VL. Combining angiogenesis inhibition and radiotherapy: a double-edged sword. Drug Resist Updat. 2012; 15(3):173-182.
189. Bruce Alberts JA, Lewis Julian, Raff Martin, Roberts Keith, Walter Peter. (2002). Molecular Biology of the Cell. (New York: Garland Science).
190. Simoes BM, O’Brien CS, Eyre R, Silva A, Yu L, Sarmiento-Castro A, Alferez DG, Spence K, Santiago-Gomez A, Chemi F, Acar A, Gandhi A, Howell A, Brennan K, Ryden L, Catalano S, et al. Anti-estrogen Resistance in Human Breast Tumors Is Driven by JAG1-NOTCH4-Dependent Cancer Stem Cell Activity. Cell Rep. 2015; 12(12):1968-1977.
191. Rizzo P, Miao H, D’Souza G, Osipo C, Song LL, Yun J, Zhao H, Mascarenhas J, Wyatt D, Antico G, Hao L, Yao K, Rajan P, Hicks C, Siziopikou K, Selvaggi S, et al. Cross-talk between notch and the estrogen receptor in breast cancer suggests novel therapeutic approaches. Cancer Res. 2008; 68(13):5226-5235.
192. Gonzalez-Angulo AM, Timms KM, Liu S, Chen H, Litton JK, Potter J, Lanchbury JS, Stemke-Hale K, Hennessy BT, Arun BK, Hortobagyi GN, Do KA, Mills GB and Meric-Bernstam F. Incidence and outcome of BRCA mutations in unselected patients with triple receptor-negative breast cancer. Clin Cancer Res. 2011; 17(5):1082-1089.
193. Nagamatsu I, Onishi H, Matsushita S, Kubo M, Kai M, Imaizumi A, Nakano K, Hattori M, Oda Y, Tanaka M and Katano M. NOTCH4 is a potential therapeutic target for triple-negative breast cancer. Anticancer Res. 2014; 34(1):69-80.
194. Harrison H, Farnie G, Howell SJ, Rock RE, Stylianou S, Brennan KR, Bundred NJ and Clarke RB. Regulation of breast cancer stem cell activity by signaling through the Notch4 receptor. Cancer Res. 2010; 70(2):709-718.
195. Kolev V, Mandinova A, Guinea-Viniegra J, Hu B, Lefort K, Lambertini C, Neel V, Dummer R, Wagner EF and Dotto GP. EGFR signalling as a negative regulator of Notch1 gene transcription and function in proliferating keratinocytes and cancer. Nature Cell Biology. 2008; 10(8).
196. Trigka EA, Levidou G, Saetta AA, Chatziandreou I, Tomos P, Thalassinos N, Anastasiou N, Spartalis E, Kavantzas N, Patsouris E and Korkolopoulou P. A detailed immunohistochemical analysis of the PI3K/AKT/mTOR pathway in lung cancer: correlation with PIK3CA, AKT1, K-RAS or PTEN mutational status and clinicopathological features. Oncol Rep. 2013; 30(2):623-636.
197. Pallis AG and Syrigos KN. Targeting tumor neovasculature in non-small-cell lung cancer. Crit Rev Oncol Hematol. 2013; 86(2):130-142.
198. Norihiko Saito JF, Shuzhen Wang, Erik P. Sulman, Frederick F. Lang, Wk Alfred Yung, and Dimpy Koul. (2013). Oncogene addiction switch from NOTCH to PI3K/AKT requires simultaneous targeting of NOTCH and PI3K pathway inhibition in glioblastoma. AACR 104th Annual Meeting of the American Association for Cancer Research. (Washington, DC.
199. Palomero T, Sulis ML, Cortina M, Real PJ, Barnes K, Ciofani M, Caparros E, Buteau J, Brown K, Perkins SL, Bhagat G, Agarwal AM, Basso G, Castillo M, Nagase S, Cordon-Cardo C, et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat Med. 2007; 13(10):1203-1210.
200. Zhang CC, Pavlicek A, Zhang Q, Lira ME, Painter CL, Yan Z, Zheng X, Lee NV, Ozeck M, Qiu M, Zong Q, Lappin PB, Wong A, Rejto PA, Smeal T and Christensen JG. Biomarker and pharmacologic evaluation of the gamma-secretase inhibitor PF-03084014 in breast cancer models. Clin Cancer Res. 2012; 18(18):5008-5019.
201. Stoeck A, Lejnine S, Truong A, Pan L, Wang H, Zang C, Yuan J, Ware C, MacLean J, Garrett-Engele PW, Kluk M, Laskey J, Haines BB, Moskaluk C, Zawel L, Fawell S, et al. Discovery of Biomarkers Predictive of GSI Response in Triple-Negative Breast Cancer and Adenoid Cystic Carcinoma. Cancer Discov. 2014; 4(10):1154-1167.
202. Aste-Amezaga M, Zhang N, Lineberger JE, Arnold BA, Toner TJ, Gu M, Huang L, Vitelli S, Vo KT, Haytko P, Zhao JZ, Baleydier F, L’Heureux S, Wang H, Gordon WR, Thoryk E, et al. Characterization of Notch1 antibodies that inhibit signaling of both normal and mutated Notch1 receptors. PLoS One. 2010; 5(2):e9094.
203. Harrison H, Farnie G, Howell SJ, Rock RE, Stylianou S, Brennan KR, Bundred NJ and Clarke RB. Regulation of Breast Cancer Stem Cell Activity by Signaling through the Notch4 Receptor. Cancer Research. 2010; 70(2):709-718.
204. Davis L, Xu, Hill, Kapoun, Dupont, Munster, Eckhardt, Patnaik. (2013). A first-in-human phase I study of the novel cancer stem cell (CSC) targeting antibody OMP-52M51 (anti-Notch1) administered intravenously to patients with certain advanced solid tumors. Mol Cancer Ther.
205. Demin Li iM, Alison H. Banham and Adrian L. Harris. The Notch ligand Jagged1 as a target for anti-tumor therapy. Front Oncol. 2014.
206. Newton RC BE, Levy RE, Doval D, Bondarde S, Sahoo TP, Lokanatha D, Julka PK, Nagarkar R and Friedman SM. Clinical benefit of INCB7839, a potent and selective ADAM inhibitor, in combination with trastuzumab in patients with metastatic HER2+breast cancer. Journal of Clinical Oncology. 15:28.
207. Infante J BH, Lewis N, Donehower R, Redman J, Friedman S, Scherle P, Fridman J, Li J, Emm T, Troy S and Eckhardt, G. A multicenter phase Ib study of the safety, pharmacokinetics, biological activity and clinical efficacy of INCB7839, a potent and selective inhibitor of ADAM10 and ADAM17. Breast Cancer Research and Treatment. 106:S269-S269.
208. Luistro L, He W, Smith M, Packman K, Vilenchik M, Carvajal D, Roberts J, Cai J, Berkofsky-Fessler W, Hilton H, Linn M, Flohr A, Jakob-Rotne R, Jacobsen H, Glenn K, Heimbrook D, et al. Preclinical profile of a potent gamma-secretase inhibitor targeting notch signaling with in vivo efficacy and pharmacodynamic properties. Cancer Res. 2009; 69(19):7672-7680.
209. Strosberg JR, Yeatman T, Weber J, Coppola D, Schell MJ, Han G, Almhanna K, Kim R, Valone T, Jump H and Sullivan D. A phase II study of RO4929097 in metastatic colorectal cancer. Eur J Cancer. 2012; 48(7):997-1003.
210. Maillard I, Weng AP, Carpenter AC, Rodriguez CG, Sai H, Xu L, Allman D, Aster JC and Pear WS. Mastermind critically regulates Notch-mediated lymphoid cell fate decisions. Blood. 2004; 104(6):1696-1702.
211. Kobia F, Duchi S, Deflorian G and Vaccari T. Pharmacologic inhibition of vacuolar H+ ATPase reduces physiologic and oncogenic Notch signaling. Mol Oncol. 2014; 8(2):207-220.