
INTRODUCTION
Epigenetic alterations including DNA methylation and covalent histone modifications are known as important mechanisms in carcinogenesis. Aberrant DNA methylation at the CpG island (CGI) promoter regions of tumor suppressor genes (TSGs) has been frequently observed in various cancers [1]. Histone marks of di- and tri-methylation of H3 lysines (K) 4 (H3K4me2 and H3K4me3) of the gene promoter regions are drastically correlated with their gene activation [1, 2]. The promoter and enhancer regions of transcriptionally active genes are marked by mono-methylation of H3K4 (H3K4me1) as well [3]. In contrast, transcriptionally inactive genes are characterized by di- and tri-methylated H3K9 (H3K9me2 and H3K9me3) and tri-methylated H3K27 (H3K27me3) [1, 2].
The histone lysine methyltransferases (KMTs) family has a highly conserved SET domain that is characterized in the Drosophila proteins Su(var) 3-9, Enhancer-of-zeste and Trithorax [2, 4]. SET7/9 (also known as SET7, SET9 and SETD7) containing the SET domain has two distinct functions for lysine methylation. First, SET7/9 has a mono-methyltransferase activity for H3K4, which is associated with transcriptional activation of genes [5, 6]. It has been reported that SET7/9 can activate expression of myogenic differentiation genes through H3K4me1 at their promoter/enhancer regions [7], implying that SET7/9 is involved in cellular differentiation. Second, SET7/9 methylates the lysine in non-histone proteins, such as K372 of p53 and K873 of pRB, and subsequently participates in the posttranslational modifications of these proteins [8–10].
Growing evidences obtained on in vitro analyses of SET7/9 functions for methylation of non-histone proteins provide the possibility that SET7/9 exerts tumor suppressor activities through p53 stabilization, pRB activation and DNMT1 degradation [8–11]. We also reported that SET7/9 suppresses SUV39H1 methyltransferase activity by methylating codons 105 and 123 in SUV39H1 in response to DNA damage, subsequently induced genomic instability and inhibited cell proliferation [12]. In contrast, activation of estrogen receptor α (ERα) by SET7/9 may be implicated in the development of hormone-dependent breast cancer [13]. Thus, the functions of SET7/9 are controversial in cancers. There have been no reports on SET7/9 alterations in primary cancers, and hence it remains unknown how SET7/9 contribute to carcinogenesis.
Gastric cancer (GC) is the second leading cause of cancer death in the world [14]. GCs are histologically classified into two major types, intestinal and diffuse, and distinct genetic and epigenetic alterations of tumor-related genes have been shown in both types of GCs [15]. Although somatic mutations and expression changes of the histone modifier genes including HMT ones are known to play critical roles in the pathogeneses of various cancers [2, 4], the relationship between alterations of the histone modifier genes and GCs is unclear.
Here we observed that SET7/9 expression was frequently reduced in GCs. The aim of this study is to characterize the clinicopathologic features of primary GCs with loss or weak SET7/9 expression, and further study the functional significances of SET7/9 alterations in gastric carcinogenesis. In addition, to elucidate a role of SET7/9 as an H3K4 mono-methyltransferase, we searched SET7/9 downstream target genes and then investigated their transcriptional regulation associated with H3K4me1.
RESULTS
SET7/9 expression and its clinicopathological relevance in primary GCs
SET7/9 protein expression was evaluated by immunohistochemistry (IHC), and then graded the SET7/9 expression as weak or loss (expression-low) and retained (expression-high) in primary GCs (Figure 1A). It was noted that SET7/9 protein was strongly expressed in noncancerous gastric epithelial tissues by IHC. Among the 376 GC cases from the formalin-fixed paraffin-embedded (FFPE) tissue microarray, 129 cases (34.3%) showed loss or weak expression of SET7/9 protein compared to matched non-cancerous tissues from the patients.
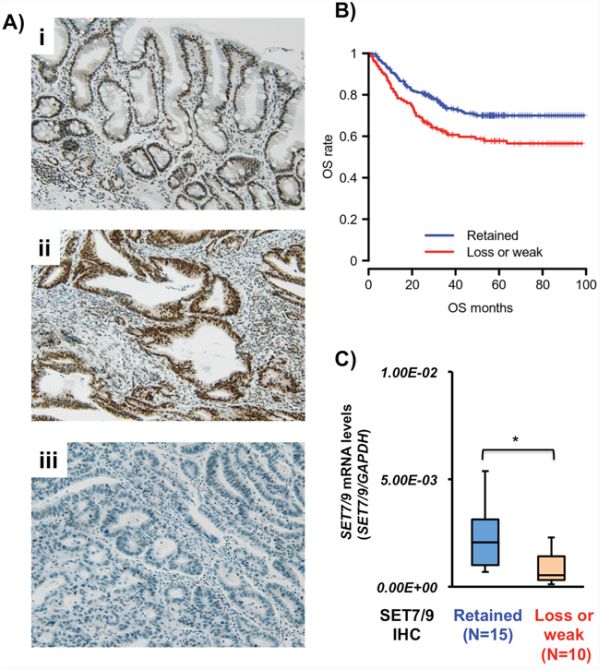
Figure 1: SET7/9 expression in primary GCs. A. Representative IHC photographs of SET7/9 in primary tissues. Non-cancerous gastric mucosa including intestinal metaplasia exhibited strong SET7/9 expression (i). Positive (ii) and negative (iii) staining of SET7/9 was detected in GCs. Original magnification x100. B. Kaplan-Meier curve of overall survival for GC patients with SET7/9 protein expression. The GC patients with loss or weak SET7/9 expression (red, N = 129) had a significantly poorer outcome than those with retained SET7/9 (blue, N = 247) (P = 0.038, logrank test). C. qRT-PCR analysis of SET7/9 expression in 25 primary GC tissues. IHC of SET7/9 was performed in these tissues, which were divided into two groups, SET7/9 protein expression-high (retained, N = 15) and -low (loss or weak, N = 10). Relative expression was calculated using GAPDH expression as an internal control. Mann—Whitney U-test, *P = 0.017.
The relationships between SET7/9 expression and clinicopathological characteristics of 376 FFPE GC cases from the tissue microarray are summarized in Table 1. Loss or weak SET7/9 expression was significantly associated with age (P = 0.028), gender (P < 0.001), Lauren’s classification (P < 0.001), Ming’s classification (P < 0.005), perineural invasion (P < 0.001), pT stage (P < 0.001), lymph node metastasis (P = 0.038), and pTNM stage (P < 0.01). The frequency of SET7/9 reduction in advanced GCs was higher than that in early GCs (P < 0.054). The patients with GCs showing loss/weak SE7/9 expression exhibited significantly shorter overall survival (OS, P = 0.038, Figure 1B) and disease-free survival (DFS, P = 0.025, Supplementary Figure S1) than ones with SET7/9-retained GCs with the logrank test. However, SET7/9 expression was not significantly correlated with OS or DFS by multivariate analyses (data not shown).
Table 1: Clinicopathological correlations of SET7/9 expression in 376 GCs1).
Clinicopathological features |
No. of cases |
SET7/9 staining2) |
P value3) |
Loss or weak |
Retained |
|---|---|---|---|---|
Age |
||||
≤60 |
198 |
78 (39.4) |
120 (60.6) |
0.028* |
>60 |
178 |
51 (28.7) |
127 (71.3) |
|
Gender |
||||
Female |
104 |
50 (48.1) |
54 (51.9) |
0.001* |
Male |
272 |
79 (29.0) |
193 (71.0) |
|
Lauren’s classification |
||||
Intestinal |
162 |
37 (22.8) |
125 (77.2) |
<0.001* |
Non-intestinal (Diffuse/Mixed) |
211 |
92 (43.6) |
119 (56.4) |
|
Ming’s classification |
||||
Expanding |
41 |
6 (14.6) |
35 (85.4) |
0.005* |
Infiltrative |
335 |
123 (36.7) |
212 (63.3) |
|
Tumor invasion |
||||
EGC (pT1) |
89 |
23 (25.8) |
66 (74.2) |
0.054 |
AGC (pT2-4) |
287 |
106 (36.9) |
181 (63.1) |
|
Lymphatic invasion |
||||
Absent |
133 |
42 (31.6) |
91 (68.4) |
0.409 |
Present |
243 |
87 (35.8) |
156 (64.2) |
|
Venous invasion |
||||
Absent |
304 |
103 (33.9) |
201 (66.1) |
0.72 |
Present |
72 |
26 (36.1) |
46 (63.9) |
|
Perineural invasion |
||||
Absent |
177 |
46 (26.0) |
131 (74.0) |
0.001* |
Present |
199 |
83 (41.7) |
116 (58.3) |
|
Radicality |
||||
R0 |
331 |
108 (32.6) |
223 (67.4) |
0.063 |
R1/2 |
45 |
21 (46.7) |
24 (53.3) |
|
pT stage |
||||
pT1 |
89 |
23 (25.8) |
66 (74.2) |
0.001* |
pT2 |
58 |
15 (25.9) |
43 (74.1) |
|
pT3 |
138 |
48 (34.8) |
90 (65.2) |
|
pT4 |
91 |
43 (47.3) |
48 (52.7) |
|
pT4a |
82 |
39 (47.6) |
43 (52.4) |
|
pT4b |
9 |
4 (44.4) |
5 (55.6) |
|
Lymph node metastasis |
||||
Absent (N0) |
143 |
42 (29.4) |
101 (70.6) |
0.114 |
Present |
233 |
87 (37.3) |
146 (62.7) |
|
N0-N1 |
194 |
57 (29.4) |
137 (70.6) |
0.038* |
N2-N3 |
182 |
72 (39.6) |
110 (60.4) |
|
pTNM stage |
||||
I |
111 |
31 (27.9) |
80 (72.1) |
0.009* |
II |
85 |
24 (28.2) |
61 (71.8) |
|
III |
129 |
51 (39.5) |
78 (60.5) |
|
IV |
51 |
23 (45.1) |
28 (54.9) |
|
1) A total of 376 primary GCs from the tissue microarray were examined by immunohistochemistry.
2) SET7/9 expression levels were divided into two groups, retained and decreased (loss or weak), as shown under Materials and Methods in Supplementary Information.
3) P values were determined by Pearson’s chi-square test. The Spearman rank correlation analysis was used for pT and pTMN stages. The TMN staging is based on the 7th edition of AJCC (44). *Statistically significant difference.
We compared the SET7/9 mRNA and protein expression in other 25 primary GC samples (frozen and matched FFPE tissues) by qRT-PCR and IHC analyses, respectively. The expression levels of SET7/9 mRNA in 10 GCs with low SET7/9 protein expression were significantly lower than those in 15 GCs with retained SET7/9 (P = 0.017, Figure 1C).
Analysis of SET7/9 expression and mutation in cancer cell lines
We examined the SET7/9 expression levels in 12 GC cell lines and three non-cancerous gastric mucosa samples by RT-PCR, quantitative RT-PCR (qRT-PCR) and western blot (WB) (Figures 2A and 2B). Non-cancerous stomach tissues strongly expressed SET7/9 mRNA and protein, which is consistent with data on IHC (Figure 1A (i)). MKN7 and HSC43 showed weak SET7/9 expression at the mRNA level (Figure 2A). Although expression of SET7/9 protein and mRNA corresponded in most cell lines, HSC60 and HSC57 cells with strong SET7/9 mRNA expression exhibited weak SET7/9 protein. Thus, four of 12 GC cell lines exhibited low SET7/9 protein expression when compared their expression levels of SET7/9 with ones in non-cancerous stomach tissues.
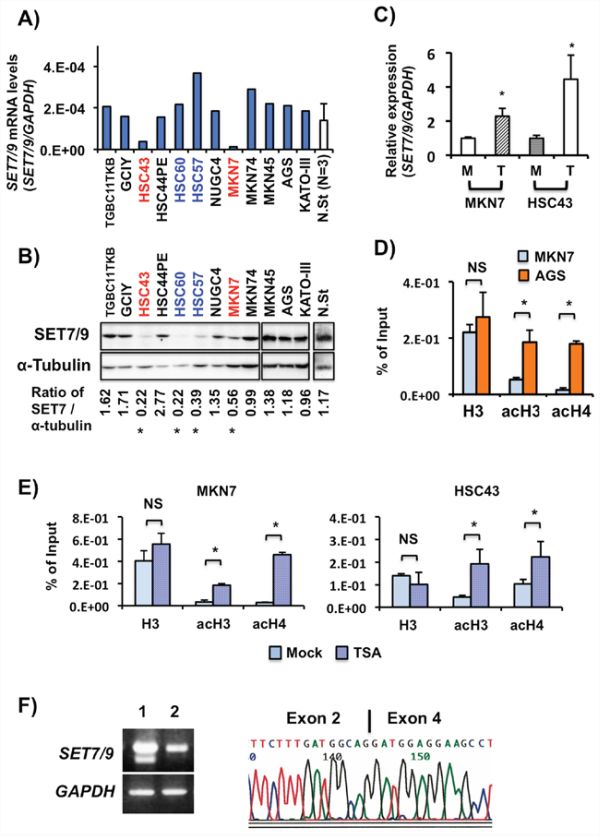
Figure 2: SET7/9 expression in GC cell lines and non-cancerous stomach tissues. A. SET7/9 mRNA expression in 12 GC cell lines and three non-cancerous stomach tissues from GC patients. Quantitative RT-PCR was performed using a LightCycler system. The 2nd Derivative Maximum method was utilized for determination of the concentrations, and relative expression was calculated using GAPDH expression as an internal control. The averages (blue columns) of two independent experiments for GC cell lines are indicated. The average (white column) and standard deviation (S.D) of the SET7/9 expression levels in the three non-cancerous stomach tissues (N.St) were also calculated. B. WB analysis of SET7/9 protein. a-Tubulin was used as an internal control. The Image-J 1.47v software software (http://imagej.nih.gov/ij/index.html) was used to calculate the SET7/9 protein expression levels. Red, GC cases with weak expression of both SET7/9 mRNA and protein; blue, GC cases with strong SET7/9 mRNA but weak SET7/9 protein expression. C. qRT-PCR analysis of SET7/9 expression in MKN7 and HSC43 cells after epigenetic drug treatment. MKN7 and HSC43 cells were treated with 0.3 mM trichostatin A (TSA; WAKO, Osaka, Japan) for 24 hrs. M, mock; T, TSA. *P < 0.01. D. qChIP assay of the SET7/9 promoter region in AGS and MKN7 cells exhibiting strong and weak SET7/9 expression, respectively. ChIP was quantitatively performed with anti-histone H3, and anti-acetylated histone H3 and H4 polyclonal antibodies. Input DNA samples were used as internal controls. E. Effects on histone H3 and H4 acetylation in MKN7 cells after TSA treatment. F. Longer RT-PCR of SET7/9 exons 1-8 in TGBC11TKB and KATO-III cells (left). Sequencing analysis of the PCR product of SET7/9 in TGBC11TKB cells. After subcloning of the RT-PCR product showing an abnormal SET7/9 transcript in TGBC11TKB cells, sequencing was performed (right). Deletion of exon 3 predicts to encode a truncated SET7/9 protein lacking the SET domain (5,6). We could not detect this predicted small SET7/9 protein (62 aa) in TGBC11TKB cells on WB using anti-SET7/9 antibodies, although wild-type SET7/9 (366 aa) was detected (see Figure 2B).
SET7/9 expression was enhanced in MKN7 and HSC43 cells after treatment with Trichostatin A (TSA) a histone deacetylase inhibitor (HDACi) (P < 0.01, Figure 2C). Treatment of SAHA, another HDACi, also showed significantly increased expression of SET7/9 in MKN7 cells (data not shown). We observed that the histone acetylation levels of H3 and H4 at the SET7/9 promoter region in AGS cells with strong SET7/9 expression were much higher than those in its expression-low MKN7 cells by quantitative chromatin immunoprecipitation (qChIP) assay (P < 0.01, Figure 2D, and Supplementary Figure S2A). The histone acetylation levels of H3 and H4 were significantly increased in MKN7 and HSC43 cells with TSA treatments (P < 0.01, Figure 2E). In contrast, conventional ChIP assays did not show any differences of the H3K4me3, H3K9me3 and H3K27me3 levels between AGS and MKN7 cells (Supplementary Figure S2B). Although SET7/9 expression was increased in MKN7 and HSC43 cells with a de-methylating reagent, 5-aza-dC treatment (P < 0.01), CGI hypermethylation at the promoter region of SET7/9 was not detected in any of the GC cell lines including them and 25 primary GC tissues examined (Supplementary Figures S2C and S2D).
Genetic alterations of SET7/9 were examined in 12 GC cell lines and 25 primary GC tissues. By long RT-PCR from exons 1-8 and PCR-SSCP, TGBC11TKB cells exhibited a deletion of exon 3 and a C to T transition at the first nucleotide of exon 3 in SET7/9 mRNA and genomic DNA, respectively (Figure 2F, and Supplementary Figures S2E and S2F), which predicts to encode a truncated SET7/9 protein. Except for TGBC11TKB cells, no mutation was detected in any cases examined.
The effects of SET7/9 knockdown in GCs
Expression of both SET7/9 mRNA and protein were inhibited in four GC cell lines (MKN74, MKN45, AGS and KATO-III) after SET7/9 siRNAs transfection (Figures 3A-3C). With the exception of MKN74 cells, these three GC cell lines with SET7/9 knockdown exhibited higher cell proliferation rates compared with ones with negative control siRNA (Figure 3D). Matrigel invasion assays demonstrated that SET7/9 knockdown significantly enhanced cell invasion (using transwells with matrigels) as well as migration (ones without matrigels) in MKN74, MKN45 (Figure 3E), and AGS cells (Figure 7D, see data on AGS cells with si7-1transfection). Similar effects on cell migration were found in AGS and MKN74 cells with SET7/9 knockdown on Scratch assay (Supplementary Figure S3).
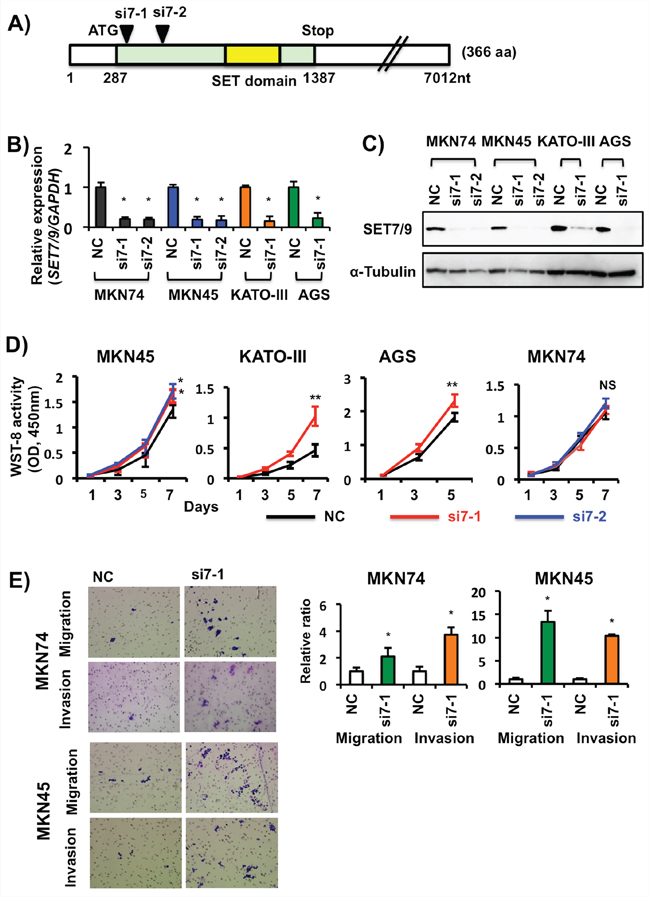
Figure 3: Effects of siRNA-based SET7/9 knockdown in GC cells. A. Locations of the two SET7/9 siRNAs used in this study. B. qRT-PCR analysis of SET7/9 expression in four GC cell lines after its siRNA transfection. The average (column) ± S.D (bar) of three independent experiments is indicated. *P < 0.01. C. WB analysis of SET7/9 protein expression in GC cell lines. a-Tubulin was used as an internal control. D. Cell proliferation assays of four GC cell lines after SET7/9 knockdown. The number of viable cells was determined using a WST-8 cell proliferation assay kit. Three independent assays were conducted. Representative data are shown and error bars indicate S.D. *P < 0.05; **P < 0.01; NS, not significant. E. Cell migration and invasion assays of MKN74 and MKN45 cells after SET7/9 knockdown. GC cells with SET7/9 (si7-1) and negative control (NC) siRNA transfection were grown in 24-transwell plates coated with (invasion) or without (migration) matrigel for 18-48 hrs. The total numbers of migrating and invading cells on the membrane were counted, and then relative migration and invasion ratios were calculated. The average (column) ± S.D (bar) of three independent experiments is indicated (*P < 0.01). Photographs show representative fields of migrating and invading cancer cells on the membrane (x100).
Analysis of SET7/9 target genes by microarray
On microarray analysis, we detected the 313 down- (≤1.5-fold) and 127 up-regulated (≥1.5-fold) probes in MKN74, and 483 and 172 probes in MKN45 cells, respectively, after SET7/9 knockdown. Moreover, expression of 37 (27 genes) and 8 (7 genes) probes were commonly decreased and increased in these two cell lines, respectively (Figures 4A and 4B). Among them, we selected five decreased (CCDC28B, SREK1IP1/P18SRP/SFRS12IP1, RASL10B, PHF21A/BHC80 and PGC) and two increased genes (IL-11 and CXCL2), by microarray, as differentiation-, splicing-, inflammation-, or tumor-related genes. The expression changes of these seven genes were confirmed in MKN74 and MKN45 cells with SET7/9 knockdown on conventional RT-PCR and qRT-PCR (Figures 4C and 4D). The expression levels of these genes were also changed in KATO-III and AGS cells after SET7/9 knockdown. As for the three matrix metalloproteinase genes selected, MMP1 was activated in all the four cell lines examined, and the expression levels of MMP9 and MMP7 were increased in MKN74 and the other three GC cell lines, respectively (Figures 4C and 4D).
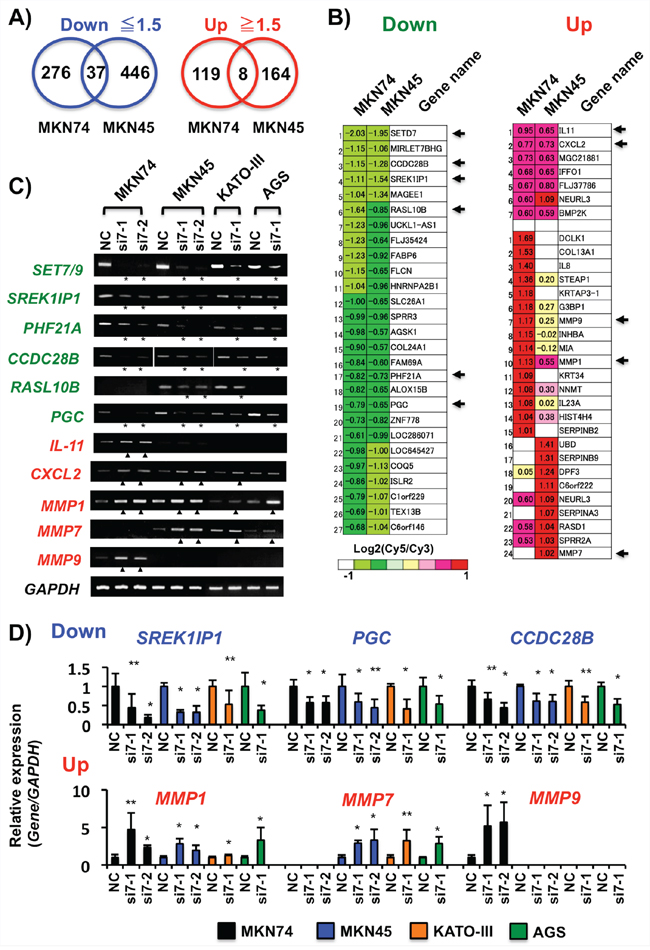
Figure 4: Analyses of the SET7/9 target genes in GC cells. A. Venn diagrams of transcriptionally down- and up-regulated gene expression of the genes in MKN74 and MKN45 cells after SET7/9 knockdown by microarray. The cut-off values of altered expression levels were determined as 1.5-fold change in GC cells with SET7/9 knockdown compared to ones with negative control transfection. B. Heat maps of transcriptionally down- and up-regulated gene expression of the genes in MKN74 and MKN45 cells after SET7/9 knockdown. The expression levels of the genes are represented by log2 ratios, and cut-off is determined as 1.5-fold changes. Six genes showing reduced expression including SET7/9 (Green), and five (Red) ones showing increased expression were selected in this study, as shown by arrows. C. RT-PCR analyses of the predicted STE7/9 target genes in 4 GC cell lines after SET7/9 siRNA transfection. MKN74, MKN45, KATO-III and AGS cells, which exhibited strong SET7/9 expression, were used in this study. At 48 hrs after transfection, RT-PCR was performed. GAPDH was used as an internal control. PCR products showing down- and up-regulation of the SET7/9 target genes are shown by asterisks and triangles, respectively. D. qRT-PCR analyses of the selected genes showing increased and decreased expression by SET7/9 knockdown in four GC cells. The average (column) ± S.D (bar) of three independent experiments is indicated. *P < 0.01; **P < 0.05.
On the contrary, SREK1IP1, CCDC28B, RASL10B and PGC were activated in MKN7 cells with exogenous SET7/9 overexpression, while PHF21A was not (Figures 5A, 5B and 5D). The SREK1IP1, PHF21A, CCDC28B and PGC genes were expressed in non-cancerous stomach tissues by RT-PCR (Figure 5B). In contrast, SET7/9 overexpression inhibited expression of IL-11, CXCL2, MMP1, MMP7 and MMP9 in MKN7 cells (Figure 5C and 5D).
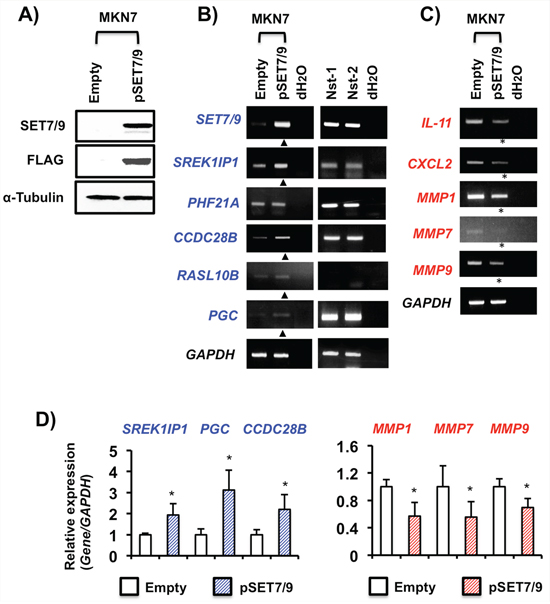
Figure 5: Effects of SET7/9 overexpression in GC cells. A. WB analysis of SET7/9 protein expression in MKN7 cells. Exogenous SET7/9 protein expression was detected using anti-SET7/9 and anti-FLAG antibodies. Empty: empty vector (p3X-FLAG-CMVTM-10), and pSET7/9: wild-type SET7/9. B. and C. RT-PCR analyses of the SET7/9 target genes in MKN7 cells after SET7/9 overexpression. The expression levels of the six genes in non-cancerous stomach tissues were also analyzed by RT-PCR. Expressions of the genes showing increased and decreased expression are shown by triangles and asterisks, respectively. D. qRT-PCR analyses of SREK1IP1, PGC, CCDC28B and three MMP genes in MKN7 cells exhibiting SET7/9 overexpression. The average (column) ± S.D (bar) of three independent experiments is indicated. *P < 0.01.
Identification of the H3K4 mono-methylated regions of SET7/9 target genes
We examined whether or not positively activated genes (SREK1IP1, PHF21A, CCDC28B and PGC) by SET7/9 are correlated with H3K4me1. Because H3K4me1 is known to be marked at transcriptionally active promoter and enhancer regions, we searched the regions by conventional ChIP assays using H3K4me1 antibodies according to the ChIP-seq data on the UCSC Genome Bioinformatics Site (http://genome.ucsc.edu/) (Supplementary Figures S4A, S5A and S6A), and further quantitatively evaluated their histone H3K4me1 levels by qChIP assays (Figure 6A).
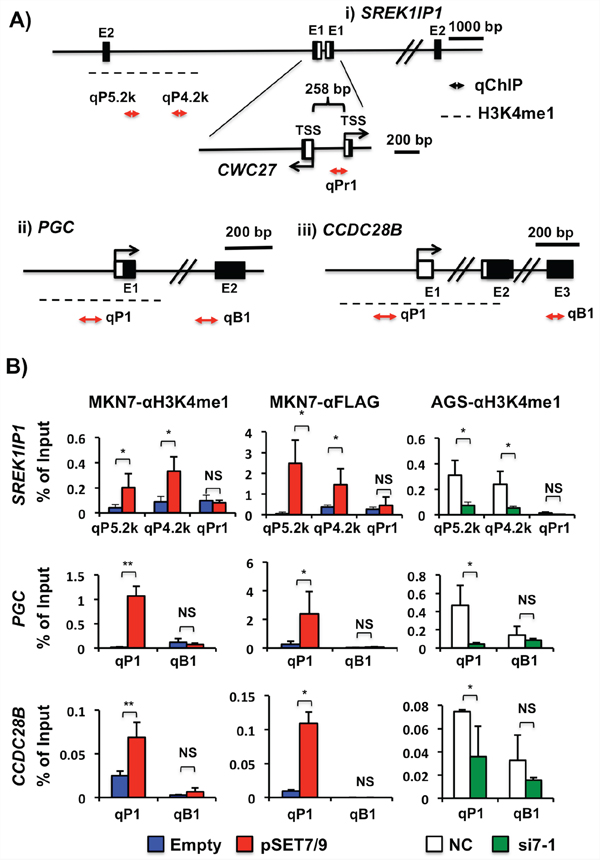
Figure 6: Transcriptional regulatory mechanisms of SREK1IP1, PGC and CCDC28B via H3K4me1 in GCs. A. Schematic representation of the 5′-regions of SREK1IP1, PGC and CCDC28B. TSS and closed boxes demonstrate the transcriptional start site and exons, respectively, and horizontal arrows are the qChIP sites in this study. A dotted line indicates an H3K4me1 enrichment region that has been shown by ChIP-seq data on the UCSC database. The SREK1IP1 and CWC27 genes are organized in a head-to-head configuration. B. The H3K4me1 levels in association with SET7/9 expression. qChIP was performed using anti-H3K4me1 antibodies in MKN7 cells with exogenous SET7/9 overexpression (left) and AGC cells with knockdown of the SET7/9 (right). As SET7/9 was overexpressed in MKN7 cells on transfection of SET7/9-3x FLAG expression vector, ChIP using anti-FLAG antibodies (middle) was performed.*P < 0.01; **P < 0.05; NS, not significant.
Computational analysis showed that the SREK1IP1 and CWC27 genes are located in a head-to-head orientation separated by a 258 bp sequence, which predicts a bidirectional promoter in this region. However, MKN7 cells with SET7/9 overexpression and AGS cells with its knockdown did not show any differences in the H3K4me1 level at this predicted promoter region compared to those with empty/negative control transfection (Figure 6B, qPr1 and Supplementary Figure S4B, Pr1). The ChIP-seq data on the UCSC demonstrates that the region located approximately 4-6 kb upstream from the SREK1IP1 transcriptional start site (TSS) is enriched with H3K4me1 in GM128 cells, a lymphoid cell line. We observed that overexpression of SET7/9 in MKN7 cells and knockdown of it in AGS cells increased and decreased the H3K4me1 level at this distal region from SREK1IP1 TSS, respectively, on conventional ChIP (Supplementary Figure S4B, P4.8k and P5.7k regions) and qChIP (Figure 6B, qP4.2k and qP5.2k regions) assays. Moreover, the H3K4me1 levels at this region tightly corresponded to expression of SREK1IP1 but not those of CWC27 (Supplementary Figure S4C).
H3K4me1 at the PGC and CCDC28B promoter regions was more enriched in MKN7 cells after transfection with the SET7/9 vector than in ones with the empty vector, while the region at exon 2 and exon 4 in PGC and CCDC28B, respectively, did not show any difference (Figure 6B and Supplementary Figure S5B). Knockdown of SET7/9 in AGC cells decreased the H3K4me1 levels at the promoters of these two genes, indicating that the H3K4me1 enrichment at their promoters may be related to their transcriptional regulation. As for PHF21A, the H3K4 levels were not changed at the regions within 5 kb upstream from its TSS in GC cells with SET7/9 overexpression and knockdown (Supplementary Figure S6B).
SET7/9 bound the H3K4 mono-methylated regions in SREK1IP1, PGC and CCDC28B
To further clarify whether or not SET7/9 directly bind the H3K4me1 enrichment region of the genes, we performed ChIP assay using anti-FLAG antibodies in MKN7 with exogenous SET7/9 overexpression. SET7/9 protein specifically bound approximately 4-6 kb upstream from the SREK1IP1 TSS, and the promoter regions of PGC and CCDC28B in MKN7 in response to SET7/9 overexpression (see MKN7-αFLAG, Figure 6B, and Supplementary Figures S4B and S5B). Thus, the SET7/9 binding regions in the target genes are consistent with data obtained using anti-H3K4me1 antibodies.
Functional analysis of the SET7/9 target genes in GC cells
To determine which functions of the genes detected are similar to those of SET7/9 in GCs, we evaluated the effects on cell migration in AGS cells. We conducted siRNA-based knockdown of SREK1IP1, PHF21A, CCDC28B and PGC in AGS cells (Supplementary Figures S7A and S7B). At 18hrs after transfection, cell migration was faster in AGS cells with SREK1IP1 siRNA than in those with a negative control and the other three siRNAs on scratch assay (Supplementary Figure S7C). Knockdown of SREK1IP1 in AGS cells enhanced cell proliferation rate (see Figure 7C), while knockdown of PHF21A, CCDC28B and PGC did not (data not shown).
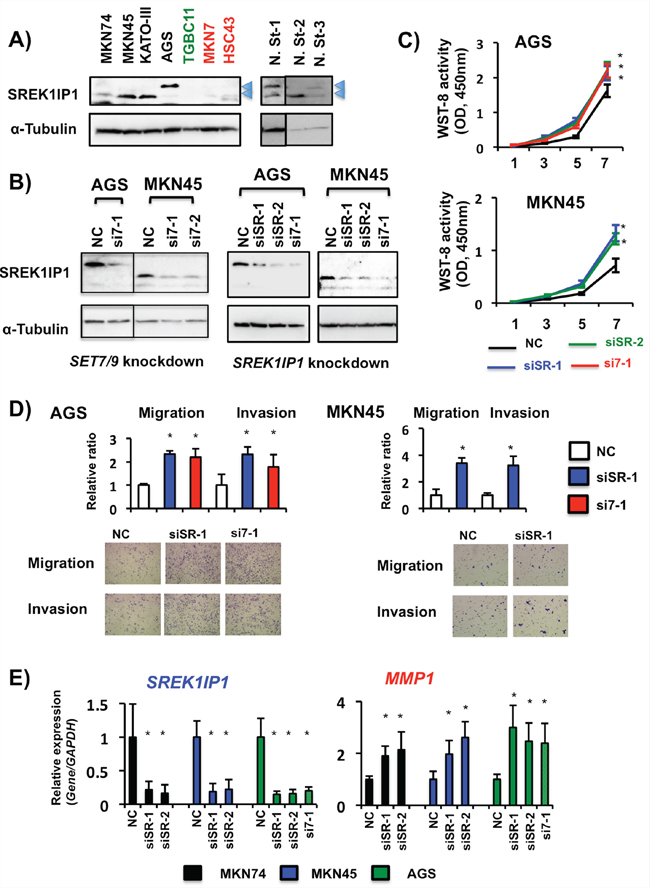
Figure 7: Effects of SREK1IP1 siRNA transfection in GC cells. A. Endogenous SREK1IP1 protein expression levels in seven GC cell lines and three non-cancerous stomach tissues. Although the size of SREK1IP1 protein is known to be 18kDa, two different sizes (approximately 18kDa and 20kDa, arrowheads) of SREK1IP1 protein were detected on WB. B. WB analyses of SREK1IP1 protein expression in AGS and MKN45 cells after SET7/9 (left, si7-1 and si7-2) and SREK1IP1 (right, siSR-1 and siSR-2) siRNA transfection. Knockdown of each gene inhibited expression of the predicted (18kDa) and large size (20kDa) proteins in MKN45 and AGS cells, respectively. C. Cell proliferation assays after SREK1IP1 knockdown in AGS and MKN45 cells. To compare the effects on SREK1IP1 and SET7/9, we also performed knockdown of SET7/9 (si7-1) in this study. Three independent assays of each knockdown were conducted. Representative data are shown and error bars indicate S.D. *P < 0.05. D. Matrigel cell invasion and migration assays of AGS and MKN45 cells. The total numbers of migrating and invading cells on the membrane in at least three fields were counted. The average (column) ± S.D (bar) of three independent experiments is indicated (*P < 0.01, top). Photographs show representative fields of migrating and invading cells on the membrane (bottom). E. The effects of SREK1IP1 knockdown by its siRNA transfection and the resultant MMP1 expression were quantitatively analyzed by qRT-PCR. The average (column) ± S.D (bar) of three independent experiments is indicated (*P < 0.01).
The biological significance of SET7/9 and SREK1IP1 in GC cell lines
We analyzed the relationship between SET7/9 and SREK1IP1 in GC cells. At least two different sizes of SREK1IP1 protein, approximately 18kDa and 20kDa, were detected in GC cell lines and non-cancerous stomach tissues by WB (Figure 7A), both of which expression were decreased by SET7/9 and SREK1IP1 knockdown in GC cells (Figure 7B). SREK1IP1 siRNA transfection in AGS and MKN45 cells promoted cell proliferation, migration and invasion (Figures 7C and 7D). Importantly, the relative effects on cell proliferation, migration and invasion in AGS cells with SREK1IP1 knockdown were similar to in those with SET7/9 knockdown.
To clarify the molecular mechanisms underlying cell invasion through SREK1IP1 and SET7/9, we examined expression of CXCL2, IL-11 and three MMPs in MKN45 and AGS cells. After SREK1IP1 siRNA transfection, MMP1 expression was enhanced in these GC cells, and expression of MMP7 and MMP9 was elevated in AGS and MKN74 cells after SREK1IP1 knockdown, respectively, which are the same effects as SET7/9 siRNAs in these cells (Figure 7E and Supplementary Figure S8A). Nevertheless, expression of IL-11 and CXCL2 were not activated by SREK1IP1 knockdown. Knockdown of PHF21A, CCDC28B and PGC in MKN74 and AGS cells did not induce any expressional changes of three MMPs (Supplementary Figure S8B). Thus, the target genes of SREK1IP1 in GC cells partially resembled to those of SET7/9.
SREK1IP1 expression analyses in GCs
We divided the 12 GC cell lines into two groups, i.e., SET7/9 alteration -positive (group A; SET7/9-mutated TGBC11TKB cells and four cell lines with low SET7/9 expression) and -negative (group B; remaining seven cases with high SET7/9 expression), and then compared the SREK1IP1 expression levels between them by qRT-PCR. The average SREK1IP1 expression level in the group A was significantly lower than that in the group B (P = 0.033, Supplementary Figure S8C). As for the 25 primary GCs, GC cases with loss or weak SET7/9 protein expression showed a tendency of weaker SREK1IP1 expression compared with ones with retained SET7/9 expression (P = 0.073, Supplementary Figure S8D).
DISCUSSION
In this study, we observed frequent SET7/9 reduction in primary GCs. Our data on SET7/9 and its target gene analyses demonstrated that SET7/9 knockdown in GC cells significantly promoted to cell proliferation, migration and invasion, and enhanced MMPs expression. We also observed that SET7/9 transcriptionally activated SREK1IP1, CCDC28B and PGC expression through H3K4me1. These data indicate that SET7/9 has tumor suppressive functions in GCs.
The characteristics of GCs with low SET7/9 expression in our study were perineural invasion-positive, Stage III-IV and lymph node metastasis-positive. Frequent SET7/9 reduction was preferentially found in diffuse and infiltrating type GCs, which are reported to be more malignant than intestinal type GCs [16]. It has been observed that SET7/9-knockout mice were viable and fertile, and did not develop tumors [17, 18]. Hence, absence of SET7/9 expression may be late event in gastric carcinogenesis and implicated in the clinical aggressiveness of GCs. In addition, reduced SET7/9 expression was significantly associated with shorter OS and DFS of our GC patients. Among the histone modification genes, overexpression of SUV39H1 and EZH2 has been suggested to correlate with poor prognosis in GCs [19, 20]. Reduced SET7/9 expression could be also used as a biomarker for predicting poor survival in GC patients.
The expression levels of SET7/9 mRNA significantly corresponded to it its protein expression in this study. SET7/9 was enhanced in MKN7 and HSC43 cells with treatment of HDACi and de-methylation reagents, indicating that reduced SET7/9 may be caused by epigenetic changes at the mRNA levels in GC cells. SET7/9 contains a dense CGI at the promoter region. The characteristics of genes silenced by CGI hypermethylation at the gene promoter region are generally known to be restoration of expression on 5-aza-dC but not TSA treatment [1], and distinct histone methylation patterns of H3K4 and H3K9 [21]. Nevertheless, neither CGI hypermethylation nor histone methylation patterns at the SET7/9 promoter region were found in GC cells with strong and low SET7/9 expression. Although we could not deny the possibility of CGI hypermethylation except the promoter region, such as a CpG shore and gene body region, the significant relationships between SET7/9 expression and histone acetylation patterns of histone H3 and H4 at its promoter region were shown in GC cells. Thus, epigenetic changes through decrease of the histone acetylation levels of SET7/9 should be considered as the factors associated with its gene silencing.
Recently, frameshift mutation of SET7/9 was reported in a castration-resistant prostate cancer by whole transcriptome sequencing [22]. We also observed an out-of-frame deletion in TGBC11TKB cells. Somatic mutations of the HMT genes including SET7/9 have been shown in several types of cancers according to the COSMIC database [23]. Hence, somatic mutations of SET7/9 as well as loss of its expression may contribute to the development of diverse cancers.
Our data on knockdown and overexpression of SET7/9 demonstrate that SET7/9 suppressed CXCL2, IL-11 and three MMP genes in GC cells, which were shown to promote not only cancer cell motility and invasion but also metastasis [24–26]. Most importantly, the phenotypes of SREK1IP1 functions were similar to those of SET7/9. First, the endogenous expression patterns of SREK1IP1 and SET7/9 were similar in primary GCs as well as GC cell lines. Second, knockdown of SREK1IP1 in GC cells promoted cell growth, migration and invasion, and increased transcriptional regulation of MMP1, MMP7 and MMP9. Thus, it is possible that SREK1IP act as a TSG as well as SET7/9, and they inversely suppressed three MMPs expression in GCs. Overexpression of these three MMPs has been reported in advanced GCs [25, 27]. MMP1 overexpression was also correlated with peritoneal metastasis and lymph node metastasis of GCs [27]. Therefore, a SET7/9-SREK1IP1-MMPs axis may be involved in GC progression.
Reduced SREK1IP1 expression was strongly associated with cell proliferation and invasiveness in our study, suggesting that SREK1IP1 may act as a TSG as well as SET7/9 in GCs. Somatic frameshift mutations at the (A)10 repeat site in SREK1IP1 exon 3 have been frequently detected in CRCs showing severe microsatellite instability [28]. It is likely that absence of SREK1IP1 is caused by not only loss of SET7/9 but also genetic alterations as a target of MSI. SREK1IP1 is predicted to participate as a splicing regulator [29], because overexpression and knockdown of SRrp86, a SREK1IP1 interacting protein, resulted in the alternative splicing of various genes, such as c-Jun and IκBβ, in cancer cells [30]. Further studies are necessary to clarify whether or not SREK1IP1 serves in the splicing process of genes.
The active histone marks of H3K4me1 and H3K27 acetylation (H3K27ac) are enrichments at the region from 4-6 kb upstream of SREK1IP1 TSS on the UCSC Genome Bioinformatics Site. In our study, overexpression and knockdown of SET7/9 in GC cells demonstrated that H3K4me1 enrichment at this distal region corresponded to the SET7/9 expression level. It has reported that Set7/9 was recruited to enhancers of the genes and mono-methylated H3K4 in a MyoD-dependent gene activation manner during muscle differentiation [31]. Therefore, the enrichment of H3K4me1 levels at this region may be important for transcriptional activation of SREK1IP1 by SET7/9. The enhancer regions of several genes are shown within the introns of neighboring genes [32]. For example, the enhancer region of the SHH gene for limb development is located within intron 5 of Lmbr1, where is more than 1 Mb far from the SHH promoter region [33]. Indeed, the distal region associated with SREK1IP1 expression is located within intron 1 of CWC28B. Thus, it is possible that the H3K4me1 enrichment region of SREK1IP1 detected in our ChIP assay may act as a transcriptional enhancer.
As for other two genes activated by SET7/9, PGC has widely been considered as a differentiation marker of gastric epithelial cells, and encodes pepsinogen C (PG-II), which is secreted from antral glands and corpus chief cells in the stomach and duodenal glands [34]. Although it remain uncertain the functions of CCDC28B, its was reported that depletion of CCDC28B in a ciliated human cell line, hTERT-RPE, established from retinal pigmented epithelial cells, resulted in defective ciliogenesis [35], suggesting that CCDC28B may paly a role in cellular differentiation. Since several pathological groups reported the ciliated metaplasia that is implicated in gastric ciliogenesis [36], it is important to analyze the relationship between CCDC28B and ciliated metaplasia further. Our data further support the observations that SET7/9 and H3K4me1 is correlated with cell-type- and differentiation-stage-specific genes [7, 31, 37], and postulate that it plays an important role in gastric cell differentiation through activating of PGC and CCDC28B.
SET7/9 is known to inhibit cell cycle and apoptosis through methylating of p53 and pRB as non-histones [9, 10, 38]. Taken together with our present finding, loss of SET7/9 expression may promote the tumor progression through absences of H3K4me1 and methylation of non-histones in carcinogenesis, and hence restoration of SET7/9 expression could contribute to new therapeutic strategies for GCs. On the contrary, SET7/9 inhibitors are suggested to be new therapeutic strategies for hormone-dependent breast cancers, such as ERa-positive ones [13, 39], even though there have been no reports on the SET7/9 mutation and expression change in primary breast cancer. Therefore, SET7/9 may play distinct roles in tissue- and cancer-specific manners.
In conclusion, our data provide evidence that SET7/9 has tumor suppressor functions in GCs, and suggest that loss of it may contribute to GC progression and aggressiveness. We also indicated the importance of abnormal gene regulatory mechanisms of loss of SET7/9, that is, a SET7/9-SREK1IP1-MMPs axis, for tumor invasion and migration. Furthermore, it is possible that loss of SET7/9 expression may serve as a potential diagnostic biomarker and a therapeutic target of GCs.
MATERIALS AND METHODS
Cell lines and tissue samples
We used 12 GC cell lines, AGS, GCIY, HSC43, HSC44PE, HSC57, HSC60, NUGC4, KATO-III, MKN7, MKN45, MKN74 and TGBC11TKB, which were obtained as described previously [40, 41]. HSC44PE, HSC57 and HSC60 cells were obtained from Dr. Kazuyoshi Yanagihara (National Cancer Center, Japan). All the cell lines were grown in Dulbecco’s modified Eagle’s medium (DMEM) or RPMI1640 medium containing 10% fetal bovine serum (FBS), and 1% penicillin and streptomycin.
A total of 376 primary GCs from formalin-fixed paraffin-embedded (FFPE) tissue microarray samples in Seoul National University (SNU) were used for immunohistochemistry. In addition, a total of other 25 primary GCs of frozen and matched FFPE tissues were obtained from the affiliated hospital of Tokyo Medical and Dental University (TMDU) in order to compare SET7/9 expression at mRNA and protein levels. Informed consent was obtained from all the patients, and these studies were approved by the Medical Research Ethics Committee for Genetic Research of TMDU and by the Institutional Review Board for Human Subject Research at SNU Hospital.
RNA extraction, reverse transcription (RT)-PCR and quantitative RT-PCR (qRT-PCR)
Total RNA was extracted using Trizol reagent (Thermo Fisher Scientific Inc., Waltham, MA) or RNeasy (QIAGEN, Valencia, CA), and then reversely transcribed to cDNA using a Superscript kit III. We amplified all genes examined in this study with multiple cycle numbers (28-38 cycles). After PCR amplification, their expression levels were semi-quantitatively determined in 2-3% agarose gels. By using a longer RT-PCR from SET7/9 exons 1-8, we searched for large deletions including exon skipping in 12 GC cell lines.
qRT-PCR was conducted with a LightCycler system (Roche Diagnostics, Mannheim, Germany) using DNA Master SYBR Green I, according to the manufacturer’s instructions. The 2nd Derivative Maximum method was performed for the determination of concentrations using LightCycler software version 3.5. GAPDH (Glyceraldehyde 3-phosphate dehydrogenase) was used as an internal control for RT-PCR. At 48 hrs after transfection, cells were harvested, and then conventional RT-PCR and qRT-PCR were performed. The representative primer sequences and their PCR conditions are shown in Supplementary Table S1.
Gene mutation and methylation analyses
The methods of PCR-single strand conformation polymorphism (SSCP) and methylation-specific PCR (MSP) are shown in Supplementary Information. The representative primer sequences and their PCR conditions are shown in Supplementary Tables S1 and S2.
Transfection
The SET7/9 expression-high GC cell lines, MKN74 (5x105 cells/well), MKN45 (3x105/well), AGS (1x105/well), and KATO-III (2x105/well), were transfected with small interfering RNA (siRNA) of SET7/9 (SASI_Hs01_00123130 and SASI_Hs01_00123132; Sigma-Aldrich Japan, Ishikari, Japan) or negative control (Mission siRNA Universal Negative Control; Sigma-Aldrich Japan) to give a final concentration of 50nM using a Neon electroporation system (Thermo Fisher Scientific Inc., Waltham, MA). In addition, SREK1IP1 (SASI_Hs01_00221119 and SASI_Hs01_00221122), PHF21A (SASI_Hs02_00322973) and CCDC28B (SASI_Hs02_00356628) siRNAs (Sigma-Aldrich Japan) and PGC siRNA (SI00683144, QIAGEN) were used.
MKN7 cells were transfected with 2 μg of the wild type-SET7/9-3xFLAG expression vector [9] or the empty vector by electroporation. At 48-72 hrs after transfection, transfected-cells were harvested, and then used for RNA and protein analyses.
Immunohistochemistry
Mouse monoclonal anti-SET7/9 antibody (clone 5F2.3, 04-0805; Merck Millipore, Darmstadt, Germany) was diluted at 1:100. Immunohistochemistry of SET7/9 protein was performed using a Histofine Simple Stain MAX PO system (Nichirei Biosciences Inc., Tokyo, Japan) and a Ventana BenchMark XT automatic immunostainer system (Ventana Medical Systems, Tucson, AZ) according to the manufacturer’s protocol. We assessed the SET7/9 staining score as to both the percentage of the extent of cell staining and intensity. Briefly, immunoreactivity for SET7/9 was scored as follows; proportional score (PS): 0 (negative), 1 (1 - 5% of tumor cells), 2 (6 - 25%), 3 (26-50%), and 4 (51~100%), and intensity score (IS): 0 (no tumor cell staining), 1 (weak), 2 (moderate), and 3 (strong). The total score (TS) was obtained by summing IS and PS. Because more than half of the tumor cells were stained in SET7/9 expression cases, TS ≥ 6 was considered to represent high expression (retained expression) of SET7/9. Thus, we made two groups, SET7/9 retained (high) and loss/weak (low).
Western blot
Western blot (WB) was performed as described previously [42]. The primary antibodies used in this study were anti-SET7/9 (rabbit monoclonal, C24B1, 1:1000; Cell Signaling Technology, Danvers, MA; and mouse monoclonal, clone 5F2.3, 1:500; Merck Millipore), anti-SREK1IP1 (rabbit polyclonal, 1:200; GeneTex Inc, Irvine, CA), anti-H3K4me1 (No. 39298, rabbit polyclonal, 1:5000; Active Motif, Carlsbad, CA), anti-FLAG (mouse monoclonal M2, 1:10,000; Sigma-Aldrich Japan), and anti-α-Tubulin (mouse monoclonal, 1:400; Santa Cruz Biotechnology, Santa Cruz, CA) antibodies.α-Tubulin was used as an internal control for WB.
Microarray analysis
At 48hrs after transfection of SET7/9 siRNA, total RNA was extracted from MKN74 and MKN45 cells using Trizol reagent. DNA microarray analysis was carried out by DNA Chip Research Inc. (Kanagawa, Japan) using the Agilent Human GE 4x44K v2 Microarray (Design ID: 026652). The raw data have been deposited in NCBI Gene Expression Omnibus (GEO) under the accession number GSE59834.
Chromatin immunoprecipitation (ChIP)
We prepared sonicated DNA samples from AGS and MKN7 cells using a ChIP-IT Express (No. 39163, Active Motif), and then incubated with 1-3 μg of three histone H3 methylation-related (H3K4me3, No. 39915; H3K9me3, No. 29766; and H3K27me3, No. 39155; Active Motif) and two histone acetylation-related antibodies (acetyl-histone H3 (K9 and K14), #06-599, acetyl-histone H4 (K5, K8, K12, K16), #06-866; Upstate). Moreover, after transfection of the SET7/9 expression vector or its siRNA into GC cells, ChIP assays were conducted using anti-H3K4me1 polyclonal (ab8895, Abcam, Cambridge, UK) and anti-FLAG monoclonal (Sigma-Aldrich Japan) antibodies. Histone H3 (No. 39163, Active Motif) and Normal Rabbit IgG (No. 2729, Cell Signaling Technology) were used as positive and negative controls, respectively. Input DNA samples were used as internal controls. The primer sequences and their conditions are shown in Supplementary Table S2.
Cell growth, migration and invasion
We transfected siRNA of SET7/9 or SREK1IP1, or a negative control into MKN74 (3x103 cells) MKN45 (3x103), AGS (0.5x103), and KATO-III (0.5x103) cells on 96-well plates. Cell proliferation was evaluated on days 1, 3, 5 and 7 after replating by determining the number of cells with a Cell Counting Kit-8 (Dojindo, Kumamoto, Japan), according to the manufacturer’s instructions [42].
The siRNA-transfected GC cells (1x104 cells) were grown in 24-well transwells (8 μm pore size)-coated with (invasion) or without (migration) matrigel (BD Biosciences, Franklin Lakes, NJ) [43]. After 18-48 hrs, invading cells were detected by Diff-Quik staining. The method of scratch assay is shown in Supplementary Information.
Statistics
All the experiments were repeated at least twice with duplicate or triplicate samples. The Mann-Whitney’s U-test, t-test and χ2-test were used to compare the values for the test and control samples. The nonparametric Mann—Whitney U-test was used to determine the differences between the cases with SET7/9-expression high and low groups. A value of P < 0.05 was taken as being significant. Patient survival was calculated from the date of surgery until death or the date of last follow-up. Survival was evaluated by the Kaplan-Meier method, and uni- and multi-variate survival analyses were performed using the Cox proportional hazard model.
ACKNOWLEDGMENTS
The authors thank Ms. Hiromi Nagasaki (TMDU) and the staff members in Kim WH’s laboratory (SNU) for their technical assistance.
CONFLICTS OF INTEREST
The authors have declared no competing intersts.
GRANT SUPPORT
This work was supported in part by Grants-in-Aid for Scientific Research (C) (24590444), (A) (25253081), and the A3 Foresight Program (AA005), from the Japan Society for the Promotion of Science (JSPS).
Author contribution
Conception, design of the study and writing of the manuscript: Y.A., Y.Y., S.T.; analysis and interpretation of data: Y.A., Y.K., S.B., S.S., T.N., A.S.; patient sample collection: W.H.K., S.B., K.K., T.K., Y.E.; drafting or revision of the manuscript: Y.A., Y.C., D.D., W.H.K., W.G.Z., Y.Y., S.T.
Abbreviations
CGI, CpG island; ChIP, Chromatin immunoprecipitation; DFS, disease-free survival; GC, gastric cancer; HMT; histone methyltransferase; H3K4me1, histone H3 lysine 4 mono-methylation; IHC, immunohistochemistry; MMP, matrix metalloproteinase; MSP, methylation-specific polymerase chain reaction; OS, overall survival; (q)RT-PCR; (quantitative) reverse transcription-polymerase chain reaction; TSGs, tumor suppressor genes; TSS, transcriptional start site.
REFERENCES
1. Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer. 2011; 11:726–734.
2. Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet. 2012; 13:343–357.
3. Zentner GE, Tesar PJ, Scacheri PC. Epigenetic signatures distinguish multiple classes of enhancers with distinct cellular functions. Genome Res. 2011; 21:1273–1283.
4. Varier RA, Timmers HT. Histone lysine methylation and demethylation pathways in cancer. Biochim Biophys Acta. 2011; 1815:75–89.
5. Wilson JR, Jing C, Walker PA, Martin SR, Howell SA, Blackburn GM, Gamblin SJ, Xiao B. Crystal structure and functional analysis of the histone methyltransferase SET7/9. Cell. 2002; 111:105–115.
6. Nishioka K, Chuikov S, Sarma K, Erdjument-Bromage H, Allis CD, Tempst P, Reinberg D. Set9, a novel histone H3 methyltransferase that facilitates transcription by precluding histone tail modifications required for heterochromatin formation. Genes Dev. 2002; 16:479–489.
7. Tao Y, Neppl RL, Huang ZP, Chen J, Tang RH, Cao R, Zhang Y, Jin SW, Wang DZ. The histone methyltransferase Set7/9 promotes myoblast differentiation and myofibril assembly. J Cell Biol. 2011; 194:551–565.
8. Pradhan S, Chin HG, Estéve PO, Jacobsen SE. SET7/9 mediated methylation of non-histone proteins in mammalian cells. Epigenetics. 2009; 4:383–387.
9. Liu X, Wang D, Zhao Y, Tu B, Zheng Z, Wang L, Wang H, Gu W, Roeder RG, Zhu WG. Methyltransferase Set7/9 regulates p53 activity by interacting with Sirtuin 1 (SIRT1). Proc Natl Acad Sci USA. 2011; 108:1925–1930.
10. Munro S, Khaire N, Inche A, Carr S, La Thangue NB. Lysine methylation regulates the pRb tumour suppressor protein. Oncogene. 2010; 29:2357–2367.
11. Estéve PO, Chin HG, Benner J, Feehery GR, Samaranayake M, Horwitz GA, Jacobsen SE, Pradhan S. Regulation of DNMT1 stability through SET7-mediated lysine methylation in mammalian cells. Proc Natl Acad Sci USA. 2009; 106:5076–5081.
12. Wang D, Zhou J, Liu X, Lu D, Shen C, Du Y, Wei FZ, Song B, Lu X, Yu Y, Wang L, Zhao Y, Wang H, et al. Methylation of SUV39H1 by SET7/9 results in heterochromatin relaxation and genome instability. Proc Natl Acad Sci USA. 2013; 110:5516–5521.
13. Subramanian K, Jia D, Kapoor-Vazirani P, Powell DR, Collins RE, Sharma D, Peng J, Cheng X, Vertino PM. Regulation of estrogen receptor alpha by the SET7 lysine methyltransferase. Mol Cell. 2008; 30:336–347.
14. Jemal A, Center MM, DeSantis C, Ward EM. Global patterns of cancer incidence and mortality rates and trends. Cancer Epidemiol Biomarkers Prev. 2010; 19:1893–1907.
15. Yuasa Y. Control of gut differentiation and intestinal-type gastric carcinogenesis. Nat Rev Cancer. 2003; 3:592–600.
16. Adachi Y, Yasuda K, Inomata M, Sato K, Shiraishi N, Kitano S. Pathology and prognosis of gastric carcinoma: well versus poorly differentiated type. Cancer. 2000; 89:1418–1424.
17. Campaner S, Spreafico F, Burgold T, Doni M, Rosato U, Amati B, Testa G. The methyltransferase Set7/9 (Setd7) is dispensable for the p53-mediated DNA damage response in vivo. Mol Cell. 2011; 43:681–688.
18. Lehnertz B, Rogalski JC, Schulze FM, Yi L, Lin S, Kast J, Rossi FM. p53-dependent transcription and tumor suppression are not affected in Set7/9-deficient mice. Mol Cell. 2011; 43:673–680.
19. Cai L, Ma X, Huang Y, Zou Y, Chen X. Aberrant histone methylation and the effect of Suv39H1 siRNA on gastric carcinoma. Oncol Rep. 2014; 31:2593–2600.
20. Matsukawa Y, Semba S, Kato H, Ito A, Yanagihara K, Yokozaki H. Expression of the enhancer of zeste homolog 2 is correlated with poor prognosis in human gastric cancer. Cancer Sci. 2006; 97:484–491.
21. Kondo Y, Shen L, Issa JP. Critical role of histone methylation in tumor suppressor gene silencing in colorectal cancer. Mol Cell Biol. 2003; 23:206–215.
22. Sowalsky AG, Xia Z, Wang L, Zhao H, Chen S, Bubley GJ, Balk SP, Li W. Whole transcriptome sequencing reveals extensive unspliced mRNA in metastatic castration-resistant prostate cancer. Mol Cancer Res. 2015; 13:98–106.
23. Kudithipudi S, Jeltsch A. Role of somatic cancer mutations in human protein lysine methyltransferases. Biochim Biophys Acta. 2014; 1846:366–379.
24. Kavandi L, Collier MA, Nguyen H, Syed V. Progesterone and calcitriol attenuate inflammatory cytokines CXCL1 and CXCL2 in ovarian and endometrial cancer cells. J Cell Biochem. 2012; 113:3143–3152.
25. Fanelli MF, Chinen LT, Begnami MD, Costa WL Jr, Fregnami JH, Soares FA, Montagnini AL. The influence of transforming growth factor-α, cyclooxygenase-2, matrix metalloproteinase (MMP)-7, MMP-9 and CXCR4 proteins involved in epithelial-mesenchymal transition on overall survival of patients with gastric cancer. Histopathology. 2012; 61:153–161.
26. Bao W, Fu HJ, Jia LT, Zhang Y, Li W, Jin BQ, Yao LB, Chen SY, Yang AG. HER2-mediated upregulation of MMP-1 is involved in gastric cancer cell invasion. Arch Biochem Biophys. 2010; 499:49–55.
27. Inoue T, Yashiro M, Nishimura S, Maeda K, Sawada T, Ogawa Y, Sowa M, Chung KH. Matrix metalloproteinase-1 expression is a prognostic factor for patients with advanced gastric cancer. Int J Mol Med. 1999; 4:73–77.
28. Williams DS, Bird MJ, Jorissen RN, Yu YL, Walker F, Zhang HH, Nice EC, Burgess AW. Nonsense mediated decay resistant mutations are a source of expressed mutant proteins in colon cancer cell lines with microsatellite instability. PLoS One. 2010; 5:e16012.
29. Heese K, Fujita M, Akatsu H, Yamamoto T, Kosaka K, Nagai Y, Sawada T. The splicing regulatory protein p18SRP is down-regulated in Alzheimer’s disease brain. J Mol Neurosci. 2004; 24:269–276.
30. Solis AS, Patton JG. Analysis of SRrp86-regulated alternative splicing: control of c-Jun and IκBβ activity. RNA Biol. 2010; 7:486–494.
31. Blum R, Vethantham V, Bowman C, Rudnicki M, Dynlacht BD. Genome-wide identification of enhancers in skeletal muscle: the role of MyoD1. Genes Dev. 2012; 26:2763–2779.
32. Levine M. Transcriptional enhancers in animal development and evolution. Curr Biol. 2010; 20:R754-R763.
33. Lettice LA, Heaney SJ, Purdie LA, Li L, de Beer P, Oostra BA, Goode D, Elgar G, Hill RE, de Graaff E. A long-range Shh enhancer regulates expression in the developing limb and fin and is associated with preaxial polydactyly. Hum Mol Genet. 2003; 12:1725–1735.
34. Samloff IM, Liebman WM. Cellular localization of the group II pepsinogens in human stomach and duodenum by immunofluorescence. Gastroenterology. 1973; 65:36–42.
35. Cardenas-Rodriguez M, Osborn DP, Irigoín F, Graña M, Romero H, Beales PL, Badano JL. Characterization of CCDC28B reveals its role in ciliogenesis and provides insight to understand its modifier effect on Bardet-Biedl syndrome. Hum Genet. 2013; 132:91–105.
36. Rubio CA, Nesi G, Zampi GC, de Ruiz PA, Jessurun J, Jónasson J, Hojman R, Kogan Z, Antonioli D, Miller ML, Hirota T, Itabashi T, Mandai K, et al. Gastric ciliated metaplasia. A study of 3406 gastrectomy specimens from dwellers of the Atlantic and the Pacific basins. J Clin Pathol. 2005; 58:605–610.
37. Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, Ching KA, Antosiewicz-Bourget JE, Liu H, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009; 459:108–112.
38. Ivanov GS, Ivanova T, Kurash J, Ivanov A, Chuikov S, Gizatullin F, Herrera-Medina EM, Rauscher F 3rd, Reinberg D, Barlev NA. Methylation-acetylation interplay activates p53 in response to DNA damage. Mol Cell Biol. 2007; 27:6756–6769.
39. Wagner T, Jung M. New lysine methyltransferase drug targets in cancer. Nat Biotechnol. 2012; 30:622–623.
40. Hashimoto Y, Akiyama Y, Yuasa Y. Multiple-to-multiple relationships between microRNAs and target genes in gastric cancer. PLoS One. 2013; 8:e62589.
41. Tani Y, Akiyama Y, Fukamachi H, Yanagihara K, Yuasa Y. Transcription factor SOX2 up-regulates stomach-specific pepsinogen A gene expression. J Cancer Res Clin Oncol. 2007; 133:263–269.
42. Wanajo A, Sasaki A, Nagasaki H, Shimada S, Otsubo T, Owaki S, Shimizu Y, Eishi Y, Kojima K, Nakajima Y, Kawano T, Yuasa Y, Akiyama Y. Methylation of the calcium channel-related gene, CACNA2D3, is frequent and a poor prognostic factor in gastric cancer. Gastroenterology. 2008; 135: 580–590.
43. Hellebrekers DM, Lentjes MH, van den Bosch SM, Melotte V, Wouters KA, Daenen KL, Smits KM, Akiyama Y, Yuasa Y, Sanduleanu S, Khalid-de Bakker CA, Jonkers D, Weijenberg MP, et al. GATA4 and GATA5 are potential tumor suppressors and biomarkers in colorectal cancer. Clin Cancer Res. 2009; 15:3990–3997.
44. Washington, K. 7th Edition of the AJCC Cancer Staging Manual: Stomach. Ann Surg Oncol. 2010; 17:3077–3079.