
INTRODUCTION
Gastroenteropancreatic neuroendocrine tumors (GEP-NETs) are relatively rare tumors accounting for about 0.5% of all human cancers [1–6]. Their incidence is significantly increasing based on data from recent population-based studies, and this phenomenon is explained by increased awareness of the disease entity and increased detection by advanced diagnostic modalities [1–6]. However, there has been no significant improvement in clinical outcome over the same period based on UK and US databases [7].
Recently, the FDA approved a few targeted agents for pancreatic NETs including sunitinib, a multi-tyrosine kinase inhibitor, and everolimus, an inhibitor of the PI3K-Akt-mTOR signal pathway [8]. In pancreatic NET, sunitinib was compared to a placebo in a phase III trial of 171 pancreatic NET patients, and the median progression-free survival was significantly prolonged in the sunitinib arm (11.4 versus 5.5 months) [9]. Based on these data, sunitinib was approved in the US for the treatment of progressive, well-differentiated pancreatic NET. In randomized controlled trials, everolimus demonstrated a 65% decrease in the risk for tumor progression in pancreatic NETs [7] and a 23% decrease in patients with non-pancreatic NETs [9]. In an analysis of 159 patients with NETs, everolimus showed a response rate of 7.7% with a progression-free survival of 12 months [10].
We recently conducted a phase II trial for pazopanib in metastatic GEP-NET patients [11]. Our phase II study demonstrated an objective response rate of 18.9% (7 of 37, 95% CI 8.0 – 35.2) and a disease control rate (CR + confirmed PR + stable disease) of 75.7% (28 of 37, 95% CI, 58.8 – 88.2) in metastatic GEP-NETs. Through this trial, we observed that a small subset of NET patients responded to pazopanib for > 6 months. Recently, pancreatic NETs were characterized as having recurrent somatic mutations in MEN1, DAXX, ATRX, TSC, and PTEN on the basis of exome sequencing of 10 pancreatic NETs [12]. Small intestinal NETs showed recurrent somatic mutations and deletions in CDKN1B by whole exome and whole genome sequencing of 50 small intestinal NETs [13]. However, in another study of 48 small intestinal NETs by exome sequencing, recurrent mutations were not identified. Rather, 197 single nucleotide variations in a preponderance of cancer-related genes were identified; 33% of small intestinal NET patients showed PIK3/Akt/mTOR pathway alteration, and 72% had therapeutically actionable genomic alterations [13]. The understanding of genomic profiles in GEP-NETs is still incomplete and the genetic alterations associated with drug responses have not been extensively studied. In this study, we performed whole exome sequencing of 12 GEP-NETs from patients enrolled in a nonrandomized, open-labeled, single-center phase II study of pazopanib [11].
RESULTS
Genomic profiling identifies the BRAF V600E mutation in pazopanib non-responder and the TP53 mutation in pazopanib responder patients
The mean number of somatic mutations varied widely from 20 to 4682, and the mutation counts for each case are shown in Figure 1. One case with 4682 somatic mutations showed a mutation in MLH1 and additional missense mutations (ATR, PARP2, RBBP8, and RIF1) and splice site (XPC) mutations in DNA repair-related genes, and was thus classified as having a “hypermutated” phenotype. Among 12 samples, eight showed mutations of more than one of the cancer-related genes (TP53, CNBD1, RB1, APC, BCOR, BRAF, CTNNB1, EGFR, EP300, ERBB3, KDM6A, KRAS, MGA, MLL3, PTEN, RASA1, SMARCB1, SPEN, TBC1D12, and VHL) described by Lawrence et al. [24] (Figure 2). TP53 was recurrently mutated in three cases, whereas CNBD1 and RB1 mutations were identified in two cases. In our data set, we found the presence of BRAF V600E mutation in one primary NET from the small intestine, which was further confirmed by Sanger direct sequencing. To exclude the possibility of the occurrence of mixed adenocarcinoma, neuroendocrine features, or both, we performed an independent pathology review in terms of architecture, tumor grade, and chromogranin, synaptophysin, and CD56 immunoreactivity by IHC (Supplementary Table 1). All of the pathological features corresponded to GEP-NET, rather than mixed adenocarcinoma with neuroendocrine features.
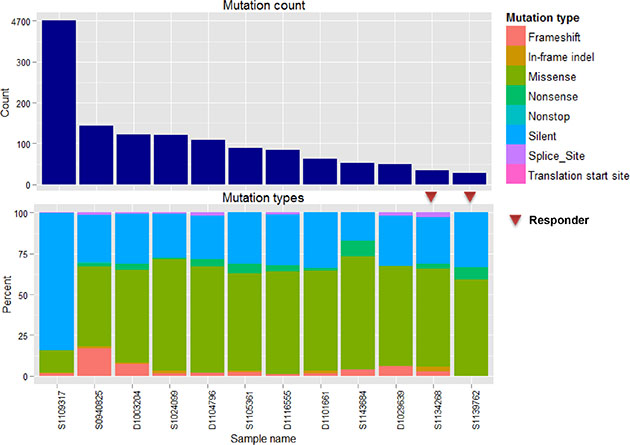
Figure 1: Mutations in 12 NET samples. (A and B) show total counts and a percentage bar plot according to mutation type.
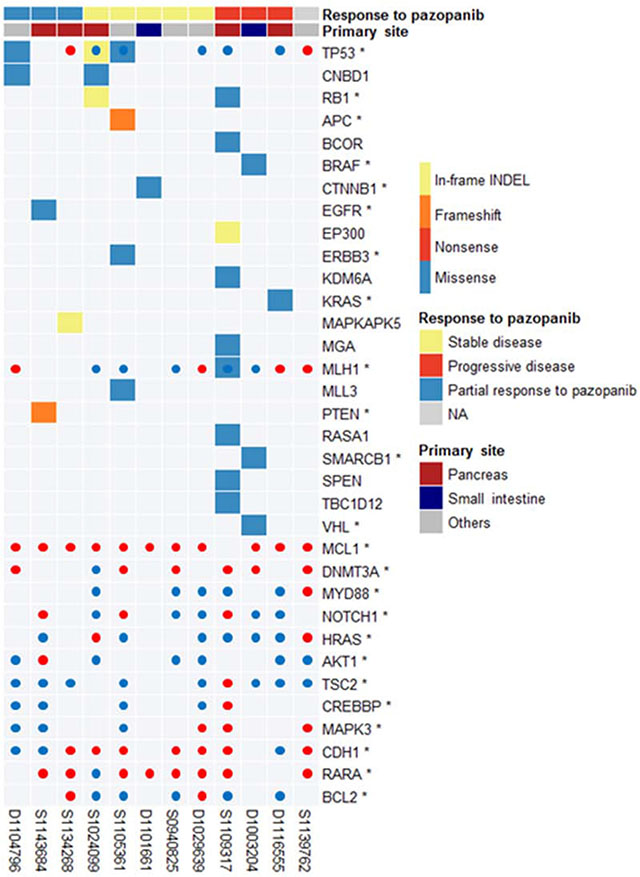
Figure 2: Landscape of cancer-related mutations found in 12 GEP-NETs.
The patient with BRAF V600E mutation was 52 years old and had a metastatic grade 1 neuroendocrine tumor. The primary mass originated from the duodenum and had metastasized to multiple and distant lymph nodes at diagnosis. The patient received capecitabine and oxaliplatin as a palliative first-line treatment. After disease progression following the first-line therapy, the patient was enrolled in the pazopanib clinical trial. Before starting pazopanib, mutational profiles of the primary tumor tissue were evaluated. After two cycles of pazopanib therapy, the follow-up computed tomography (CT) scan revealed tumor growth corresponding to disease progression based on RECIST 1.1 criteria (Figure 3A). Hence, this small intestinal NET patient with BRAF V600E mutation showed tumor progression after pazopanib treatment, although at the time of clinical trial enrollment, the genomic information was not available to the clinician because this trial was not a genome-selected trial.
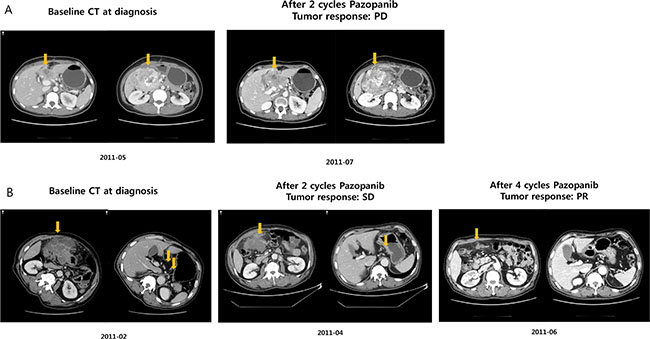
Figure 3: Response to pazopanib. (A) BRAF V600E mutant; (B) TP53 mutant NET patient.
We also identified the TP53 mutation in a patient with a dramatic response to pazopanib. The patient had a grade 3 neuroendocrine carcinoma with gastric primary tumor location and extensive abdominal lymph node and peritoneal seeding nodule involvement. After failing to respond to cytotoxic chemotherapy, the patient was treated with pazopanib. After 2 cycles of pazopanib, the patient presented stable disease per the RECIST 1.1 criteria (Figure 3B). At 4 months, CT evaluation (after 4 cycles) revealed definite radiologic tumor shrinkage corresponding to a partial response based on the RECIST 1.1 criteria (Figure 3B), which lasted for > 6 months.
BRAF mutations in an independent cohort
Next, we surveyed the presence of the BRAF V600E mutation in an independent cohort of 44 GEP-NET patients. We included GEP-NETs from the rectum (n = 26 consisting 19 G1, 4 G2 and 3 G3), colon (n = 7 consisting 6 G3 and one G1), pancreas (n = 4, G2), small intestine (n = 3 consisting one G3 and 2 G2), stomach (n = 3 consisting 2 G3 and one G1) and appendix (n = 1, adenocarcinoid). We found BRAF V600E (G1 NET from rectum and two G3 NETs from colon) and BRAF G593S (G2 NET from pancreas) missense mutations (9.1%) in an independent cohort of 44 GEP-NETs by Sanger sequencing. The cohort consisted of GEP-NET from rectum (n = 27), colon (n = 6), pancreas (n = 4), small intestine (n = 3), stomach (n = 3) and appendix (n = 1). All G1 and G2 NETs were positive for synaptophysin while rectal NETs and G3 NETs were negative for chromogranin (Figure 4). The pathology and immunohistochemical staining of the four NETs with BRAF V600E or G593S mutation are shown in Figure 4 (G1, 1-cm rectal NET; G2, 8.5 cm pancreatic NET; G3, 6.5 cm colon NET; and G3, 3.2 cm colon NET).
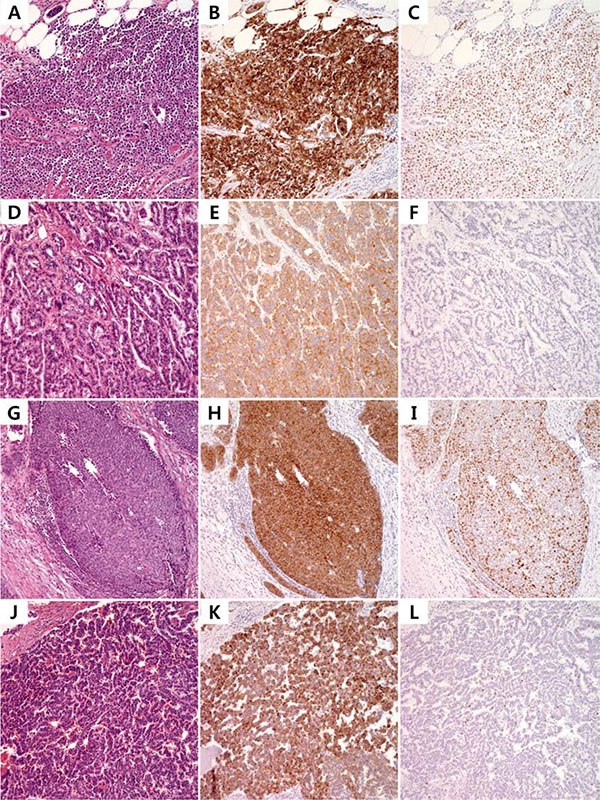
Figure 4: Photomicrograph of BRAF-mutant NETs from independent cohort. Pathologic findings of V600E mutant small intestinal G3 NET (A), V600E mutant rectal G1 NET (D), V600E mutant sigmoid colon G3 NET (G) and G593S mutant G2 pancreas NET (J) and corresponding immunohistochemistry for synaptophysin (B, E, H) and chromogranin (K) with Ki-67 (C, F, I, L).
Copy number variations in GEP-NET
The copy number profile of Korean GEP-NETs clearly demonstrates a difference in CNV pattern for each primary organ (Supplementary Figure 1). The copy number of pancreatic NET relatively fluctuated interchromosomally. In contrast, there was no concordance of arm-level copy number in small intestinal NET (Supplementary Table 2). Our copy number results are similar to those of a previous small intestinal NET study [13]. Moreover, arm-level amplification of 19q13.31 (q value, 0.49) frequently occurred in the pancreas, i.e., in three of four amplification samples (Supplementary Tables 1 and 2). Fourteen CNV genes (MCL1, DNMT3A, MLJ1, MYD88, NOTCH1, HRAS, AKT1, TSC2, CREBBP, MARK3, CDH1, TP53, RARA, and BCL2) that may potentially be actionable are illustrated in Figure 2.
MAPKAPK5 mutation in an exceptional responder to pazopanib
Whole exome sequencing results in two cases showed an exceptional response to pazopanib. In these two cases, we could not find mutations in any of the 272 recurrently mutated genes. Furthermore, we investigated the mutation status of genes in four pathways (hsa04010, MAPK signaling pathway; hsa04020, calcium signaling pathway; hsa04060, cytokine-cytokine receptor interaction; hsa05200, pathways in cancer) involved in the primary mechanism of action of pazopanib. We found a novel p.I16fs MAPKAPK5 mutation involved in the MAPK signaling pathway in a patient with pancreatic NET (Supplementary Figure 2).
Table 1: Patient characteristics
ID |
Sex |
ECOG |
Histology |
|
Sites of metastasis |
Best response |
Age |
|---|---|---|---|---|---|---|---|
D1003204 |
2 |
1 |
G1 |
1 |
liver, LN, ovary |
PD |
48 |
D1029639 |
1 |
1 |
G1 |
1 |
liver, lung, adrenal |
SD |
46 |
D1101661 |
1 |
1 |
G3 |
2 |
liver, LN |
SD |
46 |
D1104796 |
1 |
1 |
G1 |
2 |
liver, LN, peritoneal seeding |
PR |
65 |
D1116555 |
1 |
1 |
G1 |
1 |
liver, LN |
PD |
61 |
S0929052 |
1 |
1 |
G2 |
1 |
liver |
SD |
70 |
S0940825 |
2 |
1 |
G2 |
1 |
liver, LN |
SD |
53 |
S1024099 |
1 |
1 |
G3 |
2 |
liver, LN |
SD |
59 |
S1105361 |
1 |
1 |
G3 |
2 |
liver |
SD |
72 |
S1109317 |
2 |
1 |
G3 |
2 |
liver |
PD |
71 |
S1134268 |
1 |
1 |
G3 |
2 |
liver |
PR |
19 |
S1139762 |
1 |
0 |
G2 |
1 |
liver, lung |
SD |
64 |
S1143684 |
2 |
1 |
G2 |
1 |
liver |
PR |
69 |
Integrative analysis of Korean GEP-NETs with pancreatic NETs and small intestinal NETs from previously published data
As our GEP-NETs included various primary organs, we integrated our results with previously published results for pancreatic and small intestinal NETs [12, 13]. Most of the mutated genes did not overlap among the three datasets, suggesting different genomic alterations according to the primary location of the tumor (Supplementary Figure 3). In this analysis, we found 272 recurrently mutated genes irrespective of the organ, and we used those recurrently mutated genes to construct a gene interaction network.
Finally, a network of 65 genes and 89 interactions was generated after genes that did not pass the GSEA test were trimmed (Supplementary Figure 4 and Table 2). Genes related to cell cycle, Wnt signaling, E2F transcription factor network, DNA damage, p53 pathway, EGFR signaling, FGFR signaling, ERBB2 signaling, PDGFR signaling, and PI3K-Akt signaling pathways were mostly associated with these genes (Table 2). TP53 was the gene with the most recurrent mutations, as mutations were observed in 6 cases in various organs (Figure 5). CDKN1B mutation was found in five cases of small intestinal NET, whereas MEN1 mutation was found in five cases of pancreatic NET. Mutations of RB1, ATM, and TP53BP1 were each identified in four cases. Overall, 46 of 72 samples (64%) had at least one mutation in at least one cancer-related gene [24]. The overall mutational profile of all GEP-NETs, including ours, is presented in Figure 5.
Table 2: Pathway analysis
Gene set |
Ratio of protein in gene det |
Number of proteins in gene set |
Proteins from network |
FDR |
Nodes |
|---|---|---|---|---|---|
Cell cycle(K) |
0.0127 |
124 |
7 |
2.00E-04 |
TP53, RB1, CREBBP, SMAD2, CDC27, ATM, CDKN1B |
Wnt signaling pathway(P) |
0.0279 |
272 |
10 |
2.50E-04 |
PCDHA4, PCDHB6, MYH3, MYH2, TP53, EP400, FAT1, APC, CREBBP, PCDH10 |
E2F transcription factor network(N) |
0.007 |
68 |
6 |
3.33E-04 |
RB1, POLA1, TRRAP, CREBBP, ATM, CDKN1B |
Direct p53 effectors(N) |
0.0135 |
132 |
8 |
5.00E-04 |
PTEN, TP53, RB1, RFWD2, TRRAP, APC, CREBBP, TSC2 |
DNA Damage/Telomere Stress Induced Senescence(R) |
0.0027 |
26 |
5 |
1.00E-03 |
TP53, RB1, EP400, ATM, CDKN1B |
regulation of transcriptional activity by pml(B) |
0.0011 |
11 |
3 |
1.00E-03 |
DAXX, TP53, RB1 |
p53 signaling pathway(K) |
0.007 |
68 |
5 |
1.71E-03 |
PTEN, TP53, RFWD2, ATM, TSC2 |
Signaling by EGFR(R) |
0.0176 |
172 |
7 |
2.13E-03 |
PTEN, ADAM12, SRC, TNRC6B, TNRC6A, CDKN1B, TSC2 |
Pre-NOTCH Expression and Processing(R) |
0.0039 |
38 |
4 |
2.89E-03 |
TP53, TNRC6B, TNRC6A, CREBBP |
p53 pathway(P) |
0.0045 |
44 |
4 |
4.55E-03 |
PTEN, TP53, CREBBP, ATM |
Signaling by SCF-KIT(R) |
0.014 |
137 |
6 |
4.67E-03 |
PTEN, SRC, TNRC6B, TNRC6A, CDKN1B, TSC2 |
TCF dependent signaling in response to WNT(R) |
0.0153 |
149 |
6 |
4.67E-03 |
MEN1, TRRAP, CHD8, BCL9L, APC, CREBBP |
HTLV-I infection(K) |
0.0267 |
260 |
8 |
4.70E-03 |
TP53, RB1, TRRAP, APC, CREBBP, SMAD2, CDC27, ATM |
Prostate cancer(K) |
0.0091 |
89 |
5 |
4.71E-03 |
PTEN, TP53, RB1, CREBBP, CDKN1B |
Notch-mediated HES/HEY network(N) |
0.0047 |
46 |
4 |
4.77E-03 |
RB1, CREBBP, CDKN1B, NCOR2 |
Hepatitis B(K) |
0.015 |
146 |
6 |
4.81E-03 |
PTEN, TP53, RB1, SRC, CREBBP, CDKN1B |
PIP3 activates AKT signaling(R) |
0.0096 |
94 |
5 |
4.94E-03 |
PTEN, TNRC6B, TNRC6A, CDKN1B, TSC2 |
Muscle contraction(R) |
0.005 |
49 |
4 |
5.13E-03 |
MYBPC2, TTN, MYH3, NEB |
Signaling by FGFR(R) |
0.0159 |
155 |
6 |
5.35E-03 |
PTEN, SRC, TNRC6B, TNRC6A, CDKN1B, TSC2 |
Signaling by ERBB2(R) |
0.0159 |
155 |
6 |
5.35E-03 |
PTEN, SRC, TNRC6B, TNRC6A, CDKN1B, TSC2 |
p53 pathway(N) |
0.0058 |
57 |
4 |
6.00E-03 |
DAXX, TP53, CREBBP, ATM |
p53 pathway feedback loops 2(P) |
0.0025 |
24 |
3 |
6.36E-03 |
PTEN, TP53, ATM |
Glypican 1 network(N) |
0.0026 |
25 |
3 |
7.04E-03 |
SLIT2, SRC, SMAD2 |
Signaling by PDGF(R) |
0.0179 |
175 |
6 |
8.17E-03 |
PTEN, SRC, TNRC6B, TNRC6A, CDKN1B, TSC2 |
Cell Cycle Checkpoints(R) |
0.0119 |
116 |
5 |
8.40E-03 |
TP53, RFWD2, CDC27, ATM, CDKN1B |
Signaling by TGF-beta Receptor Complex(R) |
0.0072 |
70 |
4 |
9.93E-03 |
MEN1, PARD3, SMAD2, NCOR2 |
Oncogene Induced Senescence(R) |
0.0031 |
30 |
3 |
1.01E-02 |
TP53, TNRC6B, TNRC6A |
PI3K-Akt signaling pathway(K) |
0.0356 |
347 |
8 |
1.04E-02 |
PTEN, COL11A1, TP53, COL1A1, COL5A1, EIF4B, CDKN1B, TSC2 |
Adherens junction(K) |
0.0075 |
73 |
4 |
1.09E-02 |
PARD3, SRC, CREBBP, SMAD2 |
ATM pathway(N) |
0.0035 |
34 |
3 |
1.23E-02 |
RFWD2, TP53BP1, ATM |
FoxO signaling pathway(K) |
0.0136 |
133 |
5 |
1.25E-02 |
PTEN, CREBBP, SMAD2, ATM, CDKN1B |
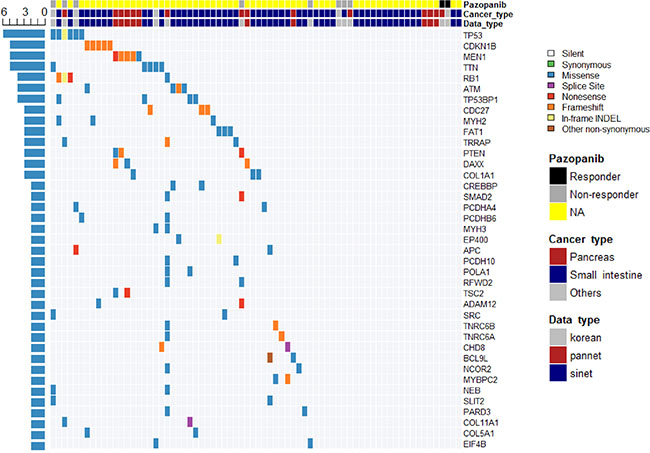
Figure 5: Overall mutational profile of all GEP-NETs revealed by whole exome and whole genome studies.
DISCUSSION
In this study, we performed whole-exome sequencing of 12 GEP-NETs from patients enrolled in a nonrandomized, phase II study of pazopanib and constructed a gene interaction network based on recurrently mutated genes with sequencing data from 72 GEP-NETs from three datasets [12, 13]. TP53 (n = 6), CDKN1B (n = 5), MEN1 (n = 5), RB1 (n = 4), ATM (n = 4), and TP53BP1 (n = 4) were frequently mutated genes. CDK1B mutation was identified only in small intestinal NET, whereas MEN1 mutation was identified in pancreatic NET. These genes are related to the cell cycle, Wnt, E2F transcription factor, and DNA damage pathways. Overall, 46 of 72 samples (64%) had at least one mutation in cancer-related genes.
Importantly, we found one BRAF V600E mutation in small intestinal NET that did not respond to pazopanib and further confirmed that 3 (6.8%) of 44 GEP-NET patients harbored the BRAF V600E mutation. BRAF V600E is an actionable mutation in melanoma with significantly prolonged survival when a BRAF inhibitor is administered in patients with this mutation [25], but it is not actionable in colorectal adenocarcinoma. To exclude the possibility of mixed histology of adenocarcinoma and NET, we performed an independent pathology review and confirmed that all of the NET cases were not adenocarcinoma with neuroendocrine differentiation. Hence, BRAF V600E is a novel mutation found in NET that could potentially confer clinical benefit in this subset of patients. BRAF V600E mutations have been reported as oncogenic mutation or resistant mutation to drugs in melanoma [26, 27] and thyroid cancer [28], GIST [29], hairy cell leukemia [30], multiple myeloma [31], and pediatric metanephric tumors [32]. The clinical implications of the BRAF V600E mutation in metastatic GEP-NET should thus be evaluated in clinical trials.
Three GEP-NET patients with TP53 mutations demonstrated a durable response to pazopanib, either as PR or achievement of stable disease (Figure 2). The tumor suppressor gene TP53 is mutated in many cancer types, and various TP53 mutations (missense, frameshift [fs], or nonsense [*], leading to gain or loss of function) have been identified during tumorigenesis and metastasis [26]. Cancer cells with mutated TP53 have accelerated tumor growth associated with increased VEGF expression and neovascularization [27], which represents an important survival pathway [7, 8], resulting in a therapeutic advantage of anti-angiogenesis inhibitors in TP53 mutant cancer patients [28]. Although our findings should be confirmed in larger patient cohort, we postulate that GEP-NET patients with TP53 mutation may have enhanced angiogenesis that may be treated using an anti-angiogenesis inhibitor, such as pazopanib.
Only a few reports have described the sequencing data of GEP-NETs, and most of them were performed for NETs from a specific organ. Jiao et al. [12] performed exome sequencing in 10 pancreatic NETs and found recurrently mutated genes as follows: MEN1 (n = 5), DAXX (n = 3), PTEN (n = 2), and TSC2 (n = 2). In a validation set of 68 cases, MEN1 was mutated in 44% cases, and DAXX and ATRX, which interacts with DAXX to form a chromatin remodeling complex, were mutated in 25% and 18% cases, respectively, with a mutually exclusive pattern. Another important finding was that at least one gene involved in the mTOR pathway, such as TSC2 or PTEN, was mutated in 14% of cases. This finding is consistent with observed clinical responses in pancreatic NET patients with the recently approved mTOR inhibitor everolimus [8]. Banck et al. [13] performed exome sequencing of 48 small intestinal NETs and identified 197 protein-altering somatic single nucleotide variations with a preponderance of cancer-related genes such as FGFR2, MEN1, HOOK3, EZH2, MLF1, CARD11, VHL, NONO, and SMAD1. However, most of these mutations were not recurrently identified. Using an integrated approach combining mutational and copy number data, Banck et al. found that 33% of small intestinal NET patients showed PIK3/Akt/mTOR pathway alteration and 72% had therapeutically actionable genomic alterations. Francis et al. [13] identified mutations of 1,230 genes by whole-exome and whole-genome sequencing of 50 small intestinal NETs. Approximately 90% of mutations were not recurrently mutated, and only CDKN1B was recurrently mutated in 10% of patients. Moreover, only a small number of mutated genes overlapped with previously reported small intestinal NETs or pancreatic NETs, suggesting different genomic alterations according to the primary location of the tumor or that many of the observed mutations were passenger and not driver mutations.
In our data sets where our results were combined with previously published results [12, 13], we confirmed that most of the mutated genes did not overlap among the three datasets, including ours. Therefore, we constructed a gene interaction network of 65 genes and 89 interactions using 272 recurrently mutated genes. As a result, traditional cancer-associated pathways, such as cell cycle, Wnt signaling pathway, E2F transcription factor network, DNA damage, p53 pathway, EGFR signaling, FGFR signaling, ERBB2 signaling, PDGFR signaling, and PI3K-Akt signaling pathway were mostly associated with the 65 genes. Notably, 46 of 72 samples (64%) showed at least one mutation of these 65 genes, suggesting that about two-thirds of GEP-NET patients may have a benefit from a drug targeting these pathways.
In this study, we found that a novel MAPKAPK5 mutation involved in the MAPK signaling pathway affected the mechanism of action of pazopanib in a pazopanib responder. MAPKAPK5 is known to be involved in tumor suppression, angiogenesis, and cytoskeletal remodeling through interaction with a variety of substrates and is associated with neurological processes, including neurosecretion [33]. However, MAPKAPK5 mutations have not been documented in GEP-NET. Our results will facilitate the identification of biomarkers for the pazopanib response and will form the basis for a further large-scale genomic study.
The limitation of this study is that it was performed in a relatively small number of samples from various primary organs. To overcome this limitation, we tried to integrate our results with previously published data of pancreatic and small intestinal NETs. To our knowledge, the current study is the first using NET samples from patients enrolled in a clinical trial to identify genetic alterations associated with drug responses.
MATERIALS AND METHODS
Patient characteristics and sample preparation
We performed whole exome sequencing of 12 GEP-NET tumor specimens from patients enrolled in a nonrandomized, open-labeled, single-center phase II study after obtaining written informed consent, and the samples were processed using protocols approved by the Institutional Review Boards [11]. We extracted DNA from fresh tumor tissue (n = 2) and formalin-fixed paraffin-embedded tumor tissue (n = 10) as previously described [14]. The primary sites of GEP-NETs included rectum (n = 3), pancreas (n = 4), small intestine (n = 2), stomach (n = 1), and unknown primary sites (n = 2), and all the cases had hepatic metastasis at presentation. An independent pathologic review by an expert gastrointestinal pathologist (G.L.) is summarized in Supplementary Table 1. The histological grade was categorized as follows: carcinoid tumors and well differentiated NETs were classified as Grade 1 tumors, atypical carcinoid and well-differentiated neuroendocrine carcinomas were classified as Grade 2 tumors, and poorly differentiated neuroendocrine carcinomas were classified as Grade 3 tumors [15]. For validation of the BRAF mutation in an independent cohort, we extracted DNA from 44 primary GEP-NETs (> 1cm in size or > Grade 2) and Sanger-sequenced them as previously described [16]. All research involving human participants have been approved by SMC Institutional Review Board (IRB), and all clinical investigation has been conducted according to the principles expressed in the Declaration of Helsinki.
Whole exome sequencing (WES) and data analysis
Sequencing data were generated using a protocol that has been detailed previously [13, 17]. Briefly, exonic regions were captured using the Agilent V2 capture probe set and sequenced by 76-bp paired-end reads using an Illumina HiSeq2000 instrument. A median of 129,621,217 total reads was generated for each sample, 97.72–99.28% reads of which were aligned to the target exome using the Burrows-Wheeler Aligner (BWA) [15], resulting in a median coverage of each base of 100X.
Downstream sequencing analysis was performed as previously described [13, 17]. Before mutation and indel calling, sequencing reads were locally realigned to improve the detection of indels and decrease the number of false-positive SNVs caused by misaligned reads, particularly at the 3′ end as previously described [13, 17]. For mutation detection, > 14 reads in the tumors and > 8 reads in the normal samples were necessary to call candidate somatic base substitutions, and indels were detected using MuTect [18]. Germline mutations were detected using the UnifiedGenotyper [19]. All somatic mutations were manually reviewed and visually confirmed using the Integrated Genomics Viewer (http://www.broadinstitute.org/igv/). Copy number variation (CNV) was analyzed by GISTIC 2.0 [20], and we investigated GEP-NET CNV genes from drug targets defined in a previous study [21].
Mutational gene interaction network and gene set enrichment analysis
To investigate the genetic characteristics of GEP-NET, we integrated our mutation profiles with previously published data for pancreatic NETs (n = 10) [12] and small intestinal NETs (n = 50) [13]. To compose the gene set, silent mutations were eliminated and then recurrently mutated genes (mutations in greater than or equal to 2) were selected. As a result, 136 genes were entered as a gene set.
The interaction network was constructed from the input gene set by ReactomeFI of Cytoscape [22, 23] [PMID: 20482850, PMID: 14597658]. Gene set enrichment analysis (GSEA) for the reactome pathway was performed within a cutoff FDR of ≤ 0.01. Finally, the interaction network was trimmed to only include genes that passed the GSEA and first neighbors of GSEA genes.
ACKNOWLEDGMENTS AND FUNDINGS
We would like to express our gratitude to Dr. Joshua Francis and Dr. Matthew Meyerson at the Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, Massachusetts 02215, USA, and Broad Institute of Harvard and MIT, Cambridge, Massachusetts 02142, USA, for genomic profiling of our tumor specimens, genomic analysis and helpful scientific advice. This work was partially supported by a grant from the Korean Health Technology R & D Project, Ministry of Health & Welfare, Republic of Korea (HI14C3418).
CONFLICTS OF INTEREST
There are no potential conflicts of interest with regard to this paper.
REFERENCES
1. Hauso O, Gustafsson BI, Kidd M, Waldum HL, Drozdov I, Chan AK, Modlin IM. Neuroendocrine tumor epidemiology: contrasting Norway and North America. Cancer. 2008; 113:2655–2664.
2. Hemminki K, Li X. Incidence trends and risk factors of carcinoid tumors: a nationwide epidemiologic study from Sweden. Cancer. 2001; 92:2204–2210.
3. Lepage C, Rachet B, Coleman MP. Survival from malignant digestive endocrine tumors in England and Wales: a population-based study. Gastroenterology. 2007; 132:899–904.
4. Modlin IM, Lye KD, Kidd M. A 5-decade analysis of 13,715 carcinoid tumors. Cancer. 2003; 97:934–959.
5. Yao JC, Hassan M, Phan A, Dagohoy C, Leary C, Mares JE, Abdalla EK, Fleming JB, Vauthey JN, Rashid A, Evans DB. One hundred years after “carcinoid”: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008; 26:3063–3072.
6. Lawrence B, Gustafsson BI, Chan A, Svejda B, Kidd M, Modlin IM. The epidemiology of gastroenteropancreatic neuroendocrine tumors. Endocrinol Metab Clin North Am. 2011; 40:1–18.
7. Modlin IM, Oberg K, Chung DC, Jensen RT, de Herder WW, Thakker RV, Caplin M, Delle Fave G, Kaltsas GA, Krenning EP, Moss SF, Nilsson O, Rindi G, et al. Gastroenteropancreatic neuroendocrine tumours. Lancet Oncol. 2008; 9:61–72.
8. Meeker A, Heaphy C. Gastroenteropancreatic endocrine tumors. Mol Cell Endocrinol. 2014; 386:101–120.
9. Raymond E, Dahan L, Raoul JL, Bang YJ, Borbath I, Lombard-Bohas C, Valle J, Metrakos P, Smith D, Vinik A, Chen JS, Horsch D, Hammel P, et al. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med. 2011; 364:501–513.
10. Panzuto F, Rinzivillo M, Fazio N, de Braud F, Luppi G, Zatelli MC, Lugli F, Tomassetti P, Riccardi F, Nuzzo C, Brizzi MP, Faggiano A, Zaniboni A, et al. Real-world study of everolimus in advanced progressive neuroendocrine tumors. Oncologist. 2014; 19:966–974.
11. Ahn HK, Choi JY, Kim KM, Kim H, Choi SH, Park SH, Park JO, Lim HY, Kang WK, Lee J, Park YS. Phase II study of pazopanib monotherapy in metastatic gastroenteropancreatic neuroendocrine tumours. Br J Cancer. 2013; 109:1414–1419.
12. Jiao Y, Shi C, Edil BH, de Wilde RF, Klimstra DS, Maitra A, Schulick RD, Tang LH, Wolfgang CL, Choti MA, Velculescu VE, Diaz LA Jr, Vogelstein B, et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science. 2011; 331:1199–1203.
13. Francis JM, Kiezun A, Ramos AH, Serra S, Pedamallu CS, Qian ZR, Banck MS, Kanwar R, Kulkarni AA, Karpathakis A, Manzo V, Contractor T, Philips J, et al. Somatic mutation of CDKN1B in small intestine neuroendocrine tumors. Nat Genet. 2013; 45:1483–1486.
14. Wong SS, Kim KM, Ting JC, Yu K, Fu J, Liu S, Cristescu R, Nebozhyn M, Gong L, Yue YG, Wang J, Ronghua C, Loboda A, et al. Genomic landscape and genetic heterogeneity in gastric adenocarcinoma revealed by whole-genome sequencing. Nat Commun. 2014; 5:5477.
15. Klimstra DS, Modlin IR, Adsay NV, Chetty R, Deshpande V, Gonen M, Jensen RT, Kidd M, Kulke MH, Lloyd RV, Moran C, Moss SF, Oberg K, et al. Pathology reporting of neuroendocrine tumors: application of the Delphic consensus process to the development of a minimum pathology data set. Am J Surg Pathol. 2010; 34:300–313.
16. Kim KM, Lee EJ, Kim YH, Chang DK, Odze RD. KRAS mutations in traditional serrated adenomas from Korea herald an aggressive phenotype. Am J Surg Pathol. 2010; 34:667–675.
17. Ojesina AI, Lichtenstein L, Freeman SS, Pedamallu CS, Imaz-Rosshandler I, Pugh TJ, Cherniack AD, Ambrogio L, Cibulskis K, Bertelsen B, Romero-Cordoba S, Trevino V, Vazquez-Santillan K, et al. Landscape of genomic alterations in cervical carcinomas. Nature. 2014; 506:371–375.
18. Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013; 31:213–219.
19. DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011; 43:491–498.
20. Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011; 12:R41.
21. Van Allen EM, Wagle N, Stojanov P, Perrin DL, Cibulskis K, Marlow S, Jane-Valbuena J, Friedrich DC, Kryukov G, Carter SL, McKenna A, Sivachenko A, Rosenberg M, et al. Whole-exome sequencing and clinical interpretation of formalin-fixed, paraffin-embedded tumor samples to guide precision cancer medicine. Nat Med. 2014; 20:682–688.
22. Wu G, Feng X, Stein L. A human functional protein interaction network and its application to cancer data analysis. Genome Biol. 2010; 11:R53.
23. Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003; 13:2498–2504.
24. Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014; 505:495–501.
25. Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011; 364:2507–2516.
26. Sabbatino F, Wang Y, Wang X, Flaherty KT, Yu L, Pepin D, Scognamiglio G, Pepe S, Kirkwood JM, Cooper ZA, Frederick DT, Wargo JA, Ferrone S, et al. PDGFRalpha up-regulation mediated by sonic hedgehog pathway activation leads to BRAF inhibitor resistance in melanoma cells with BRAF mutation. Oncotarget. 2014; 5:1926–1941. doi:10.18632/oncotarget.1878.
27. Bubolz AM, Weissinger SE, Stenzinger A, Arndt A, Steinestel K, Bruderlein S, Cario H, Lubatschofski A, Welke C, Anagnostopoulos I, Barth TF, Beer AJ, Moller P, et al. Potential clinical implications of BRAF mutations in histiocytic proliferations. Oncotarget. 2014; 5:4060–4070. doi:10.18632/oncotarget.2061.
28. Vanden Borre P, Gunda V, McFadden DG, Sadow PM, Varmeh S, Bernasconi M, Parangi S. Combined BRAF(V600E)- and SRC-inhibition induces apoptosis, evokes an immune response and reduces tumor growth in an immunocompetent orthotopic mouse model of anaplastic thyroid cancer. Oncotarget. 2014; 5:3996–4010. doi:10.18632/oncotarget.2130.
29. Miranda C, Nucifora M, Molinari F, Conca E, Anania MC, Bordoni A, Saletti P, Mazzucchelli L, Pilotti S, Pierotti MA, Tamborini E, Greco A, Frattini M. KRAS and BRAF mutations predict primary resistance to imatinib in gastrointestinal stromal tumors. Clin Cancer Res. 2012; 18:1769–1776.
30. Tiacci E, Trifonov V, Schiavoni G, Holmes A, Kern W, Martelli MP, Pucciarini A, Bigerna B, Pacini R, Wells VA, Sportoletti P, Pettirossi V, Mannucci R, et al. BRAF mutations in hairy-cell leukemia. N Engl J Med. 2011; 364:2305–2315.
31. O’Donnell E, Raje NS. Targeting BRAF in multiple myeloma. Cancer Discov. 2013; 3:840–842.
32. Chami R, Yin M, Marrano P, Teerapakpinyo C, Shuangshoti S, Thorner PS. BRAF mutations in pediatric metanephric tumors. Hum Pathol. 2015.
33. Moens U, Kostenko S. Structure and function of MK5/PRAK: the loner among the mitogen-activated protein kinase-activated protein kinases. Biol Chem. 2013; 394:1115–1132.