
INTRODUCTION
Epithelial ovarian carcinoma (EOC) accounts for the highest tumor-related mortality in women with gynecologic malignancy [1]. The current therapy for advanced ovarian cancer is debulking surgery followed by chemotherapy containing cisplatinum agent and paclitaxel with the eventual addition of bevacizumab; however, successful long-term treatment is prevented by the development of drug resistance [2]. The identification of the molecular mechanisms underlying chemoresistance is mandatory to achieve advancement in EOC therapy. An aberrant activation of endothelin-1 (ET-1) axis, which consists of the ligand ET-1 and its G-protein coupled receptors (GPCR) endothelin A receptor (ETAR) and ETBR, is now recognized as a common mechanism underlying the progression of various solid tumors, including EOC [3]. In particular, ETAR overexpression is associated with the acquisition of chemoresistance and epithelial-to-mesenchymal transition (EMT) phenotype of EOC [3–7]. The ET-1/ETAR interaction triggers the activation of the survival pathways, such as phosphoinositide-3-kinase (PI-3K)/Akt and mitogen activated protein kinase (MAPK), thus protecting EOC cells from drug-induced apoptosis [4]. All these observations suggest that ETAR expression may be a predictor of chemoresistance, and that targeting ETAR could increase the sensitivity of EOC tumors to therapeutic agents. In this regard, we recently provide evidence that ETAR/β-arrestin-1 cooperates with Wnt signaling to acquire a chemoresistant phenotype through the amplification of ET-1 autocrine loop, thus sustaining EMT, stemness features, cell invasion and metastasis [8]. In addition to ETAR, ETBR also appears to have important regulatory roles in angiogenesis, lymphangiogenesis and evasion of the immune response [3, 8–14], indicating that ETAR and ETBR could serve as key targets for EOC. The dual receptor antagonist macitentan, interfering with ETAR, and microenvironment-associated elements expressing ETBR [12–15], inhibited growth, vascularization, intravasation and progression in EOC xenografts [8].
microRNA (miRNA) are small non-coding RNAs 19-25 nucleotides in length. Mature forms of miRNA silence gene expression by binding to the 3′ UTR of target mRNA and initiate translational repression or cleavage of cognate mRNA [16]. These molecules have been recognized as critical regulators of tumor development and progression, including EOC [17–20]. Moreover, recent studies demonstrated the involvement of miRNA in chemoresistance [21–24]. Despite the growing interest around miRNA in tumor progression and chemoresistance, currently there are no studies to clarify physiologic and pathologic implications on ETAR regulation by miRNA. In this study, we aimed to identify miRNA able to control ETAR expression overcoming therapy resistance in EOC cells. miR-30a has attracted much attention due to its important role in various biological and pathological processes, including EMT and tumor progression [25, 26]. However, whether miR-30a is involved in the chemoresistance onset of EOC and the underlying mechanisms remain poorly understood. Employing the Cancer Genome Atlas (TCGA) data [27], we found that miR-30a downregulation in high-grade serous ovarian cancer (HG-SOC), which reprents a large majority of EOC cases, is downregulated compared to normal tissue and is associated with poor prognosis. Moreover, we show that miR-30a is downregulated in chemoresistant EOC cells. Functional studies demonstrate that overexpression of miR-30a decreases cellular vitality, invasion, plasticity and EMT. ETAR is identified as a direct target of miR-30a, and their expression is inversely correlated in EOC cell lines and human tissue samples. Upregulation of miR-30a re-sensitizes resistant EOC cells to cisplatinum by binding ETAR. Overexpression of miR-30a inhibits tumor growth in cisplatinum-resistant xenografts. Our findings provide a new perspective on the regulatory mechanism of endogenous ETAR gene at the post-transcriptional level. Moreover, this study suggests that miR-30a/ETAR axis regulates EOC chemoresistance onset and that interfering with ETAR function could serve as an important therapeutic approach for EOC.
RESULTS
miR-30a is downregulated in chemoresistant ovarian cancer cells
In order to identify miRNA able to bind the ETAR 3′UTR, we performed a computational target prediction analysis by using TargetScan [28], miRanda [29] and miRDB [30] bioinformatic algorithms (Supplementary Figure S1A). ETAR 3′UTR contained a perfect complementary matching region at nucleotides 1327-1334, 2200-2205, 2321-2327 for miR-30 family (Supplementary Figure S1B), consisting of six distinct mature miRNA sequences: miR-30a/miR-30c-2, miR-30d/miR-30b, and miR-30e/miR-30c-1. To search for the critical miR-30 family members involved in aberrant ETAR expression reported in chemoresistant EOC cells [4, 8], we firstly analyzed their expression levels in 2008 and A2780 human cell lines and in their cisplatinum-resistant variants. Real-time PCR (qPCR) experiments demonstrated that miR-30a resulted significantly downregulated in both 2008 CIS and A2780 CIS cells (Figure 1A and Supplementary Figure S1C), expressing higher mRNA and protein levels of ETAR compared to sensitive cells (Figure 1B and Supplementary Figure S1D). It is notable that miR-30a expression was also downregulated in paclitaxel-resistant A2780 cell line, which overexpressed ETAR [4,8] (Supplementary Figure S1D and S1E), demonstrating that overexpression of miR-30a directly binds ETAR, which is responsible for regulating the rate limiting of both paclitaxel and cisplatinum response.
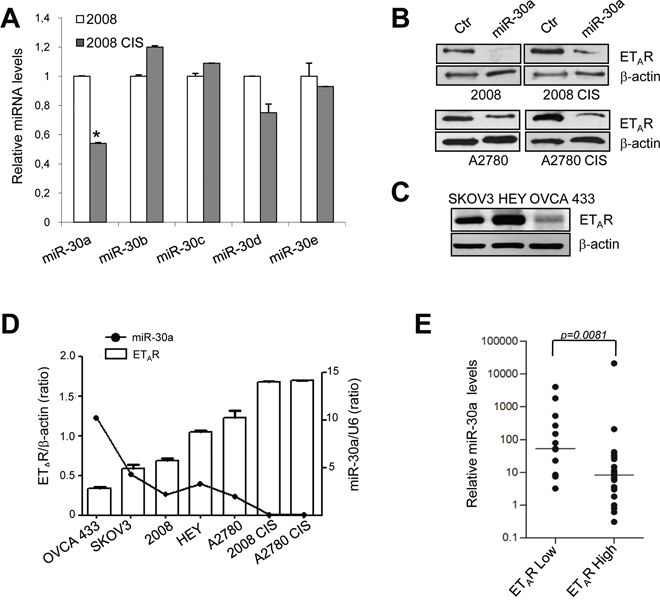
Figure 1: miR-30a is downregulated in chemoresistant EOC cells. A. Expression of miR-30 family members in sensitive and resistant 2008 cells measured by qPCR. miRNA levels are normalized using endogenous U6 snRNA. Values are the mean ± SD (n = 3; *, p < 0.005 compared to sensitive cells). Lysates from sensitive and resistant 2008 and A2780 cells transfected with mimic-miR control (Ctr) or mimic-miR-30a B. or from SKOV3, HEY and OVCA433 EOC cell lines C. are analyzed by western blotting for ETAR expression. β-actin is used as loading control. D. Expression levels of ETAR and miR-30a in a panel of seven EOC cell lines. The ratio of ETAR/ β-actin, evaluated by western blotting as shown in B and C, and miR-30a/U6 expression, evaluated by qPCR, is shown as bar and line, respectively. E. Scatter plot of the expression of miR-30a, as determined by qPCR, in the 39 EOC patients dichotomized into ETAR high or ETAR low expressing tumors. The expression levels of miR-30a were normalized to RNU44 expression. Horizontal lines indicate median n = 13 (low ETAR), n = 26 (high ETAR) and Mann-Whitney ran sum test was used in the statistical analysis (p = 0.0081).
In a panel of EOC cell lines, the expression of miR-30a was substantially decreased and was associated with a highy expression of ETAR (p = 0.02) (Figure 1B, 1C and 1D). The ectopic introduction of miR-30a significantly reduced ETAR protein levels, even in resistant cells expressing low miR-30a and high ETAR (Figure 1B). miR-30a did not affect the expression of ETAR mRNA (Supplementary Figure S1F), indicating that it unlikely inhibits transcription. Furthermore, we evaluated the expression of miR-30a by qPCR in a cohort of 39 EOC human tumor samples, whose clinical-pathological characteristics are summarized in Supplementary Table S1, and in which the ETAR expression has been previously examinated by immunohistochemistry (IHC) [8]. As show in Figure 1E, we found a significant (p = 0.0081) inverse correlation between the expression levels of ETAR and miR-30a. The median expression value of miR-30a, normalized for RNU44 expression, was significantly lower in tumors expressing high (n = 26) versus low (n = 13) ETAR levels (median=8.4, range 0.3-21,203 vs median=54 range 3.3-4,045, respectively). These data suggest that the regulation of miR-3oa/ETAR axis is involved in the pathobiology of EOC.
ETAR is a novel target of miR-30a
To assess whether ETAR is a direct target of miR-30a, we utilized a luciferase report assay. miR-30a significantly reduced ETAR 3′UTR reporter activity, confirming that miR-30a directly binds the ETAR mRNA (Figure 2A). miR-30a did not affect luciferase activity with ETAR 3′UTR possessing a mutation in the conserved miR-30a binding site (1327-1334) (Figure 2A). Taken together, these results suggest that miR-30a directly decreases ETAR expression through sequence-specific binding with 3′UTR of ETAR mRNA. In order to further confirm miR-30a specificity in ETAR regulation, we transfected chemoresistant EOC cells with the antago-miR-30a (anti-miR-30a), chemically engineered oligonucleotides used to silence endogenous miR-30a. Importantly, increased levels of ETAR protein was observed in miR-30a-silenced cells compared to control cells (Figure 2B). Next, we used miR-30a overexpression and inhibition strategies in cell proliferation. The ectopic introduction of miR-30a significantly decreased cell vitality of both sensitive and resistant EOC cells. To determine whether ETAR inhibition can recapitulate the effects of miR-30a observed in EOC cells, we explored molecular ETAR targeting treatment by using the small molecule macitentan, a potent ETAR antagonist with significant affinity for ETBR. Following treatment with macitentan, the cell vitality was significantly decreased. On the contrary, the silencing of miR-30a enhanced the proliferation of these cells (Figure 2C). Silencing of ETAR mimicked the effect of miR-30a inhibiting cell vitality (Figure 2D and 2E). Importantly, ectopic introduction of miR-30a in EOC cells overexpressing ETAR, with an expression vector encoding ETAR lacking 3′UTR, was unable to completely suppress cell proliferation (Figure 2D and 2E). Altogether, these findings demonstrate that miR-30a functionally binds the ETAR 3′UTR, thereby inhibiting ETAR expression and cell proliferation.
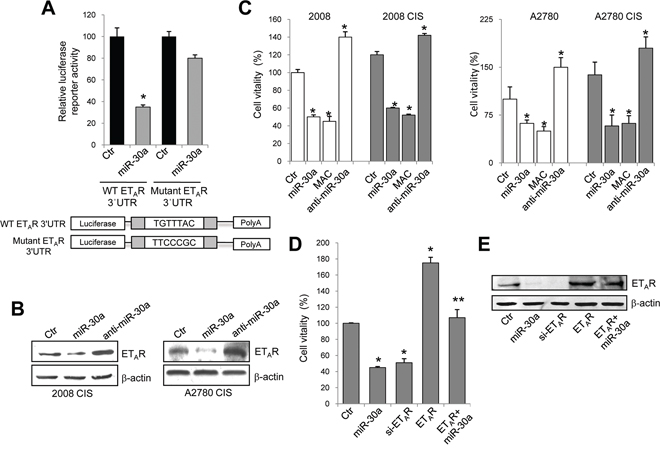
Figure 2: ETAR is a novel target of miR-30a. A. Luciferase activity in 2008 cells cotransfected with mimic-miR control (Ctr) or mimic-miR-30a and the reporter plasmid containing 3′UTR region of ETAR (WT ETAR 3′UTR) and its mutant (mutant ETAR 3′UTR). Values are the mean ± SD (n = 3; *, p < 0.05 compared to Ctr). B. Western blotting for ETAR in the lysates from 2008 CIS and A2780 CIS cells transfected for 48h with mimic-miR control, mimic-miR-30a or anti-miR-30a. β-actin is used as loading control. C. Cell viability of sensitive and resistant 2008 and A2780 cells, transfected as in B or treated with macitentan (MAC). Values are the mean ± SD (n = 3; *, p < 0.01 vs Ctr). D. Cell vitality of 2008 CIS cells, transfected with control plasmid alone (Ctr), or with mimic-miR-30a, or si-ETAR, or with ETAR expression plasmid alone or in combination with mimic-miR30a. Values are the mean ± SD (n = 3; *, p < 0.05 compared to Ctr; **, p < 0.05 compared to miR-30a). E. Lysates of cells treated as in D are analyzed by western blotting for ETAR expression. β-actin is used as loading control.
Overexpression of miR-30a sensitizes EOC cells to cisplatinum-induced apoptosis
We next evaluated whether miR-30a could inhibit the survival pathways activated by ET-1/ETAR axis to protect tumor cells from drug-induced apoptosis [4]. miR-30a-mediated ETAR inhibition was accompanied with reduced ET-1-induced phosphorylation of both p42/p44 MAPK and Akt in A2780 sensitive and cisplatinum-resistant cells (Figure 3A). The analysis of cell viability (Figure 3B and Supplementary Figure S2A) and DNA fragmentation (Figure 3C and Supplementary Figure S2B) showed that the addition of cisplatinum in cells overexpressing miR-30a, lead to an enhanced sensitivity to the chemotherapeutic drug not only in sensitive but also in resistant cells. In line with these data, by performing a TUNEL assay in A2780 CIS cells, we observed a significant increase in the number of apoptotic cells following the overexpression of miR-30a (46%) (Figure 3D). Moreover, the addition of cisplatinum in cells transfected with miR-30a sensitized chemoresistant cells to cisplatinum-induced apoptosis (82%). As shown in Figure 3E, overexpression of miR-30a in A2780 sensitive and resistant cells enhanced the cleavage of poly (ADP-ribose) polymerase (PARP) and caspase-7, compared to the control cells. Importantly, the activation of these apoptotic markers was more pronounced in the presence of cisplatinum. Together these results demonstrate that overexpression of miR-30a re-sensitizes resistant EOC cells to cisplatinum by targeting ETAR.
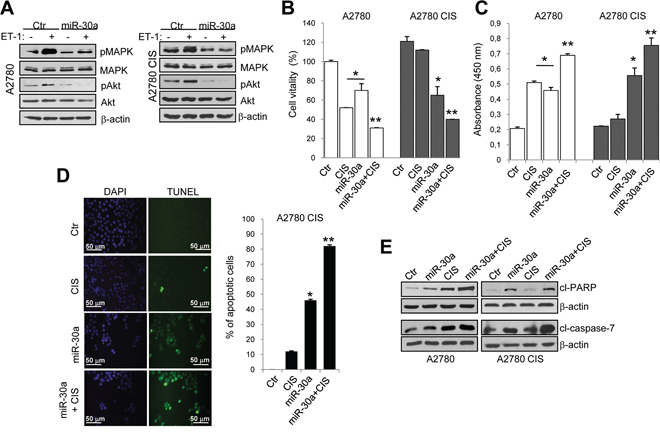
Figure 3: Ectopic expression of miR-30a sensitizes EOC cells to cisplatinum-induced apoptosis. A. Lysates from A2780 and A2780 CIS cells transfected with Ctr or mimic-miR-30a and stimulated with ET-1 as indicated, are immunoblotted with anti-pMAPK, anti-MAPK, anti-pAkt and anti-Akt Abs. β-actin is used as loading control. B. Cell vitality of sensitive and resistant A2780 cells transfected with Ctr or mimic-miR-30a and treated with cisplatinum (CIS; 1 μM) for 72 h alone or in combination. Values are the mean ± SD (n = 3; *, p < 0.05 vs Ctr; **, p < 0.05 vs cisplatinum-treated cells). C. Detection of DNA fragmentation in sensitive and resistant A2780 cells cultured for 72 h as indicated in B. Values are the mean ± SD (n = 3; *, p < 0.05 vs Ctr; **, p < 0.05 vs cisplatinum-treated cells). D. TUNEL immunofluorescence staining for apoptotic cells (green) treated for 72 h as indicated in B. Cell nuclei are counterstained with DAPI (blue). Graph represents the percentage of apoptotic cell numbers. Magnification x40. Values are the mean ± SD (n = 3; *, p < 0.05 vs Ctr; **, p < 0.05 vs cisplatinum-treated cells). E. Western blotting for cleaved PARP (cl-PARP) and caspase-7 (cl-caspase-7) in sensitive and resistant A2780 cells transfected and treated as indicated in B. β-actin is used as loading control.
Overexpression of miR-30a reverts EMT phenotype and impairs cell invasion and plasticity, and VEGF production
ET-1/ETAR overexpression, by regulating EMT and invasive behavior, endows EOC cells with an increased survival capacity and resistance to chemotherapeutic agents [4]. 2008 and A2780 resistant cells expressed enhanced mRNA levels of Snail and vimentin, associated with a concomitant decrease of E-cadherin expression (Figure 4A and Supplementary Figure S3). The overexpression of miR-30a restored E-cadherin levels, and inhibited the expression of both vimentin and Snail (Figure 4A and Supplementary Figure S3). As determined by western blotting analysis, in resistant 2008 cells miR-30a overexpression enhanced E-cadherin and reduced N-cadherin protein expression (Figure 4B), indicating that this miRNA is able to revert the EMT phenotype in EOC cells. In line with above results, transfection with miR-30a resulted in a strong inhibition of cell invasion of sensitive as well as chemoresistant 2008 cells (Figure 4C). Because in EOC cells, ET-1 through ETAR induces the release of vascular endothelial growth factor (VEGF) [31], which has been reported to have important regulatory role in drug-sensitivity [22], we evaluated whether miR-30a was also able to modulate the release of VEGF by EOC cells. Importantly, miR-30a transfection in 2008 CIS cells reduced the basal, as well as the ET-1-induced VEGF production (Figure 5A), demonstrating that regulation of the miR-30a/ETAR axis is also important in the release of the angiogenic factor VEGF. Aggressive EOC cells, expressing high levels of EMT associated markers, are capable to form vasculogenic mimicry [32]. Consistently, we found that 2008 and 2008 CIS cells plated on matrigel formed a network of interconnected tubules (Figure 5B and 5C). Notably, quantification analysis demonstrated that the length of tubes and the number of their intersections in chemoresistant EOC cells increased significantly compared to sensitive cells. Cells overexpressing miR-30a were unable to form elongated structures and formed fewer intersections (Figure 5B and 5C), indicating that ET-1/ETAR pathway is implicated in the in vitro formation of vasculogenic-like networks in chemoresistant EOC cells, and that miR-30a inhibits EMT, cell invasion and plasticity.
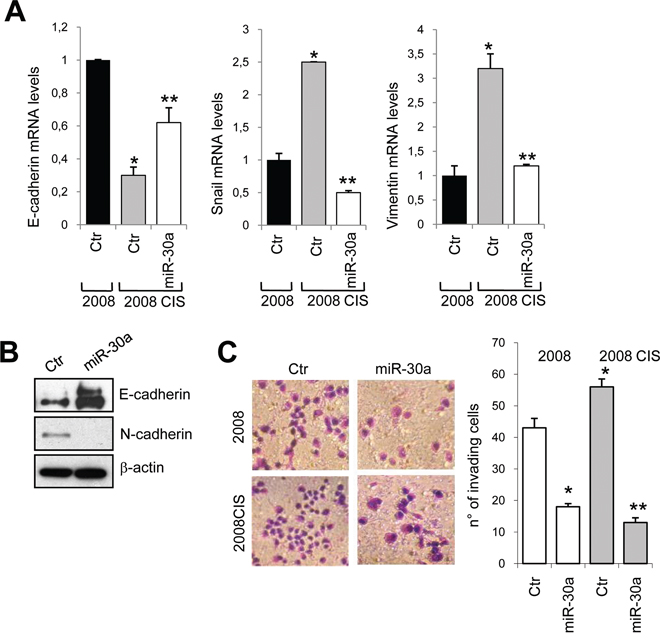
Figure 4: miR-30a inhibits EMT phenotype and cell invasion. A. E-cadherin, Snail and vimentin mRNA expression in 2008 or 2008 CIS cells transfected with Ctr or mimic-miR-30a evaluated by qPCR. Cyclophilin-A is used to normalize. Values are the mean ± SD (n = 3; *, p < 0.05 vs Ctr of sensitive cells; **, p < 0.05 vs Ctr of resistant cells). B. Lysates from resistant 2008 CIS cells transfected with Ctr or mimic-miR-30a are analyzed by western blotting for E-cadherin and N-cadherin expression. β-actin is used as loading control. C. Chemoinvasion assay of 2008 and 2008 CIS cells transfected with Ctr or mimic-miR-30a. The crystal violet-stained invasive cells are photographed (left) or counted (right). Magnification x40. Columns show the mean ± SD (n = 3; *, p < 0.001 vs Ctr of sensitive cells; **, p < 0.001 vs Ctr of resistant cells).
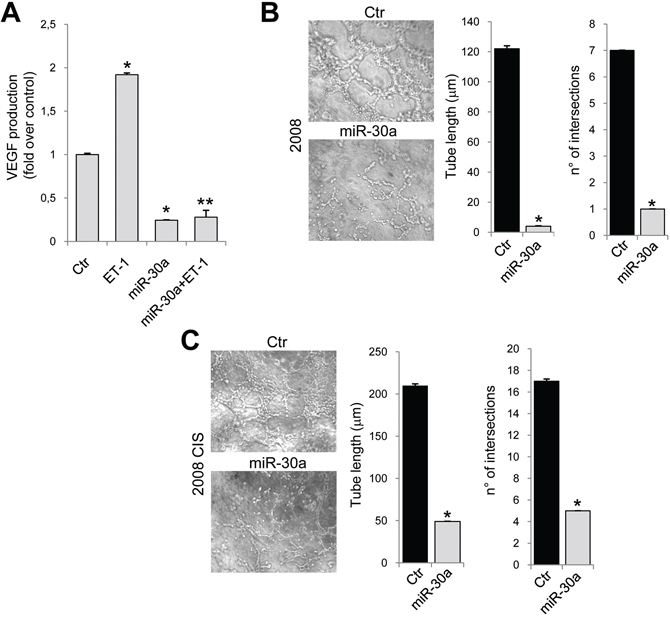
Figure 5: miR-30a inhibits the release of VEGF and vasculogenic-like tubule formation. A. VEGF release evaluated by ELISA from conditioned media of 2008 CIS cells transfected with Ctr or mimic-miR-30a in the presence or in the absence of ET-1. Values are the mean ± SD (n = 3; *, p < 0.001 vs Ctr; **, p < 0.001 vs ET-1-treated cells). Tubule-like structure formation in 2008 B. or 2008 CIS C. cells transfected with Ctr or mimic-miR-30a was photographed (left) and counted (right). Magnification x20. Graphs represent the tube length and the number of intersections. Values are the mean ± SD (n = 3; *, p < 0.05 vs Ctr).
Tumor-inhibitory effects of miR-30a in resistant EOC xenografts
Next we explored the effect of miR-30a on tumorigenesis in vivo, by using the 2008 CIS cells stably transfected with miR-30a or with vector control (Ctr) (Supplementary Figure S4). After cells were injected subcutaneously into the flank of nude mice, tumor volume was monitored every 4 days and the tumor growth curves were plotted. Tumor weight from the miR-30a overexpressing group was significantly reduced (70%) when compared with control group (p < 0.05; Figure 6A and 6B). Moreover, the control group exhibited faster growth rate. In parallel, western blotting analysis showed lower levels of ETAR and a significant reduction of MAPK and Akt activation in miR-30a overexpressing mice compared with control (Figure 6C). These findings demonstrate that overexpression of miR-30a results in decreased tumor growth in chemoresistant EOC xenografts.
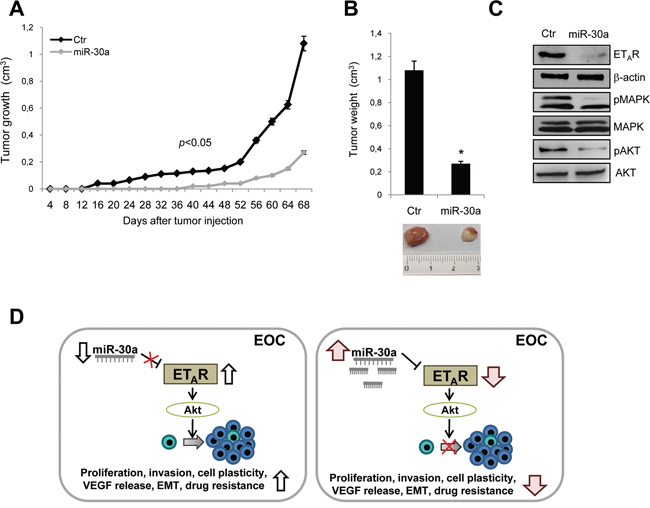
Figure 6: Tumor-inhibitory effects of miR-30a in resistant EOC xenografts. A. Growth curve of tumors from vector control (Ctr) or miR-30a-expressing resistant 2008 CIS cells s.c. injected into the mice. Data points, averages ± SD (p<0.05). B. Tumor weight of tumors grown in vector control (Ctr) and miR-30a 2008 CIS xenografts reported as the mean ± SD. *, p < 0.05 vs Ctr. Bottom, representative tumors of 2008 CIS xenografts. C. Western blotting analysis of ETAR, p-MAPK, MAPK, p-Akt and Akt expression in tumors of vector control (Ctr) and miR-30a 2008 CIS xenografts. β-actin is used as loading control. D. A schematic model describing the potential mechanism underlying the suppressive role of miR-30a in EOC. In chemoresistant EOC cells downregulated miR-30a expression has strong increased effects on ETAR expression promoting enhancement of Akt and MAPK signaling activation. Therefore miR-30a downregulation results in increased cell proliferation, invasion, plasticity, VEGF release, EMT and drug resistance by inhibiting ETAR pathway.
miR-30a correlates with poor survival in TCGA of ovarian cancer
To assess the clinical relevance of miR-30a, we analyzed its expression in TCGA of HG-SOC patient cohort. We found a strong downregulation of miR-30a when comparing tumoral (n = 567) and non tumoral (n = 8) samples in this cohort (p = 0.003; Figure 7A). To evaluate whether mir30a downregulation has a prognostic value, we subdivided patients of TCGA cohort into low or high miR-30a by using z-score higher than 1 (n = 112). As shown in Figure 7B, low miR-30a expression was documented in 57 HG-SOC patients (51%) and high miR-30a expression in 55 HG-SOC patients (49%; p = 1.8e-51). Of clinical relevance, the TCGA data set supported the association between low miR-30a expression and poor patient outcome (p = 0.01) (Figure 7C). Univariate and multivariate analysis revealed the prognostic role of low miR-30a expression that was independent from other clinical variables (tumor stage, grade and treatment response) (Figure 7C). Interestingly, ETAR gene expression inversely correlated with miR-30a expression in TCGA dataset. This anticorrelation was also present when comparing sensitive (n = 229) and resistant (n = 26) sample patients (Figure 7D). This might suggest that the overexpression of ETAR due to the loss of miR-30a activity might contribute to chemoresistance onset. In line with these observations, we observed a negative correlation between miR-30a and ETAR, evaluated by IHC in our sample set of 39 EOC (p = 0.039; Supplementary Table S2), in which patients were dichotomized into low or high miR-30a expression groups by using the median value as the cut-off. Next, we analyzed whether expression of miR-30a could correlate with survival analysis in our sample set. Recurrence/progression of disease was observed in 30 cases (76.9%), while death of disease was documented in 26 patients (66.7%); Supplementary Figure S5 shows the progression-free survival (PFS) curves according to miR-30a expression; the 3-year PFS was 38% in patients exhibiting high miR-30a expression versus 10% in cases with low miR-30a expression (p = 0.043).
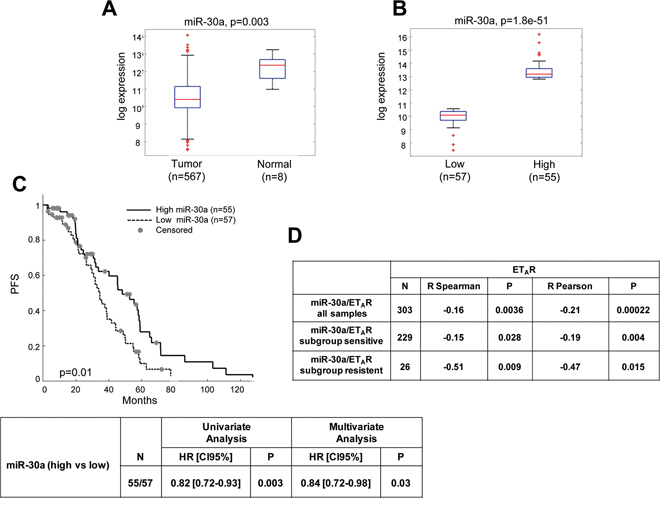
Figure 7: miR-30a correlates with poor survival in TCGA of ovarian cancer. A. Boxplot distribution of miR-30a expression in 567 tumors and 8 normal samples. B. Boxplot distribution of miR-30a expression in 112 patients subdivided into low or high miR-30a by using z-score higher than 1 (n = 55) and lower than -1 (n = 57). C. Kaplan-Meier curves of progression free survival for subgroup of 112 patients described in B. Univariate and multivariate analysis by Cox proportional hazard regression for subgroup of patients described in B, adjusted for stage, grade, and treatment response. D. ETAR gene expression resulted to be inversely correlated to miR-30a expression in the TCGA cohort of HG-SOC patients (n = 303 tumoral samples). A significative inverse correlation was found in both sensitive and resistant samples.
DISCUSSION
An in depth understanding of the molecular drivers of EOC chemoresistance is of paramount importance for improving the therapeutic regimens. We recently reported that high levels of ETAR expression are associated with poor prognosis and chemoresistance in EOC [8], however, the molecular mechanisms mediating chemoresistance remain poorly understood. In this study we provide the first evidence that downregulation of miR-30a is a feature of chemoresistant EOC cells. This decrease is likely to contribute to the chemoresistance by inhibiting chemotherapy-induced apoptosis through upregulation of ETAR and sequential activation of the MAPK and Akt pathways. Transfection of miR-30a sensitized chemoresistant cells to cisplatinum by promoting apoptosis. Importantly, we provide preclinical experiments setting the scene for a novel miR-30a function as a tumor suppressor in chemoresistant EOC by targeting ETAR signaling axis. Abundant ETAR expression and low expressed miR-30a enhance the acquisition of an aggressive chemoresistant potential of EOC cells. The investigation of the mechanism of miR-30a in EOC reveals that ETAR can be post-transcriptionally regulated by miR-30a. Several studies have demonstrated that ETAR signaling contributes to the acquisition of chemoresistance and EMT [3–5, 8, 33, 34]. In addition, ET-1 is present at high levels in tumor ascites [35] and high grade ovarian tumors overexpress ETAR [27, 36], thus making it imperative to understand the effect of ET-1/ETAR axis on ovarian tumor biology and the factors that regulate this signaling pathway. A comprehensive analysis recently highlighted that ETAR overexpression is associated with worse survival in the groups of proliferative and mesenchymal HG-SOC [27]. In this context, we recently reported that ETAR co-opts Wnt/ß-catenin signaling, which is a critical driver of EOC chemoresistance [37], to acquire a chemoresistant and mesenchymal phenotype, thus sustaining tumor progression through a self-strengthening feedback loop [8]. Here we have shown that miR-30a inhibits EOC cell invasion, plasticity, and the release of the pro-angiogenic factor VEGF, that characterize the chemoresistant behavior and EMT (Figure 6D).
Several in vitro and in vivo studies have shown that miRNA can modulate the sensitivity of EOC to chemotherapy [21–24]. The most frequently de-regulated miRNA in chemoresistant EOC are involved in EMT, as let-7 and miR-200 families [20], or in the receptor signaling pathways, as miRNA-31 that targets MET [23]. High-throughput analysis of the miRNA profile in a panel of drug-resistant EOC cells revealed that miR-30c, miR-130a and miR-335 were downregulated [21]. Moreover, a miRNA signature defines EOC chemoresistance through modulation of angiogenesis, including miR-484 as regulator of VEGF pathway [22].
miR-30 family members are located on human chromosome 6q.13 and are among the most dysregulated miRNA in metastatic cancer [38, 39]. Although recent findings suggest that miR-30 family functions as tumor suppressor in many human cancer, including gastric, anaplastic thyroid, breast, and non-small-cell lung cancers [25, 26, 40–42], its association with drug resistance has not been elucidated. Our study highlights a hitherto unappreciated role of miR-30a in EOC drug resistance and EMT and defines a unique mechanism by which it directly targets ETAR signaling axis and contributes to poor patient outcome. In parallel, inhibition of ETAR activity, by the approved small molecule macitentan recapitulated the effect of miR-30a overexpression, indicating that ETAR target therapy should represent a novel strategy to overcome chemotherapy resistance. Consistent with in vitro results, we observed that expression of miR-30a delayed tumor formation in xenograft tumors, suggesting that miR-30a is one of the tumor suppressor miRNA and that miR-30a-ETAR axis plays a crucial role in EOC cell survival. Accordingly, this miRNA was downregulated in tumor samples of the TCGA HG-SOC cohort relative to non-tumoral ovarian tissues. Of translational relevance, in this TCGA dataset low miR-30a expression correlates with poor outcome. Multivariante analysis showed that miR-30a expression had predictive values for overall survival identifying low expression of miR-30a as an independent prognostic factor. In line with these results, analysis performed in our set of EOC tissues suggest that low miR-30a is associated with poor prognosis. Moreover, the inverse correlation between miR-30a and ETAR expression further highlights the pathobiologic relevance of miR-30/ETAR axis in EOC. These findings are highly supported by our previous analysis performed on the same cohort of EOC patients demonstrating that the association between ETAR expression and poor survival is to be ascribed to the unfavorable prognostic role of ETAR in the subset of platinum-resistant cases [8]. In conclusion, our results reveal a novel mechanism in which miR-30a suppresses EOC aggressiveness and chemoresistance by inhibiting ETAR expression at the post-transcriptional level, and widening the horizon about the regulatory mechanism of ETAR. These findings also suggest that miR-30a/ETAR axis may serve as possible target to improve EOC therapeutic outcome. In light of our findings, the use of miR-30a to predict prognosis and clinical response to chemotherapy warrants future evaluation in a large cohort of patients with EOC.
MATERIALS AND METHODS
Cells and cell culture conditions
The human ovarian carcinoma cell line A2780 was obtained from European Collection of Cell Cultures (Salisbury, UK). To retain cisplatinum (CIS) and taxol (TAX) resistance, 1 μM cisplatinum (Pfizer, Italy) and 50 nM paclitaxel (Bristol Myers, Italy) were added to the culture medium A2780 CIS and A2780 TAX, respectively, every two passages. The 2008 cell line and its cisplatinum-resistant subclone 2008C13 (CIS) were kindly provided by Dr. S.B. Howell (University of San Diego, La Jolla, CA, USA). Human ovarian carcinoma cell lines, HEY and OVCA433 were kindly provided by Prof. G. Scambia (Catholic University School of Medicine, Rome, Italy). The SKOV-3 cell line was obtained from the American Type Culture Collection (VA, USA). All cells, cultured as previously described [4], were passed in our laboratory for fewer than 3 months after resuscitation and were tested routinely for cell proliferation as well as mycoplasma contamination, and they showed similar growth rate and negative mycoplasma during the experiments. Cells were serum starved by incubation in serum-free medium for 24 h. ET-1 was used at 100 nM and was purchased from Bachem (Switzerland). Macitentan, also called ACT-064992 or N-(5-[4-bromophenyl]-6-{2-[5-bromopyrimidin-2-yloxy]ethoxy}pyrimidin-4-yl)-N’-propylsulfamide, was added 30 min before ET-1 at a dose of 1 μM and was kindly provided by Actelion Pharmaceuticals, Ltd. (Switzerland).
RNA isolation and quantitative real-time polymerase chain reaction (qPCR)
Total RNA was extracted using the Trizol reagent (Life Technologies, Italy) according to the manufacturer’s protocol. First-strand complementary DNA was synthesized using SuperScript® VILO™ cDNA synthesis kit (Life Technologies). qPCR was performed using Power SYBR Green PCR Master Mix (Applied Biosystems) with a 7500 Fast Real-Time PCR System (Applied Biosystems) according to the manufacturer’s instructions. The expression levels of ETAR, E-cadherin, Snail, and vimentin were determined by normalizing to cyclophilin-A mRNA expression. The primers employed for qPCR were as follows: ETAR Fw: 5′-GGGATCACCGTCCTCAACCT-3′; ETAR Rev: 5′-CAGGAATGGCCAGGATAAAGG-3′; E-cadherin Fw: 5′-CCCACCACGTACAAGGGTC-3′; E-cadherin Rev: 5′-ATGCCATCGTTGTTCACTGGA-3′; Snail Fw: 5′-CTTCCAGCAGCC-CTACGAC-3′; Snail Rev: 5′-CGGTGGGGTTGAGGATCT-3′; vimentin Fw: 5′-TTTGAAG-AAACTCCACGAAGAGGA-3′; vimentin Rev: 5′-CCACATCGATTTGGACATGCT-3′; cyclophilin-A Fw: 5′-TTCATCTGCACTGCCAAGAC-3′; cyclophilin-A Rev: 5′-TCGAGTT-GTCCACAGT CAGC-3′. Isolation of total RNA or miRNA fraction from human cryostatic EOC sections was performed by using the Isolation of small and large RNA kit (Macherey-Nagel, Germany). For miRNA analysis RNA was reverse transcribed using the hsa-miR-30a, hsa-miR-30b, hsa-miR-30c, hsa-miR-30d, hsa-miR-30e, U6 snRNA and RNU44 TaqMan™ MicroRNA Assay systems (Applied Biosystems, NJ, USA) and pPCR was performed by using Kapa Probe Fast qPCR Master Mix (Kapa Biosystems, MA, USA). miRNA levels were normalized to U6 snRNA or RNU44 expression. Final data were obtained by using 2−ΔΔCt method [43].
Western blot analysis
Whole cell lysates were prepared using a modified RIPA buffer (50 mM Tris-HCl pH 7.4, 250 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS) containing a mixture of protease and phosphatase inhibitors. Total proteins were subjected to SDS-PAGE, and the antibodies (Abs) used for the study were as follows: anti-ETAR and anti-β-actin (C-11), purchased from Abcam (UK) and Santa Cruz Biotechnology (CA, USA), respectively; anti-p-p44/42 MAPK (Thr202/Tyr204), anti-p44/42 MAPK, anti-pAkt (S473), anti-Akt, anti-cleaved-PARP (Asp214), and anti-cleaved-caspase 7 (Asp198), were from Cell Signaling Technology (MA, USA). Anti-E-cadherin and anti-N-cadherin were purchased from BD Biosciences (CA, USA). Western blotting filters were developed using the ECL-plus detection system or, otherwise, the SuperSignal West Femto kit (Thermo Scientific, IL, USA). Western blots for ETAR were quantified by densitometric analysis using ImageJ software.
Transient and stable transfection
Cells were transiently transfected with Syn-hsa-miR-30a-5p (mimic-miR-30a), anti-hsa-miR-30a-5p (anti-miR-30a), hs-EDNRA FlexiTube siRNA (pool of 4 siRNAs; si-ETAR) or AllStars Negative Control siRNA (Qiagen, Germany) using RNAi/MAX Lipofectamine (Life Technologies) at a final concentration of 50 nM according to the manufacturer’s suggestions. Otherwise, cells were transiently transfected with 1 μM of EDNRA plasmid (Origene, MD, USA) or pGL3-Basic Control plasmid (Promega, WI, USA) using Lipofectamine 2000 (Life Technologies) according to the manufacturer’s instructions. Stable clones were generated by transfecting cells with pCMV-miR vector control (Ctr) or miR-30a expression vector (Origene) using Lipofectamine 2000 and cells were then selected with 700 μg/ml G418 sulfate (Calbiochem-Novabiochem Corporation, CA, USA). The exogenous miR-30a over the endogenous counterpart (vector alone) was monitored by qPCR (Supplementary Figure S3).
Luciferase reporter gene assay
Cells cultured in 12-well plate were transiently transfected with 50 nM of mimic-miR-30a or miR-negative control together with 0.5 μg of ETAR 3′UTR reporter plasmid (Origene) or its mutant (ETAR 3′UTR 1328-1332) by using Lipofectamine 2000 (Life Technologies) according to manufacturer’s instructions. Transfection efficiency was monitored by cotransfection with 0.1 μg of pmiR-Report Beta-gal (Life Technologies), which was used as an internal control. Reporter activity was measured after 48h using the Luciferase assay system (Promega) and normalized to β-galactosidase activity. ETAR 3′UTR reporter plasmid mutagenesis was performed using the QuikChange Site-Directed Mutagenesis Kit (Stratagene, CA, USA) and the following primers: Fw: 5′-ACAGTGACTTTTGCTGGGCA TTTTCCCAGATTCCCGCAGACTGTGAGTACAGCA G-3′; Rev: 5′-CTGCTGTACTCACAGTCTGCGGGAATCTGGGAAAATGCCCAGCAAAAGTCACT-GT-3′.
Cell viability analysis
Sensitive or cisplatinum resistant cells were seeded in twelve-well plates in triplicate. After 24 h cells were transfected with miRNA mimics (miR-negative control, mimic-miR-30a, anti-mimic-miR-30a, si-ETAR or ETAR plasmid) and/or treated with cisplatinum and macitentan. After 48-72 h, cells were harvested and counted by using trypan blue dye exclusion method.
Apoptosis assays
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay was performed using a commercial apoptosis detection kit (Roche, Italy) according to the manufacturer’s suggestions. Breafly, treated cells were washed in PBS, fixed in 4% Paraformaldehyde (PFA), and permabilized with 0.1% sodium citrate, 0.1% Triton X-100 in 1xPBS for 2 minutes at 4°C. Nuclei were stained with DAPI (Vector Laboratories, CA, USA) and slides were mounted with Vectashield mounting medium for fluorescence (Vector laboratories). Fluorescence signals were captured at 40X magnitude by using a Leica DMIRE2 microscope equipped with a Leica DFC 350FX camera and elaborated by a Leica FW4000 deconvolution software (Leica, Germany). The percentage of apoptotic cells was calculated by the ratio of TUNEL-positive nuclei and total nuclei stained with DAPI. Quantitative detection of apoptotic cells was also performed on 1x106 cells using TiterTACS colorimetric apoptosis detection kit (Trevigen, MD, USA) according to the manufacturer’s instructions.
Chemoinvasion assay
Chemoinvasion assays were carried out using modified Boyden chamber consisting of transwell membrane filter inserts with 8 μm size polycarbonate membrane precoated with polymerized collagen placed in a 24-well plate (BD Biosciences). The cells (5 × 105 cells) were stimulated with serum-free RPMI, or RPMI containing ET-1, added to the lower chamber. The cells were left to migrate for 12 h at 37°C. Cells on the upper part of the membrane were scraped and the migrated cells were fixed in 3.7% PFA and stained with 0.4% Crystal Violet in 10% ethanol. From every transwell, several images were taken under a phase-contrast with Olympus Ix70 microscope (Olympus Corporation, Japan) at 10x magnification and two broad fields were considered for quantification.
Tubule-like structure formation
The ability of tumor cells to form capillary-like structure formation has been assessed on cells cultured on Cultrex (basal membrane extract; Trevigen, MD, USA), as previously described [32]. Images were analyzed with ImageJ v.1.34s (http://rsb.info.nih.gov/ij/) for determining the length of the tubes and the number of intersections. Representative images were captured with Olympus Ix70 microscope at 20x magnification.
ELISA
Cells were plated at 4 × 105 per 60 mm dish and transfected for 48h with mimic-miR control or mimic-miR-30a in serum-free medium. Then, cells were treated or not with ET-1 for 24h. The VEGF protein levels in the cell-conditioned medium were determined in triplicate by ELISA using the Quantikine human VEGF immunoassay kit (R&D Systems, Minneapolis, MN, USA). The sensitivity of the assay is less than 5.0 pg/ml.
Xenografts in nude mice
2 × 106 viable 2008 cisplatinum-resistant control vector or miR-30a stable expressing cells were randomly injected subcutaneously into the flank of female athymic (nu+/nu+) of 5 mice for group, 4–6 week of age (Charles River Laboratories, Italy), following the guidelines for animal experimentation of the Italian Ministry of Health. Tumor volume was measured with caliper and was monitored every 4 days and the tumor growth curves were plotted. Tumor volume was calculated using the formula: π/6 larger diameter x (smaller diameter)2. At the end of experiments all the mice were euthanized, the site of tumors was noted, and the removed tumors were frozen and analyzed for western blotting.
Samples from human EOC tissues
Human tumor and normal ovarian specimens were kindly provided by Prof. G. Ferrandina from patients admitted to the Gynecologic Oncology Unit, Catholic University of Campobasso and Rome (Italy). Human EOC specimens were collected at time of initial diagnosis both in patients undergoing maximal cytoreduction and in patients deemed unresectable and therefore triaged to neo-adjuvant chemotherapy; therefore all tissues specimens were naive to chemotherapy. Tissues specimens were immediately frozen in liquid nitrogen and then stored at -80°C until the assay. Written informed consent to tumor tissue collection was obtained by each patient according to the research protocol approved by the local ethical committee.
Immunohistochemistry
Immunohistochemical analysis of ovarian cancers was performed on archival from 39 frozen tumors collected from patient population above described. Avidin-biotin indirect immunoperoxidase staining was performed by using the polyclonal antibody anti-ETAR (Abbott Laboratories, IL, USA). The avidin-biotin assays were performed using the Vectastain Elite kit (Vector Laboratories). Sections in which the incubation with the primary Ab was substituted by isotype-matched immunoglobulins were used as control. AEC was used as chromogenic substrate and Mayer’s haematoxylin as nuclear counterstain [8].
Bioinformatic analysis
Normalized miRNA and gene expression were obtained from TCGA of HG-SOC. Data from different platforms were interrogated (Agilent, RNAseqv2 and Illumina miRNA-Seq). All the expression value differences between subgroups of samples were evaluated applying unpaired t-test and permutation test. A false discovery rate procedure [44] was also included for multiple comparisons. Pearson’s correlation coefficient and Spearman coefficient were calculated between miRNA and gene target expression. Survival and progression free survival were evaluated by Kaplan-Meier method [45] and a log-rank test used to establish the statistical significance of the distance between curves [46]. The impact of clinical variables on the survival curves was investigated by a multivariate Cox proportional hazard regression model [47]. Subgroup of samples in survival analyses were individuated basing on the z-scores of the signal, and considering as the most separate subgroups of samples those with absolute z-score higher than 1.
Statistical analysis
Student’s t-test (unpaired, two-tailed) was used for comparing statistical differences. Differences were considered statistically significant when p<0.05. The time course of tumor growth was compared across the groups using two-way ANOVA, with group and time as variables. The χ2 test or Fisher’s exact test for proportion were used to analyze the distribution of clinical-pathological variables according to different subgroups. PFS was calculated from the date of surgery to the date of relapse or the date of the last follow-up. Medians and life tables were computed using the product limit estimate by Kaplan–Meier method. Statistical analysis was carried out using SPSS (SPSS II, SPSS Inc., IL, USA), SOLO (BMDP Statistical Software, CA, USA) and MATLAB (The MathWorks, MA, USA) software. The Mann-Whitney’s U test was used to analyze differences in miRNA expression levels.
ACKNOWLEDGMENTS
We gratefully acknowledge Aldo Lupo for technical assistance and Maria Vincenza Sarcone for secretarial support. This work was supported by Associazione Italiana Ricerca sul Cancro (AIRC) to A.B. (AIRC 14199) and L.R. (AIRC 12852).
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
REFERENCES
1. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012; 62: 10–29.
2. Banerjee S, Kaye SB. New strategies in the treatment of ovarian cancer: current clinical perspectives and future potential. Clin Cancer Res. 2013; 19: 961–968.
3. Rosanò L, Spinella F, Bagnato A. Endothelin 1 in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2013; 13: 637–651.
4. Rosanò L, Cianfrocca R, Spinella F, Di Castro V, Nicotra MR, Lucidi A, Ferrandina G, Natali PG, Bagnato A. Acquisition of chemoresistance and EMT phenotype is linked with activation of the endothelin A receptor pathway in ovarian carcinoma cells. Clin Cancer Res. 2011; 17: 2350–2360.
5. Rosanò L, Spinella F, Di Castro V, Nicotra MR, Dedhar S, de Herreros AG, Natali PG, Bagnato A. Endothelin-1 promotes epithelial-to-mesenchymal transition in human ovarian cancer cells. Cancer Res. 2005; 65: 11649–11657.
6. Jazaeri AA, Awtrey CS, Chandramouli GV, Awtrey CS, Chandramouli GV, Chuang YE, Khan J, Sotiriou C, Aprelikova O, Yee CJ, Zorn KK, Birrer MJ, Barrett JC, et al. Gene expression profiles associated with response to chemotherapy in epithelial ovarian cancers. Clin Cancer Res. 2005; 11: 6300–6310.
7. Helleman J, Smid M, Jansen MP, van der Burg ME, Berns EM. Pathway analysis of gene lists associated with platinum-based chemotherapy resistance in ovarian cancer: the big picture. Gynecol Oncol. 2010; 117: 170–176.
8. Rosanò L, Cianfrocca R, Tocci P, Spinella F, Di Castro V, Caprara V, Semprucci E, Ferrandina G, Natali PG, Bagnato A. Endothelin A receptor/β-arrestin signaling to the Wnt pathway renders ovarian cancer cells resistant to chemotherapy. Cancer Res. 2014; 74: 7453–7464.
9. Salani D, Taraboletti G, Rosanò L, Di Castro V, Borsotti P, Giavazzi R, Bagnato A. Endothelin-1 induces an angiogenic phenotype in cultured endothelial cells and stimulates neovascularization in vivo. Am J Pathol. 2000; 157: 1703–1711.
10. Spinella F, Garrafa E, Di Castro V, Rosanò L, Nicotra MR, Caruso A, Natali PG, Bagnato A. Endothelin-1 stimulates lymphatic endothelial cells and lymphatic vessels to grow and invade. Cancer Res. 2009; 69: 2669–2676.
11. Buckanovich RJ, Facciabene A, Kim S, Benencia F, Sasaroli D, Balint K, Katsaros D, O’Brien-Jenkins A, Gimotty PA, Coukos G. Endothelin B receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy. Nat Med. 2008; 14: 28–36.
12. Kim SJ, Kim JS, Kim SW, Brantley E, Yun SJ, He J, Maya M, Zhang F, Wu Q, Lehembre F, Regenass U, Fidler IJ. Macitentan (ACT-064992), a tissue-targeting endothelin receptor antagonist, enhances therapeutic efficacy of Paclitaxel by modulating survival pathways in orthototic models of metastatic human ovarian cancer. Neoplasia. 2011; 13: 167–179.
13. Kim SJ, Kim JS, Kim SW, Yun SJ, He J, Brantley E, Fan D, Strickner P, Lehembre F, Regenass U, Fidler IJ. Antivascular therapy for multidrug-resistant ovarian tumors by macitentan, a dual endothelin receptor antagonist. Transl Oncol. 2012; 5: 39–47.
14. Coffman L, Mooney C, Lim J, Bai S, Silva I, Gong Y, Yang K, Buckanovich RJ. Endothelin receptor-A is required for the recruitment of antitumor T cells and modulates chemotherapy induction of cancer stem cells. Cancer Biol Ther. 2013; 14: 184–192.
15. Kim SJ, Lee HJ, Kim SW, Choi HJ, He J, Wu Q, Aldape KD, Weinberg JS, Yung WK, Conrad CA, Langley RR, Lehembre F, Regenass U, et al. Macitentan, a dual endothelin receptor antagonist, in combination with temozolomide leads to glioblastoma regression and long-term survival in mice. Clin Cancer Res. 2015; e-pub ahead of print 23 June 2015; DOI: 10.1158/1078–0432.
16. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009; 136: 215–233.
17. Hata A, Lieberman J. Dysregulation of microRNA biogenesis and gene silencing in cancer. Sci Signal. 2015; 8: re3.
18. Dahiya N, Morin PJ. MicroRNAs in ovarian carcinomas. Endocr Relat Cancer. 2010; 17: F77-F89.
19. Iorio MV, Visone R, Di Leva G, Donati V, Petrocca F, Casalini P, Taccioli C, Volinia S, Liu CG, Alder H, Calin GA, Ménard S, Croce CM. MicroRNA signatures in human ovarian cancer. Cancer Res. 2007; 67: 8699–8707.
20. Llauradó M, Majem B, Altadill T, Lanau L, Castellví J, Sánchez-Iglesias JL, Cabrera S, De la Torre J, Díaz-Feijoo B, Pérez-Benavente A, Colás E, Olivan M, Doll A, et al. MicroRNAs as prognostic markers in ovarian cancer. Mol Cell Endocrinol. 2014; 390: 73–84.
21. Sorrentino A, Liu CG, Addario A, Peschle C, Scambia G, Ferlini C. Role of microRNAs in drug-resistant ovarian cancer cells. Gynecol Oncol. 2008; 111: 478–486.
22. Vecchione A, Belletti B, Lovat F, Volinia S, Chiappetta G, Giglio S, Sonego M, Cirombella R, Onesti EC, Pellegrini P, Califano D, Pignata S, Losito S, et al. A microRNA signature defines chemoresistance in ovarian cancer through modulation of angiogenesis. Proc Natl Acad Sci USA. 2013; 110: 9845–9850.
23. Mitamura T, Watari H, Wang L, Kanno H, Hassan MK, Miyazaki M, Katoh Y, Kimura T, Tanino M, Nishihara H, Tanaka S, Sakuragi N. Downregulation of miRNA-31 induces taxane resistance in ovarian cancer cells through increase of receptor tyrosine kinase MET. Oncogenesis. 2013; 2: e40.
24. Xiang Y, Ma N, Wang D, Zhang Y, Zhou J, Wu G, Zhao R, Huang H, Wang X, Qiao Y, Li F, Han D, Wang L, et al. MiR-152 and miR-185 co-contribute to ovarian cancer cells cisplatin sensitivity by targeting DNMT1 directly: a novel epigenetic therapy independent of decitabine. Oncogene. 2014; 33: 378–386.
25. Liu Z, Chen L, Zhang X, Xu X, Xing H, Zhang Y, Li W, Yu H, Zeng J, Jia J. RUNX3 regulates vimentin expression via miR-30a during epithelial-mesenchymal transition. in gastric cancer cells. J Cell Mol Med. 2014; 18: 610–623.
26. Kumarswamy R, Mudduluru G, Ceppi P, Muppala S, Kozlowski M, Niklinski J, Papotti M, Allgayer H. MicroRNA-30a inhibits epithelial-to-mesenchymal transition by targeting Snai1 and is downregulated in non-small cell lung cancer. Int J Cancer. 2012; 130: 2044–2053.
27. Bell D, Berchuck A, Birrer M, Chien J, Cramer D, Dao F, Dhir R, Di Saia P, Gabra H, Glenn P, Godwin A, Gross J, Hartmann L, et al. Integrated genomic analyses of ovarian carcinoma. Nature. 2011; 474: 609–615.
28. Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005; 120: 15–20.
29. John B, Enright AJ, Aravin A, Tuschl T, Sander C, Marks DS. Human MicroRNA targets. PLoS Biol. 2004; 2: e363.
30. Wang X. miRDB: a microRNA target prediction and functional annotation database with a wiki interface. RNA. 2008; 14: 1012–1017.
31. Spinella F, Rosanò, Di Castro V, Natali PG, Bagnato A. Endothelin-1 induces vascular endothelial growth factor by increasing hypoxia-inducible factor-1alpha in ovarian carcinoma cells. J Biol Chem. 2002; 277: 27850–27885.
32. Sood AK, Seftor EA, Fletcher MS, Gardner LM, Heidger PM, Buller RE, Seftor RE, Hendrix MJ. Molecular determinants of ovarian cancer plasticity. Am J Pathol. 2001; 158: 1279–1288.
33. Rosanò, Varmi M, Salani D, Di Castro V, Spinella F, Natali PG, Bagnato A. Endothelin-1 induces tumor proteinase activation and invasiveness of ovarian carcinoma cells. Cancer Res. 2001; 61: 8340–8346.
34. Rosanò, Spinella F, Di Castro V, Dedhar S, Nicotra MR, Natali PG, Bagnato A. Integrin-linked kinase functions as a downstream mediator of endothelin-1 to promote invasive behavior in ovarian carcinoma. Mol Cancer Ther. 2006; 5: 833–842.
35. Salani D, Di Castro V, Nicotra MR, Rosanò, Tecce R, Venuti A, Natali PG, Bagnato A. Role of endothelin-1 in neovascularization of ovarian carcinoma. Am J Pathol. 2000; 157: 1537–1547.
36. Bagnato A, Salani D, Di Castro V, Wu-Wong JR, Tecce R, Nicotra MR, Venuti A, Natali PG. Expression of endothelin 1 and endothelin A receptor in ovarian carcinoma: evidence for an autocrine role in tumor growth. Cancer Res. 1999; 59: 720–727.
37. Nagaraj AB, Joseph P, Kovalenko O, Singh S, Armstrong A, Redline R, Resnick K, Zanotti K, Waggoner S, DiFeo A. Critical role of Wnt/β-catenin signaling in driving epithelial ovarian cancer platinum resistance. Oncotarget. 2015; 6:23720–23734 doi: 10.18632/oncotarget.4690.
38. Rodriguez A, Griffiths-Jones S, Ashurst JL, Bradley A. Identification of mammalian microRNA host genes and transcription units. Genome Res. 2004; 14: 1902–1910.
39. Baffa R, Fassan M, Volinia S, O’Hara B, Liu CG, Palazzo JP, Gardiman M, Rugge M, Gomella LG, Croce CM, Rosenberg A. MicroRNA expression profiling of human metastatic cancers identifies cancer gene targets. J Pathol. 2009; 219: 214–221.
40. Boufraqech M, Nilubol N, Zhang L, Gara SK, Sadowski SM, Mehta A, He M, Davis S, Dreiling J, Copland JA, Smallridge RC, Quezado MM, Kebebew E. miR30a inhibits LOX expression and anaplastic thyroid cancer progression. Cancer Res. 2015; 75: 367–377.
41. Cheng CW, Wang HW, Chang CW, Chu HW, Chen CY, Yu JC, Chao JI, Liu HF, Ding SL, Shen CY. MicroRNA-30a inhibits cell migration and invasion by downregulating vimentin expression and is a potential prognostic marker in breast cancer. Breast Cancer Res Treat. 2012; 134: 1081–1093.
42. Qi F, He T, Jia L, Song N, Guo L, Ma X, Wang C, Xu M, Fu Y, Li L, Luo Y. The miR-30 family inhibits pulmonary vascular hypermeability in the premetastatic phase by direct targeting of Skp2 in mice. Clin Cancer Res. 2015; 21: 3071–3080.
43. Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008; 3: 1101–1108.
44. Storey JD. A direct approach to false discovery rates. J. R. Statist. Soc. B 2002; 64: 479–498.
45. Kaplan FL, Meier P. Non parametric estimation from incomplete observations. Am J Stat Assoc. 1958; 53: 457–481.
46. Mantel N. Evaluation of survival data and two new rank order statistics arising in its consideration. Cancer Chemother Rep. 1966; 50:163–170.
47. Cox DR. Regression models and life tables. J R Stat Soc. 1972; 34: 197–220.