
INTRODUCTION
Uveal melanoma (UM) is the most common primary intraocular malignancy in adults with an incidence of 5.6 cases per million per year between 1973 and 2008 reported in the United States [1]. The number of newly reported cases of UM over this time frame remained at a relatively steady rate, suggesting that the influence of ultraviolet radiation on UM is considerably smaller than the association observed with cutaneous melanoma (CMM).
In UM a recurrent somatic mutation occurs frequently in GNAQ, encoding an alpha subunit of heterotrimeric G proteins, located at chromosome band 9q21 [2]. GNA11, a paralog of GNAQ located in chromosome band 19p13.3, is also recurrently mutated at the same codon [3]. Hotspot GNAQ p.Q209 mutations are found in 45% of primary UM and 22% of metastases, while GNA11 p.Q209 mutations are found in 32% of primary tumors and 57% of UM metastases. A second mutation hotspot has also been identified at codon p.R183 in both genes. Overall, 83% of UM acquire mutations in either GNAQ or GNA11. Unlike some other somatic mutations in UM these are not associated with prognosis.
There are also commonly occurring loss-of-function mutations in the tumor suppressor gene BAP1 (BRCA1 associated protein-1) located on chromosome 3 [4]. These mutations are associated with worse prognosis. Approximately 40% of UM harbor inactivating somatic mutations in BAP1, which occur along the length of the gene and generally result in protein truncations. Metastasizing tumors are also associated with monosomy 3, suggesting loss of both copies of BAP1 is necessary [5]. There are also commonly occurring gene expression profiles whereby metastatic tumors present with clusters of up-regulated genes on chromosome 8q, and down-regulated genes on chromosomes 3 and 6q [6, 7].
There are two further frequently mutated genes in UM. Recurrent mutations in SF3B1 occur at codon 625 in approximately 18% of tumors and are associated with better prognosis [8], as are mutations in EIF1AX [9].
RESULTS AND DISCUSSION
Protein altering single nucleotide variants and small insertion/deletions
We performed WGS or WES on 28 untreated UM samples and identified a total of 297 non-synonymous mutations (mean 10.6, range 0 to 53, Supplementary Table 1), including both single nucleotide variants (SNVs) and indels (Supplementary Table 2).
We first assessed mutations in known UM drivers and detected 11 mutations in BAP1 (7 frameshifting indels, 2 splice mutations, 1 nonsense mutation and 1 missense mutation), 14 mutations occurred at GNA11 p.Q209P, 7 mutations occurred at GNAQ p.Q209P and a single mutation at GNAQ p.G48L. As expected the mutations in GNA11 and GNAQ were mutually exclusive. We detected 4 mutations occurring in EIF1AX (p.P2L, p.G6V, p.G8R and a splice mutation) which were found to be mutually exclusive with BAP1 mutations. We also detected 3 mutations in SFB31: p.R625C, p.R625H and p.K666T (Table 1, Supplementary Table 2).
Table 1: Driver mutations in UM
Tumor ID |
BAP1 |
EIF1AX |
GNA11 |
GNAQ |
PLCB4 |
SF3B1 |
|---|---|---|---|---|---|---|
00028-001-CL |
p.D73Vfs*4 |
p.Q209L |
p.R625C |
|||
00038-001-CL |
p.Q209L |
|||||
00061-001-CL |
p.R699Qfs*6 |
p.Q209L |
||||
00085-001-CL |
p.H224Qfs*14 |
p.Q209L |
||||
00099-001-CL |
p.Q209L |
|||||
141378RG-T |
p.R625H |
|||||
258 |
p.F170Lfs*13 |
p.D630Y |
p.K666T |
|||
531 |
p.G8R |
p.Q209L |
||||
533 |
p.G579Efs*63 |
p.Q209L |
||||
534 |
p.Q209L |
|||||
535 |
splice |
p.Q209P |
||||
537 |
p.Q209L |
|||||
538 |
p.G6V |
p.Q209L |
||||
539 |
p.Q209L |
|||||
550 |
p.V346Sfs*51 |
p.Q209P |
||||
552 |
p.G41_54del |
p.G48L |
||||
553 |
p.Q209L |
|||||
554 |
||||||
556 |
Splice |
p.Q209P |
||||
557 |
p.P2L |
p.Q209P |
||||
C0622943-T |
p.Q209L |
|||||
ETB-0002-T |
Splice |
p.Q209P |
||||
J2217960PR-T |
p.Q209P |
|||||
K0111890AC-T |
p.Q209P |
|||||
MM1488-T |
||||||
MM1551-T |
p.Q684X |
p.D630Y |
||||
MM1563-T |
||||||
MM639-T |
p.G128R |
p.Q209L |
To identify novel UM driver genes we searched for additional recurrent mutations. PLCB4, phospholipase C, beta 4, was the only other gene that had a recurrent mutation (c.G1888T, p.D630Y, chr20:9389753, NM_000933), which occurred in 2 of 28 samples. This mutation is predicted to be functionally deleterious by SIFT (http://string-db.org/ and probably damaging by PolyPhen2. It is also at a highly conserved site across species (Supplementary Figure 1) with a GERP++ score of 5.69. This mutation is located in the Y-domain of the highly conserved catalytic core of PLCB4, which plays a vital role in the intracellular transduction of extracellular signals in the retina. Notably, this protein is a downstream target of GNAQ/GNA11 and is activated by direct interaction with GNAQ [10]. A STRING interaction network (http://string-db.org/) shows direct binding of PLCB4 to GNAQ and GNA11 (Supplementary Figure 2). The PLCB4 interactome also shows binding of PLCB3 to GNAQ and GNA11. Interestingly, in addition to the hotspot PLCB4 mutation, 1 of 28 UM samples had a novel mutation in PLCB3, phospholipase C beta 3 (c.G2694C, p.K898N, chr11:64032834, NM_001184883). The location of this mutation is within the CTD linker which plays a significant role in GNAQ activation [10]. The two samples we identified with PLCB4 mutations did not have mutations in either GNAQ or GNA11. A search of mutations in other UM WGS/WES data sets [8, 9, 11, 12] identified the same PLCB4 mutation in 1 of 56 samples, which also occurred mutually exclusive to GNAQ and GNA11 mutations. Taken together these data suggest that the PLCB4 hotspot mutation is similarly a gain-of-function mutation leading to activation of the same signaling pathway. Of note, PLCB4 is highly mutated (21% to 28%) in CMM [11, 13-15] but in contrast to UM, these mutations occur along the length of the gene, suggestive of them being loss-of-function. Consistent with this notion, none of the 159 reported non-synonymous PLCB4 mutations in CMM occur at the recurrent hotspot we observe in UM. While, speculative, this suggests context specific roles where PLCB4 is an oncogene in UM and a tumor suppressor in CMM.
Other than PLCB4 and the previously characterized UM driver genes, only 4 additional genes were mutated in more than one sample (Supplementary Table 2). Two missense mutations were found in each of MUC3A, TCHH, TTN and LLGL1. Only the latter has previously been associated with cancer, being a tumor suppressor in glioblastoma and oesophageal squamous cell carcinoma [16, 17], aberrantly spliced in hepatocellular carcinoma [18] and with reduced expression contributing to disease progression in CMM [19]. This makes it a plausible candidate driver gene for UM but further studies are required to determine its potential contribution to the development of this cancer type.
Genomic landscape
Mutation burden
The 14 samples on which we performed WGS allowed a more global assessment of the genomic landscape of UM. Firstly, to assess the mutation burden we calculated the number of mutations per megabase (Mb). The mean mutation rate across the UM genomes was 0.50/Mb (range 0.22-0.66/Mb) and the mutation load was similar in protein coding regions (mean 0.53, range 0.06-2.52/Mb, Supplementary Table 1).
Mutation signatures
To explore the underlying mutational processes in UM, we compared the mutational spectra with the signatures previously identified by Alexandrov et al. [20]. As expected, most samples showed signatures 1 and 5, which occur ubiquitously in all cancer types (Figure 1). We also noticed significant contributions of signatures 12 or 16 in almost every sample. The majority (79%) of samples also had signature 3, which is associated with defects in DNA double-strand break-repair (the ‘BRCA’ signature) but there is no significant association between BAP1 mutation and this signature in the samples analyzed here. Notably, no sample showed signature 7, which is frequently observed in CMM and associated with ultraviolet light exposure.
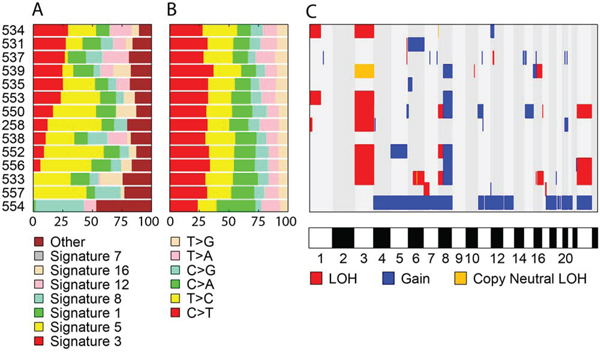
Figure 1: Genomic landscape of UM. A. Each color represents a known mutation signature as defined by Alexandrov et al. [20]. B. Proportion of base changes observed in each sample. C. Summary of chromosomal aberrations.
Copy number aberrations
Chromosomal segments showing aberrant copy number variation (CNV) or loss of heterozygosity (LOH) are shown in Figure 1. The most common event was monosomy 3 or copy neutral LOH of the entire chromosome 3, which was seen in 8 of 14 samples (57%). Notably, all samples with BAP1 mutation (n=6) were hemizygous for chromosome 3. Other frequent aberrations included 8q gain (n=6), 8p loss (n=3), 16q loss (n=2), and X loss (n=3). These common CNVs were only observed in samples with monosomy 3 (n=7) or copy neutral chromosome 3 LOH (n=1), whereas samples with diploid chromosome 3 tended to have fewer large chromosomal aberrations. The chromosomal profile of sample 554 looked quite different compared to the others and had gained copies of almost every chromosome; specifically triploid chromosomes 4, 5, 6, 11, 12, 13, 17q, and 21, and tetraploid chromosomes 7, 8, 18, 20, and X.
Chromosome 8q duplication is a late event
We observed six tumors with gained chromosome 8q. To assess whether this aberration occurred early or late in the tumor development, we examined the variant allele frequency (VAF) for the somatic mutations on chromosome 8q. If the duplication is an early event, somatic mutations are only observed in one of the copies, whereas if the duplication occurs later, half the mutations are duplicated and the VAF distribution is bimodal. Using this observation we examined the six samples with gained chromosome 8q, and as seen in Figure 2 the VAF distribution in these samples was distinctly bimodal. On average 93% (range 87-97%) of the mutations were estimated to occur before the duplication showing that gained chromosome 8q is a late event.
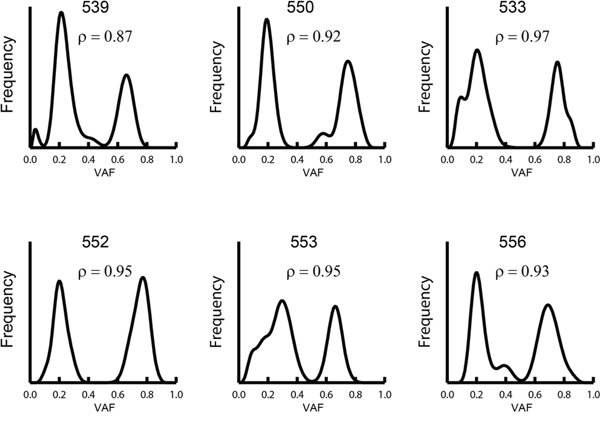
Figure 2: Distribution of somatic VAF on chromosome 8q in samples with gained copies of chromosome 8q. The fraction of mutations, ρ, occurring prior chromosomal gain was estimated using a mix-model (see Methods). The curve was generated using a Bayesian smoother in which the likelihood for the data was calculated for each mutation and the sum of these likelihoods was used as the estimate of the VAF distribution.
Structural variants
We detected a total of 297 SVs in the 14 samples subjected to whole-genome sequencing (mean 273, range 9 to 42, Supplementary Tables 3 and 4). These consisted of large deletions, gene fusions, regions of tandem duplication and truncated genes. Of these, there were 7 genes that were affected more than once (CDH13, FAM135B, GFRAL, LRRC16A, MOK, SEMA3E and VPS13B). We observed only two in-frame fusion events. Sample 553 has a fusion between GSPT1 and HSD17B3 and 550 has a fusion between FAM135B and PDSS2 (Figure 3). The latter occurs between chromosomes 6 and 8 which are two of the most frequently aberrant chromosomes in UM. In sample 539 we observed an intronic deletion of unknown consequence encompassing 154bp of BAP1. Interestingly, 539 has copy-neutral LOH of chromosome 3.
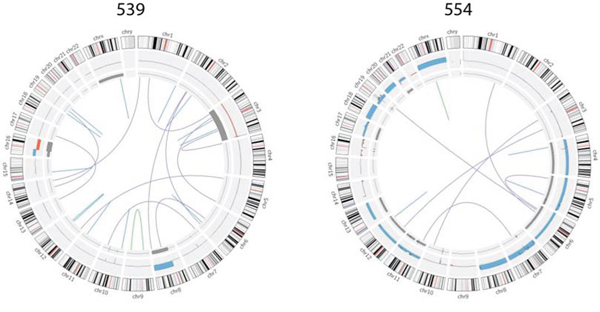
Figure 3: Summary of large genomic variations in two typical samples. Structural variants are illustrated by an edge between its two breakpoints, where the color describes the type of variant including interchromosomal (purple), deletion (blue), translocation/tandem repeat (red), and inversion (green). The middle layer illustrates allelic imbalance and LOH; the width of the segments describes how much the minor allele frequency of the segment deviate from normal (0.5). The outer layer illustrates chromosomal loss (red) and gain (blue).
CONCLUSION
Next generation sequencing of 28 UM samples shows that overall these tumors have a low SNV and SV mutation burden. The majority of tumors presented with a BRCA mutation signature, and as expected these sun-shielded melanomas had no ultraviolet radiation signature. Analysis of SNVs identified a novel recurrent mutation in PLCB4 (p.D630Y). The identical mutation is also seen in 1 of 56 UM tumors with published sequence data. PLCB4 is the canonical downstream target of GNAQ/GNA11 and is activated by direct interaction with GNAQ. This novel mutation is a likely driver in UM and occurs mutually exclusively with GNAQ/GNA11 mutations. Taken together these data suggest that the PLCB4 hotspot mutation is a gain-of-function mutation leading to activation of the same signaling pathway. Functional studies are warranted to further characterize the role of PLCB4 in UM.
MATERIALS AND METHODS
Ethics
Ethics approvals were obtained from the Human Research Ethics Committees of the QIMR Berghofer Medical Research Institute, the University of Western Australia, the Capital Region of Denmark, and Lund University. Written informed consent was obtained from each participant in this study.
Next generation sequencing
Whole-genome sequencing (WGS) was performed on 14 primary UM tumors and whole-exome sequencing (WES) was performed on 9 UM tumors and 5 cell lines established from primary UM tumors. Supplementary Table 5 details the histology of the samples, which were sequenced along with matching germline samples extracted from blood or saliva.
Sequence reads were aligned against the human reference genome (build 19) using BWA [21], duplicate reads were marked with Picard, and reads were realigned against known insertion/deletions (indels) and base qualities were recalibrated using GATK [22]. Somatic mutations and short indels were identified using Samtools/bcftools [23] and further annotated using ANNOVAR [24]. A stringent set of filtering criteria was applied to the WGS/WES output to reduce the false positive rate. Firstly the germline samples were filtered by eliminating all population variants reported in dbSNP (unless also reported in COSMIC). The germline variants were further filtered so only variants with no reads for the variant allele and ≥ 8 reads for the reference allele were included. The variants in the tumor were filtered so that only those with Phred-scaled genotype likelihood (PL) for wildtype ≥ 60 and ≥ 4 variant reads were included. Furthermore, any variants with quality warnings regarding regions of duplication, tri-allelic variants, and variants located at the tail end of reads were eliminated. Lastly the constrained log likelihood ratio (CLR) score was required to be ≥ 40; the higher the score the more likely that a variant is truly somatic. Non-frameshift variants that passed this set of filtering criteria were hand-curated, whereby variants in regions of trinucleotide expansions or reductions were removed from the dataset as they are likely due to poor mapping.
Structural variants (SV) were identified using Janda v0.8.1 (http://janda.sourceforge.net/). Janda uses both discordant read pairs and clipped reads to detect breakpoints. These were then filtered using the following criteria: SVs were required to be supported by at least three junction reads; the junction reads aligned against the junction with at most two mismatches on average. Breakpoints with extremely high coverage (>1000x) in either sample were discarded as well as breakpoints with low coverage (<2x) in the normal sample. To avoid artifacts due to segmental duplication and erroneous mapping, we removed breakpoints that overlapped with the Genomic Super Dups Database (http://humanparalogy.gs.washington.edu/SDD), or if the 20bp regions around the two breakpoints were too similar (≥90%), as well as breakpoints that were detected in multiple samples. Lastly, if the junction contained an insertion greater than 3bp inserted between the breakpoints, the variant was removed.
Copy number aberrations and/or allelic imbalances were called using binocular v0.2 (http://binocular .sourceforge.net). Binocular uses read coverage and variant allele frequencies to build likelihood that is used to segment chromosomes using a variant of the Circular Binary Segmentation algorithm. A Phred-scaled genotype likelihood (PL) cutoff ≥ 90 was used to define heterozygous germline loci. Segments were identified requiring a log likelihood ratio > 90, and that either ratio between copy numbers was > 1.1, or that difference in minor allele frequency was > 0.05 between the two segments. A p-value threshold of 0.001 was used to define segments with allelic imbalance. Copy numbers were normalized by multiplying them with a factor such that after normalization the 20th percentile (weighted with respect to segment size) across segments with allelic balance was 2.
To infer the fraction of mutations on chromosome 8q that occurred before the arm was duplicated, we built a mixed model allowing for different fractions, copy numbers, cn, and tumor content, tc. For the fraction of mutations occurring before the duplication, number of variant reads was modeled as a binomial with mean tc/cn. For mutations occurring prior to the duplication half are not duplicated and follow the same binomial distribution as above, and for the remaining half the number of variant reads follows a binomial distribution with mean tc*(1 – 1/cn). Using this model and maximum likelihood we estimated fractions of mutations occurring before the chromosomal duplication.
Analysis of mutational signatures
To estimate mutational signatures, we used a modified version of the methodology developed by Alexandrov and colleagues [20]. Given that this method optimally requires a larger number of samples than we have sequenced here, we modified it to use the 30 signatures that have been previously inferred from > 12,000 samples. We approximated the number of mutations of each signature type as M ≈ P x E, where P is the probability distribution for each signature and E describes the sample’s exposure of each signature. We used the signatures available from COSMIC, and found the E that minimized the mean squared error with the condition that all elements are non-negative. To estimate the confidence interval of the estimated exposure the procedure above was repeated with added noise. Rather than using the number of observed mutations, Mij, we used a Poisson distributed random number with mean Mij. This was repeated 1,000 times, and the range from the 5th to 95th percentile defined the confidence interval.
Sanger sequencing
Sanger sequencing was used to confirm BAP1, SF3B1 and EIF1AX variants found by WGS/WES. Primers are listed in Supplementary Table 6. Sequencing reactions were performed using Big Dye V3.1 and run on an ABI 3730xl (Applied Biosystems). Sequencing results were analyzed using the chromatogram viewer, Chromas (version 1.45; Technelysium Pty Ltd).
ACKNOWLEDGMENTS
We are indebted to the uveal melanoma patients for their participation in this study as well as the medical clinic and pathology laboratory staff for their ongoing support of this work.
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
GRANT SUPPORT
The work was funded by the National Health and Medical Research Council of Australia (NHMRC). L.G.A is supported by the Rio Tinto Ride to Conquer Cancer. N.K.H. and A.W.H are supported by fellowships from the NHMRC.
NOTE ADDED IN PROOF
While this manuscript was under review, the TCGA UM data were released (11/14/2015), which identified 2/80 samples with mutations in codon p.D630 of PLCB4. Sample TCGA-VD-A8KD carries two mutations at adjacent bases in the same codon, c.G1888T and c.A1889T, which if they are biallelic result in amino acid changes p.D630Y (the same mutation we describe here) and p.D630V, respectively, or if they occur on the same allele translate to p.D630F. Sample TCGA-YZ-A985 carries a c.G1888A mutation resulting in amino acid change p.D630N.
REFERENCES
1. Singh AD, Turell ME, Topham AK. Uveal melanoma: trends in incidence, treatment, and survival. Ophthalmology. 2011; 118:1881-1885.
2. Van Raamsdonk CD, Bezrookove V, Green G, Bauer J, Gaugler L, O'Brien JM, Simpson EM, Barsh GS, Bastian BC. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. 2009; 457:599-602.
3. Van Raamsdonk CD, Griewank KG, Crosby MB, Garrido MC, Vemula S, Wiesner T, Obenauf AC, Wackernagel W, Green G, Bouvier N, Sozen MM, Baimukanova G, Roy R, et al. Mutations in GNA11 in uveal melanoma. N Engl J Med. 2010; 363:2191-2199.
4. Harbour JW, Onken MD, Roberson ED, Duan S, Cao L, Worley LA, Council ML, Matatall KA, Helms C, Bowcock AM. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science. 2010; 330:1410-1413.
5. Worley LA, Onken MD, Person E, Robirds D, Branson J, Char DH, Perry A, Harbour JW. Transcriptomic versus chromosomal prognostic markers and clinical outcome in uveal melanoma. Clin Cancer Res. 2007; 13:1466-1471.
6. Woodman SE. BAP1tism of a Tumor Suppressor. Clin Cancer Res. 2012; 18:323-325.
7. Onken MD, Worley LA, Ehlers JP, Harbour JW. Gene expression profiling in uveal melanoma reveals two molecular classes and predicts metastatic death. Cancer Res. 2004; 64:7205-7209.
8. Harbour JW, Roberson ED, Anbunathan H, Onken MD, Worley LA, Bowcock AM. Recurrent mutations at codon 625 of the splicing factor SF3B1 in uveal melanoma. Nat Genet. 2013; 45:133-135.
9. Martin M, Masshofer L, Temming P, Rahmann S, Metz C, Bornfeld N, van de Nes J, Klein-Hitpass L, Hinnebusch AG, Horsthemke B, Lohmann DR, Zeschnigk M. Exome sequencing identifies recurrent somatic mutations in EIF1AX and SF3B1 in uveal melanoma with disomy 3. Nat Genet. 2013; 45:933-936.
10. Lyon AM, Tesmer JJ. Structural insights into phospholipase C-beta function. Molecular pharmacology. 2013; 84:488-500.
11. Krauthammer M, Kong Y, Bacchiocchi A, Evans P, Pornputtapong N, Wu C, McCusker JP, Ma S, Cheng E, Straub R, Serin M, Bosenberg M, Ariyan S, et al. Exome sequencing identifies recurrent mutations in NF1 and RASopathy genes in sun-exposed melanomas. Nat Genet. 2015; 47:996-1002.
12. Furney SJ, Pedersen M, Gentien D, Dumont AG, Rapinat A, Desjardins L, Turajlic S, Piperno-Neumann S, de la Grange P, Roman-Roman S, Stern MH, Marais R. SF3B1 mutations are associated with alternative splicing in uveal melanoma. Cancer discovery. 2013; 3:1122-1129.
13. Wei X, Walia V, Lin JC, Teer JK, Prickett TD, Gartner J, Davis S, Stemke-Hale K, Davies MA, Gershenwald JE, Robinson W, Robinson S, Rosenberg SA, Samuels Y. Exome sequencing identifies GRIN2A as frequently mutated in melanoma. Nat Genet. 2011; 43:442-446.
14. Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, Nickerson E, Auclair D, Li L, Place C, Dicara D, Ramos AH, Lawrence MS, et al. A landscape of driver mutations in melanoma. Cell. 2012; 150:251-263.
15. Cancer Genome Atlas N. Genomic Classification of Cutaneous Melanoma. Cell. 2015; 161:1681-1696.
16. Song J, Peng XL, Ji MY, Ai MH, Zhang JX, Dong WG. Hugl-1 induces apoptosis in esophageal carcinoma cells both in vitro and in vivo. World journal of gastroenterology. 2013; 19:4127-4136.
17. Gont A, Hanson JE, Lavictoire SJ, Daneshmand M, Nicholas G, Woulfe J, Kassam A, Da Silva VF, Lorimer IA. Inhibition of glioblastoma malignancy by Lgl1. Oncotarget. 2014; 5:11541-11551. doi: 10.18632/oncotarget.2580.
18. Lu X, Feng X, Man X, Yang G, Tang L, Du D, Zhang F, Yuan H, Huang Q, Zhang Z, Liu Y, Strand D, Chen Z. Aberrant splicing of Hugl-1 is associated with hepatocellular carcinoma progression. Clin Cancer Res. 2009; 15:3287-3296.
19. Kuphal S, Wallner S, Schimanski CC, Bataille F, Hofer P, Strand S, Strand D, Bosserhoff AK. Expression of Hugl-1 is strongly reduced in malignant melanoma. Oncogene. 2006; 25:103-110.
20. Alexandrov LB, Nik-Zainal S, Wedge DC, Campbell PJ, Stratton MR. Deciphering signatures of mutational processes operative in human cancer. Cell reports. 2013; 3:246-259.
21. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 25:1754-1760.
22. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010; 20:1297-1303.
23. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009; 25:2078-2079.
24. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic acids research. 2010; 38:e164.