
INTRODUCTION
As prostate cancer is already notorious for substantial heterogeneity on the morphologic level, it is conceivable, that substantial heterogeneity may also occur for molecular features. On the one hand heterogeneity may limit the applicability of diagnostic, prognostic, and predictive molecular markers, but, on the other hand, it may help to understand the chronology of molecular events during tumor progression.
Studies by others and us have demonstrated, that the TMPRSS2:ERG fusion status is highly heterogeneous within individual prostate cancers [1–3]. Deletions on chromosome 6q are the second most common deletions in prostate cancer and occur in 18–62% [4–9]. Data suggest a minimally deleted area on 6q15 with a length of 3–4 Mb containing only few genes, including MAP3K7/TAK1. A tumor-suppressive role in prostate cancer cells has recently been demonstrated for this gene [10]. The TGF-β and BMP activated kinase MAP3K7 is essential for a series of cancer associated signaling pathways like the p38 and JNK [11–13]. In a recent study, we found a strong association of MAP3K7 deletion with early PSA recurrence in a series of 2,289 cancers analyzed by FISH. Remarkably, there was also a striking inverse relationship between MAP3K7 deletions and presence of TMPRSS2:ERG fusions [9]. This finding raises the question whether MAP3K7 deletion prevents cancers cells from developing TMPRSS2:ERG fusions or vice versa.
To thoroughly evaluate the heterogeneity for MAP3K7 deletions and TMPRSS2:ERG fusion status and their interrelationship in prostate cancer, we took advantage of a newly developed heterogeneity tissue microarray (TMA) containing samples from 10 different tumor blocks of 189 large prostate cancers. This approach enables a high-throughput mapping of molecular features across entire tumors. The data show substantial heterogeneity for MAP3K7 deletion in prostate cancer and suggest that MAP3K7 deletion largely prevents development of TMPRSS2:ERG fusions.
RESULTS
Heterogeneity of MAP3K7 deletion in prostate cancer
The 6q15 FISH analysis of our heterogeneity TMA revealed 924 informative cancer spots (48.9% of 1,890 samples arrayed). 966 of 1,890 tissue spots were non-informative due to lack of tissue samples, absence of unequivocal cancer tissue or to lacking FISH signals for centromere 6, MAP3K7 or both. A MAP3K7 deletion was found in 165 (17.8%) of 924 cancers spots. All of these deletions were heterozygous and 140 patients had at least three informative cancer spots. Of these, 15 were homogeneously and 21 were heterogeneously MAP3K7 deleted. 104 of 140 patients showed no MAP3K7 deletion. To exclude interfocal tumor heterogeneity in patients with more than one tumor focus, we analyzed individual cancer foci for heterogeneity. There were 133 patients (95.0%) with 138 tumor foci large enough to create 3 informative cancer-containing tissue spots. Of these 138 tumor foci, there were 16 with homogeneous 6q15 deletion, 18 with heterogeneous 6q15 deletion, and 104 with a normal 6q15 result in all spots (Figure 1).
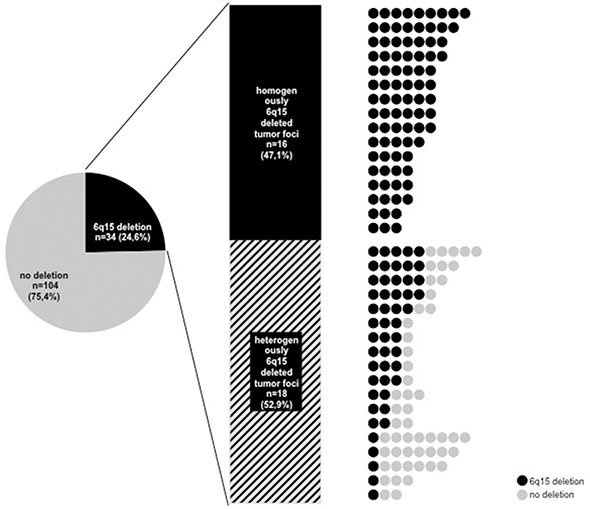
Figure 1: Heterogeneity of 6q15 deletion in all tumor foci with at least three analyzable tumor spots. Right: All circles in one line corresponds to one tumor foci, black circle: 6q15 deleted tumor spot, grey circle: 6q15 non-deleted tumor spot.
MAP3K7 deletion vs. ERG-expression
ERG data were available from a previous study in 774 of 924 tissue spots with informative 6q15 deletion status. Positive ERG status was strongly linked to absence of 6q15 deletion in these samples. 6q15 deletion was seen in 125 (28.4%) of 440 ERG-negative, but only in 22 (6.6%) of 334 ERG-positive samples (p < 0.0001, Figure 2). The evaluation of the above mentioned 138 tumor foci revealed 42 tumor foci with homogeneous and 38 tumor foci with heterogeneous ERG positivity, while 56 tumor foci were ERG-negative.
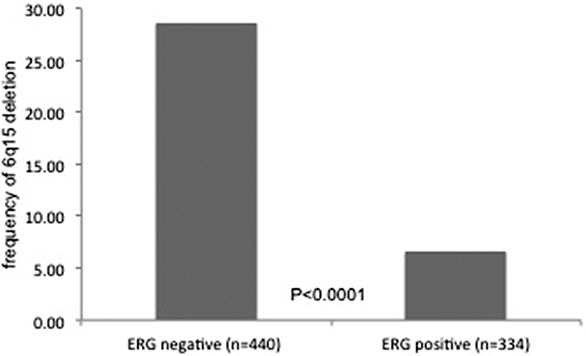
Figure 2: Relationship between MAP3K7 (6q15) deletion status and ERG-fusion status measured by immunohistochemistry.
Sequel of MAP3K7 deletion and TMPRSS2:ERG fusion development
To evaluate the chronological development of TMPRSS2:ERG fusion and 6q15 deletions we searched for tumors with focal 6q15 deletions arising in homogeneously ERG-positive cancers and tumors with focal ERG positivity arising in homogeneously or heterogeneously 6q15 deleted cancers. None of 34 cancers revealed focal ERG positivity in a 6q15 deleted setting, while 6 of 42 cancers had a small area of 6q15 deletion in an ERG-positive background (p = 0.022, Figure 3). Comparison of homogenously 6q15 deleted cancers lacking focal ERG positivity (n = 16) and homogenously ERG-positive cancers with focal 6q15 deletion (n = 6 of 42) reaches no statistical significance (p = 0.129, data not shown) most likely due to the small number of cases with homogenous alterations.
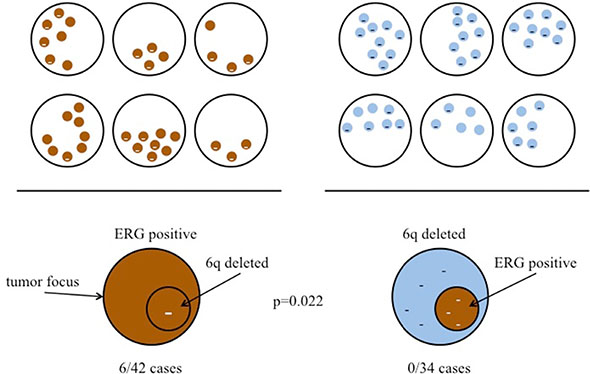
Figure 3: Sequel of MAP3K7 (6q15) deletion and TMPRSS2:ERG fusion development. Left: Cases with ERG-positive tumor focus and focal 6q15 deletion. Right: Cases with 6q15 deleted tumor focus and focal ERG positivity in the same tumor area.
DISCUSSION
Our data revealed that MAP3K7 deletion is often heterogeneous in prostate cancer and prevents TMPRSS2:ERG fusion, while TMPRSS2:ERG fusion allows MAP3K7 deletion.
To thoroughly evaluate the heterogeneity for MAP3K7 deletions and TMPRSS2:ERG fusion status and their interrelationship in prostate cancer, we took advantage of a newly developed heterogeneity tissue microarray (TMA) containing samples from 10 different tumor blocks of 189 large prostate cancers. This approach enables a high-throughput mapping of molecular features across entire tumors.
The frequency of 25.7% MAP3K7 deletions observed by FISH is somewhat higher than in our previous study describing 18.5% MAP3K7 deletions on a TMA containing 2,289 different prostate cancers [9]. This is likely to be caused by the analysis of multiple samples per tumor in this study, enabling the identification of higher number of heterogeneous 6q15 deletions. Earlier studies using CGH, PCR or LOH analysis reported between 13% and 62% 6q15 deletion in prostate cancers [4–8, 14, 15]. Only one earlier study also used FISH with a probe specific for MAP3K7 and found a deletion in 32% of 95 advanced stage tumors. As 6q15 deletions increase substantially with grade and stage, selection bias may have led to this slightly higher figure [7]. FISH represents the gold standard for the analysis of gene copy number changes. FISH enables a cell by cell analysis making the method independent of a possible pollution by non-neoplastic cells and genomic cancer heterogeneity. To fully exploit the strength of the FISH approach, stringent criteria were applied for assuring presence of cancer cells in each analyzed tissue spot and also for defining MAP3K7 deletions. In an earlier study we had successfully validated our cut-off level of ≥ 60% deleted cell nuclei by comparing PTEN deletions detected by FISH and CGH arrays and demonstrating an 100% concordance [16].
Accumulating evidence suggests a critically important role of 6q15 deletions in prostate cancer. With a frequency of 20–30% observed in FISH studies, 6q15 deletions are among the most frequent genomic alterations in prostate cancer. The strong inverse association with TMPRSS2:ERG fusions identifies 6q15 deleted cancers as a significant subgroup among “non fusion-type” prostate cancers [8, 9]. Moreover, the strong association of these deletions with early PSA recurrence potentially assigns clinical relevance to this molecular subgroup [9], which, however, may be limited if the analyzed biomarker is only present in a fraction of a tumor.
Considering its importance, the number of studies systematically analyzing target heterogeneity in cancer is relatively small. Even studies addressing the issue of tumor heterogeneity often limit themselves to the analysis of one slide/block per tumor/patient. However, one tissue section may not completely represent the biology of a large cancer. “Heterogeneity TMAs” were thus manufactured by our group as new tools for studying molecular cancer heterogeneity [1, 17]. A TMA analysis of one sample each from ten different tumor-containing blocks distributed across the entire tumor enables a comprehensive three-dimensional high-throughput analysis of molecular features in large series of tumors. This heterogeneity TMA concept differs markedly from previous attempts to increase the representativity of prostate cancer TMAs, namely by sampling multiple cores from just one tumor block [18, 19]. The data of our study demonstrate that heterogeneity of 6q15 deletion occurs in about half of 6q15 deleted prostate cancers. This clearly limits the potential of treatment decisions based on the 6q15 deletion status determined on one single cancer biopsy.
While cancer heterogeneity impairs the applicability of diagnostic, prognostic, and predictive molecular markers, it can be utilized to understand the sequel of two or more molecular features occurring in one cancer. Depending on the sequel of development of MAP3K7 deletions and ERG-expression, one can expect a small area of cancer having both alterations within a larger area having only the first one of these changes. Our observation that 6 of 42 homogeneously ERG-positive cancer foci had focal 6q15 deletions strongly suggests, that presence of TMPRSS2:ERG fusions does not prevent the development of 6q15 deletions. This is all the more the case as the frequency of 6q15 deletions (14.3%) in this subgroup did not differ significantly from the 6q15 deletion rate in the entire (unselected) cohort (36/140; 25.7%; p > 0.1). The complete absence of focal ERG positivity in 34 cancer foci with 6q15 deletions strongly supports the concept of 6q15 deletions either directly or indirectly preventing tumor cells from developing TMPRSS2:ERG fusions.
TMPRSS2:ERG fusion, and other rearrangements involving ETS family members, is facilitated through androgen-mediated activation of transcriptional programs generating inter- or intra-chromosomal ‘transcriptional hubs’ [20]. Androgen-induced, DNA double-strand breaks followed by low-fidelity DNA repair can then lead to genome alterations involving androgen-regulated genes (such as TMPRSS2), which can be fused to transcription factors with oncogenic activity (such as ERG) [21, 22]. It is well known, that the likelihood of such rearrangement events can be impacted by an altered function of chromatin modifying genes. The results of whole genome sequencing studies have highlighted a pivotal role of various chromatin regulating genes in prostate cancer [5, 23–25]. In an earlier study, we had demonstrated a striking association of 5q21 deletions with absence of TMPRSS2:ERG fusions and implicated CHD1, a chromatin modifying gene, as the target gene of this deletion. We were able to show that a reduced expression of CHD1 prevents the development of TMPRSS2:ERG fusions [26]. Also the 6q21–q22 region contains several genes involved in histone and chromatin modification, such as ASF1A, BEND3 and HDAC2. Of note, HDAC2 interacts with Topoisomerase IIB (TOP2B) in a protein complex that is essential for ERG-fusion development [27, 28]. It could, thus, be speculated, that the reduced expression of one or several genes involved in 6q deletion could affect the likelihood of TMPRSS2:ERG fusion development.
In summary, the results of this study show that MAP3K7 deletion is often heterogeneous in prostate cancer. The data also suggest that presence of a 6q15 deletion including MAP3K7 - either directly or indirectly - prevents tumor cells from developing TMPRSS2:ERG fusions.
MATERIALS AND METHODS
Patient samples and tissue microarray (TMA) construction
A total of 189 prostate cancers were used for manufacturing a prostate cancer heterogeneity TMA, as previously described [1]. All patients underwent radical prostatectomy at the Martini Clinic, Prostate Cancer Center, University Medical Center Hamburg-Eppendorf according to our institutional standard.
In brief, all prostates were prepared in a standardized way [29, 30]. Macroscopic images were taken from each tumor. Prostate cancer patients were selected for this study, if they had a tumor involvement of at least 10 different tissue blocks. These cancers had an average volume of 3.4 cm3 (maximum 60 cm3). For each cancer, the number of independent tumor foci was determined according to Wise et al. [31]. In brief, tumor areas were defined as part of a single focus if they were within 3 mm of each other in any section or within 4 mm on adjacent sections. This method identified 1–6 independent tumor foci in our prostate cancers. Seventy-six prostates had one tumor focus, 48 prostates had 2 tumor foci, 28 prostates had 3 tumor foci, and 38 prostates had 4 or more tumor foci. The latter group also included 10 prostates that contained multiple small and very small tumor foci rather than one or several clearly distinguishable tumor masses. From each of our 189 tumors ten different tumor containing tissue blocks were selected for TMA manufacturing. If more than 10 blocks were available, blocks were selected to obtain an optimal representation of the entire tumor mass (i.e. blocks were selected that enabled maximal distances between selected tumor areas). One core from each selected block was then taken and placed in 10 different TMA blocks. This resulted in 10 different TMA blocks, each containing one tissue sample from each of our 189 selected patients. For subsequent mapping of molecular findings, the exact position from where each arrayed tumor sample had been retrieved was recorded in a database also containing all macroscopical images of our tumors. Each tissue sample was assigned to a defined tumor focus. The clinical and pathological features of our tumor cohort are provided in Table 1.
Table 1: Composition of the heterogeneity tissue micro array
|
Study cohort on tissue microarray (n = 189) |
|---|---|
Age (years) |
|
< 50 |
3 |
50–60 |
43 |
60–70 |
120 |
> 70 |
23 |
Tumor stage (AJCC 2002) |
|
pT2 |
127 |
pT3a |
32 |
pT3b |
29 |
pT4 |
1 |
Gleason Score |
|
≤ 3 + 3 |
4 |
3 + 4 |
144 |
4 + 3 |
33 |
≥ 4 + 4 |
8 |
Lymph node metastasis |
|
N0 |
130 |
N1 |
10 |
Surgical margin |
|
negative |
160 |
positive |
26 |
NOTE: Numbers do not always add up to 189 in the different categories because of cases with missing data. Abbreviation: AJCC, American Joint Committee on Cancer.
Validation for presence of cancer on TMA spots
Technical issues represent a significant problem in studies analyzing heterogeneity because every false positive or false negative result will lead to a false classification as “heterogeneous”. Every effort was thus taken in this study to avoid false interpretations including immunohistochemical confirmation of presence of cancer for each sample. For this purpose, the antibody 34βE12 (clone MA903, Dako; 1:12.5; pH7.8) was used for basal cell detection and p504s (clone 13H4, Dako; 1:200; pH9.0) was utilized for AMACR detection. The EnVision™ Kit (DAKO, Glostrup) was used to visualize the immunostainings. For each tissue spot, presence or absence of normal prostate epithelium and the proportion of cancer tissue was recorded and quantified.
Fluorescence in-situ hybridization
A 4 µm TMA section was used for dual color fluorescence in-situ hybridization (FISH). Before hybridization, sections were deparaffinized and proteolytically pretreated with a commercial kit (paraffin pretreatment reagent kit; Abbott Molecular, Wiesbaden, Germany), followed by dehydration in 70%, 80% and 96% ethanol, air drying and denaturation for 10 min at 72°C in 70% formamide-2x SSC solution. For MAP3K7 deletion analysis, a dual color FISH probe set consisting of two spectrum green-labeled BAC probes (BAC RP3–470J8, BAC P11–501P02; Imergenes, Germany) containing the MAP3K7 gene and an adjacent up- and downstream region and one spectrum orange-labeled commercial centromere 6 probe (#06J36–06; Abbott), which was used as a reference. Hybridization was done overnight at 37°C in a humidified chamber, slides were washed and counterstained with 0.2 µmol/L 4′-6-diamidino-2-phenylindole in an antifade solution. For MAP3K7 deletion analysis, each spot was evaluated and the predominant signal was recorded for each FISH probe. Tissue spots with lack of MAP3K7 signals in all (tumor and normal cells) or lack of any normal cells as an internal control for successful hybridization of the MAP3K7 probe were excluded from analysis. Heterozygous deletion of MAP3K7 was defined as presence of fewer MAP3K7 signals than centromere 6 probe signals of ≥ 60% tumor nuclei. Representative cases with and without MAP3K7 deletion are shown in figure 4.
Figure 4: Examples of FISH findings showing (A) normal 6q15 signal numbers and (B) heterozygous 6q15 deletion. Orange signals correspond to centromere 6, green signals correspond to the MAP3K7 (6q15) gene locus.
ERG immunohistochemistry
ERG immunohistochemistry data were available for each individual cancer spot from a previous study [1]. The antibody ERG (clone EPR3864, dilution 1:450, Epitomics) had been used for ERG protein detection in this study. Only nuclear ERG staining was considered. For each tumor sample the staining intensity was judged from negative to strong. Tumors with at least a weak ERG immunostaining were considered ERG-positive.
Large section validation
To validate heterogeneity of MAP3K7 deletion and ERG-fusion status, two cases with documented intrafocal heterogeneity and homogeneity for ERG-fusion status and intrafocal heterogeneity for MAP3K7 deletion on our TMA were also subjected to large section analysis. In these cases, relevant cancer-containing tissue blocks (based on which TMA spots suggested heterogeneity or homogeneity) were cut and MAP3K7 and ERG status were analyzed as described.
Statistics
Statistical calculations were performed using JMP statistical software (SAS Institute, Cary, NC). Contingency tables were calculated with the chi2-test to investigate the relationship between the degrees of ERG heterogeneity.
ACKNOWLEDGMENTS
We are grateful to Christina Koop, Janett Lütgens, Inge Brandt and Bianca Kelp for excellent technical assistance.
FUNDINGS
We certify that there are no sources of any support for the work, received in the form of grants, equipment, and/or drugs in relation to this article.
CONFLICTS OF INTEREST
We certify that there is no actual or potential conflicts of interest in relation to this article.
REFERENCES
1. Minner S, Gartner M, Freudenthaler F, Bauer M, Kluth M, Salomon G, Heinzer H, Graefen M, Bokemeyer C, Simon R, Sauter G, Schlomm T, Wilczak W. Marked heterogeneity of ERG expression in large primary prostate cancers. Modern pathology. 2013; 26:106–116.
2. Mehra R, Han B, Tomlins SA, Wang L, Menon A, Wasco MJ, Shen R, Montie JE, Chinnaiyan AM, Shah RB. Heterogeneity of TMPRSS2 gene rearrangements in multifocal prostate adenocarcinoma: molecular evidence for an independent group of diseases. Cancer research. 2007; 67:7991–7995.
3. Furusato B, Tan SH, Young D, Dobi A, Sun C, Mohamed AA, Thangapazham R, Chen Y, McMaster G, Sreenath T, Petrovics G, McLeod DG, Srivastava S, et al. ERG oncoprotein expression in prostate cancer: clonal progression of ERG-positive tumor cells and potential for ERG-based stratification. Prostate cancer and prostatic diseases. 2010; 13:228–237.
4. Sun J, Liu W, Adams TS, Li X, Turner AR, Chang B, Kim JW, Zheng SL, Isaacs WB, Xu J. DNA copy number alterations in prostate cancers: a combined analysis of published CGH studies. The Prostate. 2007; 67:692–700.
5. Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, Antipin Y, Mitsiades N, Landers T, et al. Integrative genomic profiling of human prostate cancer. Cancer cell. 2010; 18:11–22.
6. Ishkanian AS, Mallof CA, Ho J, Meng A, Albert M, Syed A, van der Kwast T, Milosevic M, Yoshimoto M, Squire JA, Lam WL, Bristow RG. High-resolution array CGH identifies novel regions of genomic alteration in intermediate-risk prostate cancer. The Prostate. 2009; 69:1091–1100.
7. Liu W, Chang BL, Cramer S, Koty PP, Li T, Sun J, Turner AR, Von Kap-Herr C, Bobby P, Rao J, Zheng SL, Isaacs WB, Xu J. Deletion of a small consensus region at 6q15, including the MAP3K7 gene, is significantly associated with high-grade prostate cancers. Clinical cancer research. 2007; 13:5028–5033.
8. Lapointe J, Li C, Giacomini CP, Salari K, Huang S, Wang P, Ferrari M, Hernandez-Boussard T, Brooks JD, Pollack JR. Genomic profiling reveals alternative genetic pathways of prostate tumorigenesis. Cancer research. 2007; 67:8504–8510.
9. Kluth M, Hesse J, Heinl A, Krohn A, Steurer S, Sirma H, Simon R, Mayer PS, Schumacher U, Grupp K, Izbicki JR, Pantel K, Dikomey E, et al. Genomic deletion of MAP3K7 at 6q12–22 is associated with early PSA recurrence in prostate cancer and absence of TMPRSS2:ERG fusions. Modern pathology. 2013; 26:975–983.
10. Wu M, Shi L, Cimic A, Romero L, Sui G, Lees CJ, Cline JM, Seals DF, Sirintrapun JS, McCoy TP, Liu W, Kim JW, Hawkins GA, et al. Suppression of Tak1 promotes prostate tumorigenesis. Cancer research. 2012; 72:2833–2843.
11. Edlund S, Bu S, Schuster N, Aspenstrom P, Heuchel R, Heldin NE, ten Dijke P, Heldin CH, Landstrom M. Transforming growth factor-beta1 (TGF-beta)-induced apoptosis of prostate cancer cells involves Smad7-dependent activation of p38 by TGF-beta-activated kinase 1 and mitogen-activated protein kinase kinase 3. Molecular biology of the cell. 2003; 14:529–544.
12. Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000; 103:239–252.
13. Cuadrado A, Nebreda AR. Mechanisms and functions of p38 MAPK signalling. The Biochemical journal. 2010; 429:403–417.
14. Saric T, Brkanac Z, Troyer DA, Padalecki SS, Sarosdy M, Williams K, Abadesco L, Leach RJ, O’Connell P. Genetic pattern of prostate cancer progression. International journal of cancer. 1999; 81:219–224.
15. Cunningham JM, Shan A, Wick MJ, McDonnell SK, Schaid DJ, Tester DJ, Qian J, Takahashi S, Jenkins RB, Bostwick DG, Thibodeau SN. Allelic imbalance and microsatellite instability in prostatic adenocarcinoma. Cancer research. 1996; 56:4475–4482.
16. Krohn A, Diedler T, Burkhardt L, Mayer PS, De Silva C, Meyer-Kornblum M, Kotschau D, Tennstedt P, Huang J, Gerhauser C, Mader M, Kurtz S, Sirma H, et al. Genomic deletion of PTEN is associated with tumor progression and early PSA recurrence in ERG fusion-positive and fusion-negative prostate cancer. The American journal of pathology. 2012; 181:401–412.
17. Krohn A, Freudenthaler F, Harasimowicz S, Kluth M, Fuchs S, Burkhardt L, Stahl P, M CT, Bauer M, Tennstedt P, Graefen M, Steurer S, Sirma H, et al. Heterogeneity and chronology of PTEN deletion and ERG fusion in prostate cancer. Modern pathology. 2014; 27:1612–1620.
18. Rubin MA, Dunn R, Strawderman M, Pienta KJ. Tissue microarray sampling strategy for prostate cancer biomarker analysis. Am J Surg Pathol. 2002; 26:312–319.
19. Kristiansen G, Fritzsche FR, Wassermann K, Jager C, Tolls A, Lein M, Stephan C, Jung K, Pilarsky C, Dietel M, Moch H. GOLPH2 protein expression as a novel tissue biomarker for prostate cancer: implications for tissue-based diagnostics. British journal of cancer. 2008; 99:939–948.
20. Mani RS, Tomlins SA, Callahan K, Ghosh A, Nyati MK, Varambally S, Palanisamy N, Chinnaiyan AM. Induced chromosomal proximity and gene fusions in prostate cancer. Science. 2009; 326:1230.
21. Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, Varambally S, Cao X, Tchinda J, Kuefer R, Lee C, Montie JE, Shah RB, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005; 310:644–648.
22. Kumar-Sinha C, Tomlins SA, Chinnaiyan AM. Recurrent gene fusions in prostate cancer. Nature reviews Cancer. 2008; 8:497–511.
23. Berger MF, Lawrence MS, Demichelis F, Drier Y, Cibulskis K, Sivachenko AY, Sboner A, Esgueva R, Pflueger D, Sougnez C, Onofrio R, Carter SL, Park K, et al. The genomic complexity of primary human prostate cancer. Nature. 2011; 470:214–220.
24. Kumar A, Shendure J, Nelson PS. Genome interrupted: sequencing of prostate cancer reveals the importance of chromosomal rearrangements. Genome medicine. 2011; 3:23.
25. Wilson BG, Roberts CW. SWI/SNF nucleosome remodellers and cancer. Nature reviews Cancer. 2011; 11:481–492.
26. Burkhardt L, Fuchs S, Krohn A, Masser S, Mader M, Kluth M, Bachmann F, Huland H, Steuber T, Graefen M, Schlomm T, Minner S, Sauter G, et al. CHD1 Is a 5q21 Tumor Suppressor Required for ERG Rearrangement in Prostate Cancer. Cancer research. 2013; 73:2795–2805.
27. Tsai SC, Valkov N, Yang WM, Gump J, Sullivan D, Seto E. Histone deacetylase interacts directly with DNA topoisomerase II. Nature genetics. 2000; 26:349–353.
28. Haffner MC, Aryee MJ, Toubaji A, Esopi DM, Albadine R, Gurel B, Isaacs WB, Bova GS, Liu W, Xu J, Meeker AK, Netto G, De Marzo AM, et al. Androgen-induced TOP2B-mediated double-strand breaks and prostate cancer gene rearrangements. Nature genetics. 2010; 42:668–675.
29. McNeal JE, Redwine EA, Freiha FS, Stamey TA. Zonal distribution of prostatic adenocarcinoma. Correlation with histologic pattern and direction of spread. Am J Surg Pathol. 1988; 12:897–906.
30. Erbersdobler A, Hammerer P, Huland H, Henke RP. Numerical chromosomal aberrations in transition-zone carcinomas of the prostate. The Journal of urology. 1997; 158:1594–1598.
31. Wise AM, Stamey TA, McNeal JE, Clayton JL. Morphologic and clinical significance of multifocal prostate cancers in radical prostatectomy specimens. Urology. 2002; 60:264–269.