
INTRODUCTION
Hepatocellular carcinoma (HCC) is the second most common cause of cancer-related mortality. An estimated 782,500 new liver cancer cases and 745,500 deaths occurred in 2012 worldwide [1]. Alcohol is one of the most important risk factor for the cause of HCC especially in developed countries, and the incidence of alcohol-related HCC has recently tended to increase in Japan [2]. In the recent monograph published by the International Agency for Research on Cancer (IARC), there is ‘sufficient evidence’ that demonstrate carcinogenicity of alcoholic beverage in humans (classified as Group 1), and the report concluded that the occurrence of primary liver cancer is casually related to alcohol intake [3]. Excessive alcohol consumption of >40 to 60 g/day for more than 5 years is a well-known factor that increases the risk of HCC by nearly 5-folds [4].
Several animal experiments have revealed that ethanol (EtOH) treatment promotes chemically-induced hepatocarcinogenesis [5–10], however, conflicting results regarding the mode of action of EtOH in hepatocarcinogenesis have also been reported [11]. Lifelong exposure to 3% EtOH did not induce HCC in rodents [12] whereas 10% EtOH ingestion for 18 months induced HCC development [13]. These results suggest that EtOH is an established carcinogen, which is consistent with the IARC evaluation of EtOH. However, the detailed molecular mechanisms by which EtOH contributes to hepatocarcinogenesis have not been fully elucidated to date.
Gap junctions formed by connexin hemichannnels exchange small molecules (<1kDa) between adjacent cells and play important roles in the maintenance of tissue homeostasis, the control of cell growth and differentiation [14, 15]. Hepatocytes express two connexin proteins, Cx32 which is the major protein and broadly expressed, and Cx26 which is localized at the periportal zone [16–18]. The expression of Cx32 was down-regulated in preneoplastic and HCC lesions in rats [19], and gradually decreased during progression of chronic liver disease including viral hepatitis, cirrhosis and HCC in humans [20, 21]. Reduction of Cx32 expression and gap junctional intercellular communication (GJIC) capacity also occurred in an age-dependent manner in rat [22]. We previously established transgenic rats, which carried a dominant negative mutant of Cx32 under the control of the albumin promoter (Tg) and harbored broadly disturbed membrane localization of endogenous Cx32 and Cx26 proteins and decreased GJIC as measured by gap junction assay. Tg rats are characterized as having higher susceptibility to DEN-induced hepatocarcinogenesis as compared with wild-type (Wt) littermate rats [23, 24]. These findings suggest that Cx32 has a protective role in hepatocarcinogenesis in rodents and humans. However, the relationship between Cx32 and EtOH-related hepatotoxicity including hepatocarcinogenesis has not been clarified.
In the present study, we examined the role of Cx32 in alcohol-related hepatocarcinogenesis using a Tg rat model to investigate the molecular mechanisms in preneoplastic and neoplastic lesions. Further, to assess the genes responsible for alcohol-related hepatocarcinogenesis, cDNA microarray analysis was performed.
RESULTS
EtOH does not induce liver injury in rats of both genotypes
EtOH drinking did not affect body weight in rats of both genotypes, and there was no significant difference in final body, liver and kidney weights among the groups. Average EtOH consumption was not significantly different between Tg and Wt rats (Table 1). In addition, EtOH did not affect serum levels of hepatic enzymes, lipids and albumin (Supplementary Table 1). Histological examination revealed that slight fat deposition was observed in the livers of Tg and Wt rats with EtOH treatment, however, there was no significant difference between the different genotypes (Figure 1A). EtOH induced expression of cytochrome P450 2E1 (Cyp2e1), which is one of the metabolic enzymes for EtOH, in the centrilobular region in a dose-dependent manner in both Tg and Wt rats, and protein expression of Cyp2e1 in this area was higher in Tg rats than that in Wt rats (Supplementary Figure 1A and 1B). The expression of Cx32 and Cx26, which are hepatocyte gap junction protein, were diffusely decreased in Tg rats as compared to those in Wt rats (Supplementary Figure 2A), and the expression of Cx32 and Cx26 on the cell membrane in Wt rats were not altered by EtOH intake (Figure 1B). The dye loading assay also revealed that there was no significant difference in the capacity of gap junction between each group in Wt rats (Supplementary Figure 2B and 2C).
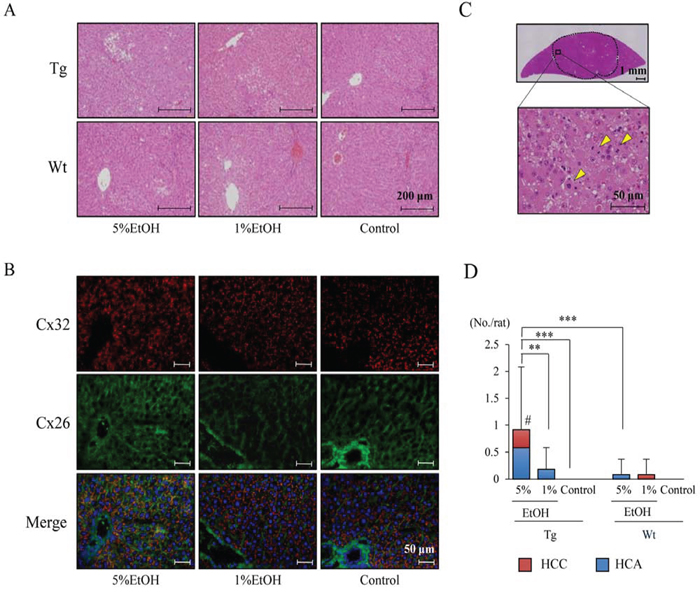
Figure 1. EtOH intake after DEN injection enhances hepatocarcinogenesis in Tg rats. A. H&E staining of non-tumor region of the liver in each group. B. Immunofluorescence staining for Cx32 (red) and Cx26 (green) in livers of Wt rats. C. H&E staining of HCC in Tg rats given 5% EtOH. Mitotic cells are indicated by arrowheads. D. Multiplicity of HCC, HCA and total tumors. Data are presented as mean ± SD, n = 11–12 per group. **, *** P < 0.01 and 0.001, respectively. # P < 0.05 indicates statistical significance in HCC as compared to Tg-Control and Wt-5%EtOH.
Table 1. Final body, liver and kidney weights, and average EtOH intake in Tg and Wt rats
No of rats |
Alcohol intake |
Liver |
Kidney |
|||||
|---|---|---|---|---|---|---|---|---|
Body(g) |
(g/kg/day) |
Absolute(g) |
Relative(%) |
Absolute(g) |
Relative(%) |
|||
Tg |
5%EtOH |
12 |
491.7 ± 18.7 |
2.51 ± 0.45 |
13.7 ± 0.9 |
2.8 ± 0.1 |
1.2 ± 0.1 |
0.25 ± 0.02 |
1%EtOH |
11 |
509.6 ± 27.1 |
0.59 ± 0.07 |
14.6 ± 1.3 |
2.9 ± 0.2 |
1.2 ± 0.1 |
0.24 ± 0.01 |
|
Control |
12 |
522.4 ± 23.7 |
- |
15.0 ± 1.1 |
2.9 ± 0.2 |
1.2 ± 0.1 |
0.24 ± 0.01 |
|
Wt |
5%EtOH |
12 |
507.2 ± 32.9 |
2.58 ± 0.30 |
14.0 ± 1.2 |
2.8 ± 0.2 |
1.2 ± 0.1 |
0.24 ± 0.02 |
1%EtOH |
12 |
535.4 ± 46.6 |
0.59 ± 0.07 |
15.1 ± 1.9 |
2.8 ± 0.2 |
1.3 ± 0.1 |
0.23 ± 0.01 |
|
Control |
12 |
542.3 ± 45.5 |
- |
15.4 ± 1.4 |
2.8 ± 0.1 |
1.3 ± 0.1 |
0.23 ± 0.01 |
|
EtOH promotes hepatocarcinogenesis in Tg rats
Induction of liver tumors was observed in both Tg and Wt rats that received EtOH. The liver tumors, which lost normal lobular architecture with solid cell growth and high mitotic counts in histology, were defined as HCC (Figure 1C). Most of the HCCs were found in the Tg-5%EtOH group. The incidence and multiplicity of HCC and/or HCA are shown in Table 2 and Figure 1D. The incidence of HCC and total tumors (HCA+HCA) was significantly increased by EtOH intake in a dose-dependent manner in Tg rats. On the other hand, induction of tumors was also observed in Wt rats that ingested EtOH, however, there was no significant correlation between tumor incidence and dosage of EtOH. Consequently, the incidence of HCC and total tumors was significantly higher in Tg rats as compared to that in Wt rats (Table 2). The multiplicity of HCC and total tumors was also significantly increased by EtOH intake only in Tg rats (Figure 1D).
Table 2. Incidence of hepatocellular carcinomas and adenomas in Tg and Wt rats
Incidence of tumors |
||||
|---|---|---|---|---|
HCC |
HCA |
HCC + HCA |
||
Tg |
5%EtOH |
3 (25%)* # |
3 (25%)* |
6 (50%)*** ## |
1%EtOH |
0 |
2 (18%) |
2 (18%) |
|
Control |
0 |
0 |
0 |
|
Wt |
5%EtOH |
0 |
1 (8%) |
1 (8%) |
1%EtOH |
1 (8%) |
0 |
1 (8%) |
|
Control |
0 |
0 |
0 |
|
Data are presented as mean ± SD. *, *** P < 0.05, 0.001 indicates statistical significance as compared to Tg-Control group. #, ## P < 0.05, 0.01 indicates statistical significance as compared to Wt-5%EtOH.
EtOH enhances cell proliferation in preneoplastic lesions in Tg rats
Total glutathione S-transferase placental form (GST-P) positive areas including GST-P positive foci (diameter > 200 μm) as preneoplastic lesions, HCA and HCC were increased in Tg rats as compared to those in Wt rats regardless of EtOH intake, and tended to increase with EtOH intake only in Tg rats. However, EtOH intake did not affect the number and area of GST-P positive foci among each group in both Tg and Wt rats (Figure 2A). According to these results, we hypothesized that EtOH promotes hepatocarcinogenesis by elevation of proliferation in hepatic preneoplastic foci. To verify the hypothesis, we measured Ki-67 labeling index in GST-P positive foci of each group. The index were significantly increased by EtOH in a dose-dependent manner in Tg rats, and correlated with tumor incidence and multiplicity. In contrast, EtOH did not affect the Ki-67 labeling indices in GST-P positive foci of Wt rats, indicating that EtOH increased proliferation activity only in Tg rats (Figure 2B and 2C). The Ki-67 labeling index was substantially higher in HCC than in GST-P positive foci, and correlation between GST-P positive area and Ki-67 labeling index was observed (Pearson correlation coefficient, r=0.39, p=0.0001, n=71) (Figure 2D). These results suggest that EtOH may contribute to the promotion phase during hepatocarcinogenesis.
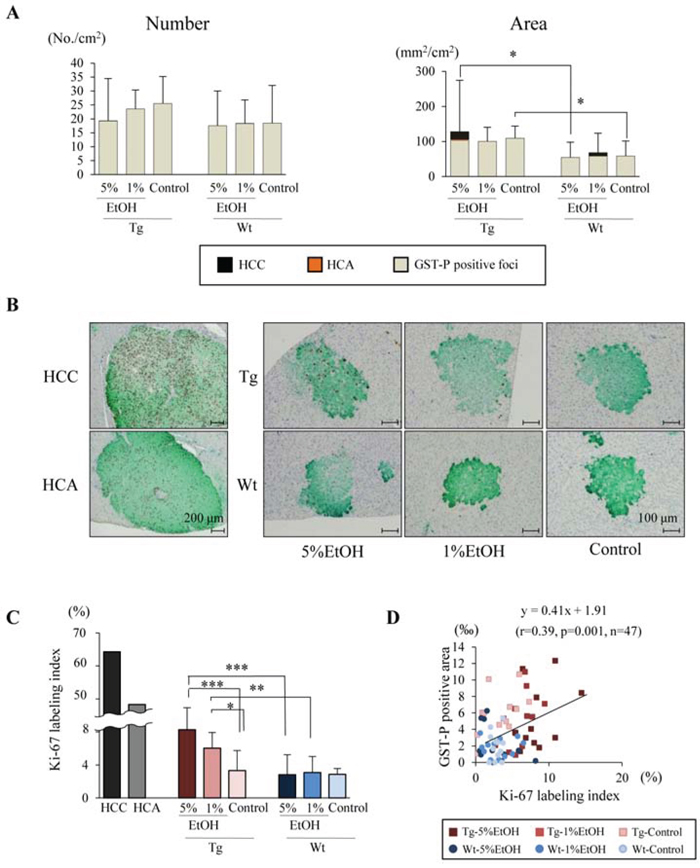
Figure 2. EtOH intake promotes cell proliferation in GST-P positive foci of Tg rats. A. Number and area of GST-P positive lesions including foci, HCA and HCC. B. Double immunohistochemical staining for Ki-67 (brown) and GST-P (green). C. Ki-67 labeling indices in HCC, HCA and GST-P positive foci of each group. D. Calculation of the regression line based on the relationship between Ki-67 labeling index and GST-P positive area by Pearson’s correlation (r = 0.39, p = 0.001, n = 47). Data are presented as mean ± SD, n = 11–12 per group. *, **, ***: P < 0.05, 0.01, and 0.001 respectively.
EtOH activates Erk signaling pathway in Tg rats
To elucidate how EtOH promotes increased cell proliferation during hepatocarcinogenesis in Tg rats, we examined the expression of proteins in the mitogen-activated protein kinase (MAPK) pathway that play crucial roles in tumorigenesis. Western blot analyses revealed that Erk1/2 were activated in the liver of Tg and Wt rats given EtOH, although c-Raf was not affected. In addition, phosphorylated Elk1 (pElk1) and cyclin D1 expression were increased by EtOH intake in Tg rats, but not in Wt rats (Figure 3A). These results suggest that EtOH promotes cell proliferation in Tg rats through the Erk signaling pathway. Next, we analyzed the localization of activated Erk in HCC, HCA, GST-P positive foci and adjacent non-tumor tissue in the liver of each group. Immunohistochemical staining revealed that phosphorylated Erk 1/2 (pErk) was found in the nucleoli of neoplastic lesions as well as GST-P positive foci in each group although nucleolar pErk was not observed in normal hepatocyte (Figure 3B). The frequency of nucleoli-localized pErk in GST-P positive foci was significantly increased by EtOH intake only in Tg rats (Figure 3C and 3D). We also examined the expression of activated protein 1 (AP-1) transcription factors including c-Fos and c-Jun, which are downstream targets of Erk signaling and known to play pro-oncogenic roles in HCC [25]. Quantitative RT-PCR revealed that there was no significant difference in the mRNA expression of these two molecules between each group (Supplementary Figure 3A). Phosphorylated c-Jun (p-c-Jun) was increased in GST-P positive foci as compared to that in surrounding non-tumor tissues, however, EtOH did not affect p-c-Jun levels in GST-P positive foci in both Tg and Wt rats (Supplementary Figure 3B). These observations suggest that other molecules rather than c-Jun and c-Fos contribute to the promotion of cell proliferation by Erk during alcohol-related hepatocarcinogenesis.
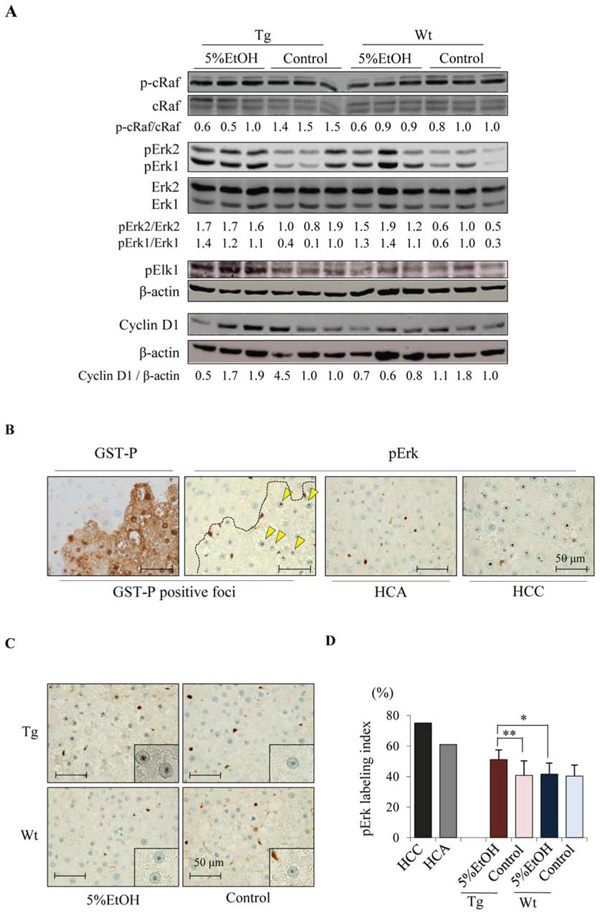
Figure 3. EtOH intake activates Erk signaling pathway in Tg rats. A. Western blotting analysis for components of the Erk pathway including p-c-Raf (Ser338), c-Raf, Erk1/2, pErk1/2 (Thr202/Tyr204), pElk1 (Ser383), and cyclin D1. B. GST-P and pErk in liver of Tg rat given EtOH. Nucleolar pErk is indicated by arrowheads. C. Immunohistochemical staining for pErk in GST-P positive foci of each group. D. The percentage of nucleolar pErk positive hepatocytes in HCC, HCA of Tg-5%EtOH group and GST-P positive foci of each group. Date are presented as mean ± SD, n = 11–12 per group. *,**: P < 0.05 and 0.01 respectively.
EtOH and dysfunction of Cx32 down-regulate Dusp1 expression in preneoplastic foci
To further clarify the mechanisms of alcohol-related hepatocarcinogenesis, cDNA microarray analysis was performed using liver tissues of 5%EtOH and control groups of each genotype. Genes that were up-regulated or down-regulated more than 2-folds by EtOH treatment in Tg rats are listed in Table 3. Among these genes, we focused on Dusp1 and Dusp4, whose expression levels were decreased by EtOH in Tg rats, because some members of Dusp family are known to be inhibitors of MAPK signaling including the Erk pathway. Quantitative RT-PCR confirmed that Dusp1 mRNA expression was down-regulated by EtOH treatment in both genotypes, and immunoblot analyses demonstrated that Dusp1 protein expression tended to be decreased by EtOH in both genotypes, especially in Tg rats (Figure 4A and 4B). On the other hand, the mRNA expression of Dusp4, another member of the Dusp family, was too low and no significant difference was detected among the groups by quantitative RT-PCR (data not shown). Therefore, further analyses focused only on Dusp1 expression especially in GST-P positive foci, since EtOH increased nucleoli-located pErk in GST-P positive foci of Tg rats compared with that of Wt rats. The mRNA and protein levels of Dusp1 in GST-P positive foci were significantly decreased in Tg rats treated with EtOH as compared to those in treatment-matched Wt rats. EtOH intake also significantly decreased the Dusp1 protein expression as compared to the no treatment group of Tg rats (Figure 4C and 4D). To elucidate the relationship between localization of pErk and Dusp1 in HCC and GST-P positive foci, we performed triple immunofluorescence staining for pErk, Dusp1 and GST-P. Expression of Dusp1 was lower in HCC than in normal hepatocytes (Figure 5A). In addition, lower Dusp1 expression was observed in GST-P positive foci than that in normal hepatocytes in the Tg-5%EtOH group. Moreover, the Dusp1 expression level was inversely correlated with pErk expression in nucleoli in both HCC and GST-P positive foci of Tg rats that received EtOH (Figure 5B). These results suggest that EtOH treatment and Cx32 dysfunction decreased Dusp1 expression which led to Erk activation in GST-P positive foci. Therefore, the interaction of Erk and Dusp1 may be involved in the promotion of EtOH-related hepatocarcinogenesis in Tg rats.
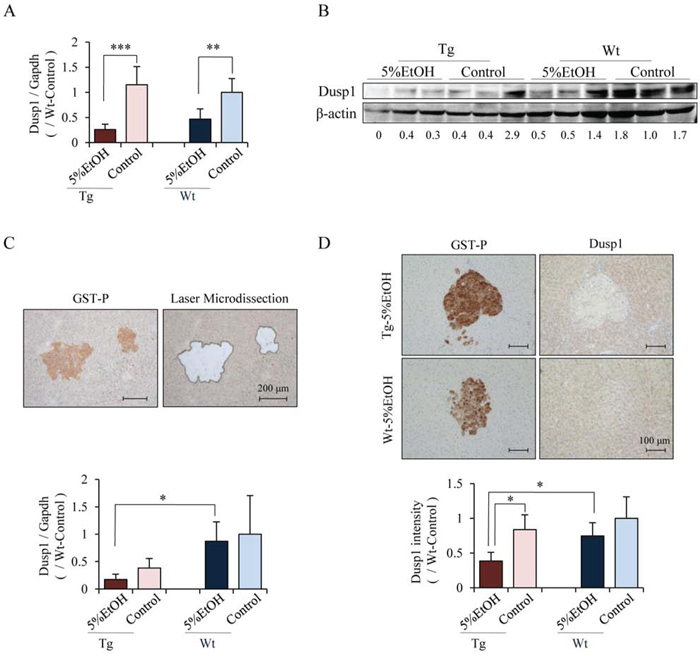
Figure 4. The mRNA and protein levels of Dusp1 were decreased by EtOH in Tg rats especially in GST-P positive foci. Levels of Dusp1 mRNA A. and protein B. in whole liver tissues measured by quantitative RT-PCR and western blotting, respectively. Data of quantitative RT-PCR are presented as mean ± SD, n = 4 per groups. **, ***: P < 0.01 and 0.001 respectively. Representative image of laser microdissected tissue (upper) and Dusp1 mRNA level in GST-P positive foci as determined by quantitative RT-PCR (lower) C. Immunohistochemical staining for Dusp1 in GST-P positive foci of Tg and Wt rats that received 5%EtOH (upper) and the intensity score of Dusp1 in GST-P positive foci as compared to Wt-control rats (lower) D. Data are presented as mean ± SD, n = 5 per group, 10 foci/rat. *: P < 0.05.
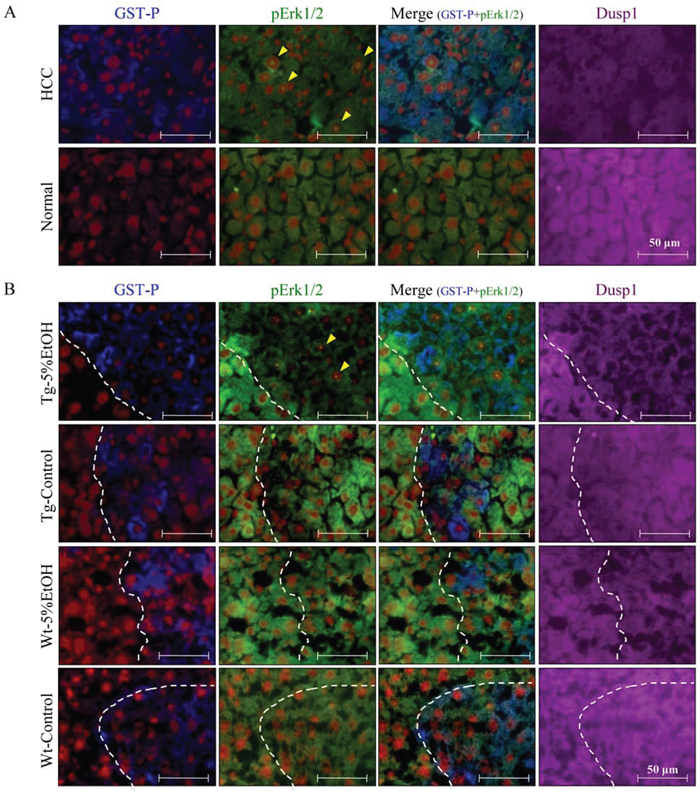
Figure 5. Localization of GST-P (blue), pErk (green), Dusp1 (purple), and nuclei (red) by triple immunofluorescence staining in HCC and normal tissue of the Tg-5%EtOH group A. and GST-P positive foci of each group B. Nucleolar pErk positive hepatocytes are indicated by arrowheads.
Table 3. Up-regulated and down-regulated genes in the liver of EtOH-treated Tg rats
Reference ID |
Symbol |
Description |
Ratio |
|---|---|---|---|
Up-regulated genes |
(Tg-5%EtOH/Tg-Control) |
||
NM_019156.2 |
Vtn |
Vitronectin |
13.58 |
Down-regulated genes |
|||
NM_024385.1 |
Hhex |
Hematopoietically expressed homeobox |
0.23 |
DISCUSSION
In the present study, we clearly demonstrated the enhancing effects of EtOH on DEN-induced hepatocarcinogenesis via Cx32 dysfunction. These results are considered to reflect the strong association of alcohol intake with HCC development in patients with chronic hepatitis since Cx32 is down-regulated in hepatitis, cirrhosis and aging. EtOH intake in rats of the 5% EtOH group was approximately 2.5 g/kg/day which is equivalent to 150 g/day for a human who is 70 kg in weight. However, since the activity of the alcohol metabolizing enzyme, alcohol dehydrogenase, in rat liver has been reported to be 4-5 times higher than that in human liver [26], the equivalent ingested amount would be 30-37.5 g/day/70 kg person after adjusting for the difference in metabolizing enzyme activity. This is speculated to be the marginal level of EtOH intake for elevating the risk of alcohol-related HCC development in human. In fact, only a small number of tumors were observed in Wt rats in contrast to Tg rats that were susceptible to hepatocarcinogenesis.
One effect of EtOH in the whole livers of both genotypes was up-regulation of pErk protein expression, while pElk1 and cyclin D1 were increased by EtOH treatment only in Tg rats. Since a previous paper suggests that alcohol consumption increases the percentage of pErk-positive sinusoidal lining cells in rats [13], the labeling index in sinusoidal lining cells was measured in this study. The pErk-positive sinusoidal lining cells were significantly increased by EtOH treatment in both Tg and Wt rats, and there was no significant difference between genotypes (Supplementary Figure 4A), which was consistent with the results by western blot analysis. The Ki-67 labeling index in sinusoidal lining cells was also analyzed to investigate whether pErk induction affects proliferative activity in these cells, and we found that it was not altered among the groups (Supplementary Figure 4B). Concerning to the expression of pErk in GST-P positive foci, EtOH increased the percentage of nucleoli-localized pErk and Ki-67 positivity in hepatocytes of GST-P positive foci in Tg rats, and there effects were not observed in Wt rats. Elk1 is known to be activated by phosphorylated Erk, p38 Mapk or Sapk/Jnk [27], and only Erk was activated among these MAPK (Supplemantary Figure 5) in our study. Several reports have described the relationship of Cx32 with cell proliferation and cyclin D1 expression: (a) proteome analysis indicated that Cx32 indirectly interacted with Erk [28], (b) radiation-induced liver tumors in Cx32 knock-out mice had increased pErk staining compared with that in Wt mice [29], and (c) overexpression of Cx32 decreased cyclin D1 expression and caused G1 arrest in liver cancer cells [30]. These reports and our data in the present study suggest that nucleolar pErk may induce expression of pElk1 and cyclin D1, promote cell proliferation in GST-P positive foci and increase incidence of HCC only in Tg but not Wt rats although the detailed mechanisms and intermediary factors are not known.
Therefore, microarray analysis was performed to elucidate these factors, and we focused on Dusp1 as a candidate regulator of pErk via Cx32 dysfunction in EtOH-related hepatocarcinogenesis, because Dusp1 is known to inhibit MAPKs including Erk. Dusp1 is known to inhibit MAPKs including Erk, and its down-regulation was reported to be a valuable factor for poor prognosis in HCC patients [31]. It has been demonstrated that Dusp1 expression in the liver is associated with the difference in susceptibility of different strains of rats to hepatocarcinogenesis [32]. Recently, Lawan et al. demonstrated an increase in hepatic lipogenesis and inhibition of CREB-mediated glucogenesis in liver-specific Dusp1 knock-out mice, which indicate that Dusp1 also contributes to steatosis including ALD and nonalcoholic fatty liver disease (NAFLD) [33]. Therefore, Dusp1 may be a potential target of prevention of the development of EtOH-related HCC as well as ALD and NAFLD.
It has been reported that the expression levels of both Dusp1 and Cx32 are positively regulated by S-adenosylmethionine (SAM) in hepatocytes [34, 35]. Methionine adenosyltransferase 1 alpha (MAT1A), which encodes the SAM synthesizing enzyme, was demonstrated to be reduced in the patients with ALD, and MAT1A knock-out mice showed increased susceptibility to hepatocarcinogenesis leading to development of HCC [36]. SAM exerted preventive effects in chemically-induced rat hepatocarcinogenesis and in an orthotopic-inoculated HCC rat model [37, 38]. These findings suggest that the chemopreventive and therapeutic effects of SAM are through Dusp1-Erk and Cx32, and it is likely that the Cx32-Dusp1-Erk signaling pathway was deeply involved in the promotion of hepatocarcinogenesis by EtOH in our Tg rat model.
With regard to the molecular mechanisms by which EtOH contributes to hepatocarcinogenesis in vivo models, Mercer et al. demonstrated that the Wnt/β-catenin signal pathway was activated in EtOH-related DEN-induced hepatocarcinogenesis in mice [9]. They reported that multiplicities of both eosinophilic liver cell foci and hepatocellular adenoma were significantly increased by EtOH treatment whereas HCC was not increased. These observations suggested that the Wnt/β-catenin signal pathway was not associated with malignant transformation in EtOH-related hepatocarcinogenesis. In contrast to their findings, it is likely that the Cx32-Dusp1-Erk signaling axis is profoundly involved in the entire carcinogenic process throughout all of the progression steps; namely, Dusp-Erk signaling is activated even in preneoplastic lesion in our present study.
In conclusion, Cx32-Dusp1-Erk interaction may contribute to the tumor promoting activity of EtOH and subsequent development of hepatocarcinogenesis. The data in the present study provide evidence that the Cx32-Dusp1-Erk signaling pathway is a potential target for chemoprevention and alternative therapy in EtOH-related hepatocarcinogenesis. In addition, the Cx32 dominant negative transgenic rats used in this study may be a useful in vivo model to study alcohol-related hepatocarcinogenesis because HCC can be induced by EtOH in a short period of time.
MATERIALS AND METHODS
Animal experiment
The establishment, production and screening of Tg rats carrying the mutated Cx32 gene were as previously described in detail [23]. Male Tg rats were produced by mating heterozygous males with Wt Sprague-Dawley females (Japan SLC, Shizuoka, Japan). Rats were maintained in plastic cages on hardwood chips in an air-conditioned specific pathogen-free animal room at 22 ± 2°C and 50% humidity with 12h/12h light-dark cycle. All animal experiments were performed under protocols approved by the Institutional Animal Care and use Committee of Nagoya City School of Medical Sciences. All heterozygous male Tg and Wt littermate rats were administrated a single intraperitoneal injection of 200 mg/kg DEN (Tokyo Kasei Kogyo Co, Ltd., Tokyo, Japan) dissolved in saline at 9 weeks of age. Thereafter they received 1 % or 5 % EtOH (Wako Pure Chemical Industries, Ltd., Osaka, Japan) or water ad libitum for 16 weeks: Tg-5%EtOH, Tg rats drinking 5% EtOH (n=12); Tg-1%EtOH, Tg rats drinking 1% EtOH (n=12); Tg-Control, Tg rats drinking water (n=12); Wt-5%EtOH, Wt rats drinking 5% EtOH (n=12); Wt-1%EtOH, Wt rats drinking 1% EtOH (n=12); and Wt-Control, Wt rats drinking water (n=12). All rats were sacrificed at the sixteenth week following the initiation of treatment.
Biochemical analysis
Blood was collected by puncture of the abdominal aorta in anesthetized rats and separated serum by centrifugation (3,000 rpm) was transferred into tubes. Plasma albumin, alkaline phosphatase, aspartate aminotransferase, alanine aminotransferase, gamma-glutamyl transpeptidase, lactate dehydrogenase, and total cholesterol were determined by The Tohkai Cytopathology Institute: Cancer Research and Prevention (Gifu, Japan).
Histological analysis of the livers
The livers were immediately excised, weighed and cut into slices 3 to 4 mm thick. They were then fixed in 10% buffered formalin, embedded in paraffin and routinely processed for histological evaluation (3 μm thick). Sections were stained with hematoxylin and eosin (H&E), and were also used for immunohistochemistry using anti-Cyp2e1 (Enzo Biochem Inc., New York, NY), anti-GST-P (Medical & Biological Laboratories, Nagoya, Japan), anti-pErk (Thr202/Tyr204), anti-p-c-Jun (Ser73) (Cell Signaling Technology, Danvers, MA), anti-Ki-67 (Abcam plc, Cambridge, UK) antibodies and anti-Dusp1 (Santa Cruz Biotechnology Inc., Santa Cruz, CA) antibodies. Double immunostaining for Ki-67 and GST-P or Ki-67 and p-c-Jun were performed using a previous method with modifications [39]. The section was incubated with a primary antibody against Ki-67 and visualized with DAB, and then the primary antibody was inactivated by heat treatment (95°c) for 10 min in 10 mM citrate buffer (pH 6.0). Thereafter the section was incubated with a second antibody against GST-P or p-c-Jun and visualized with the Vina Green Chromogen Kit (Biocare Medical, LLC. Concord, CA). The average number and area of GST-P positive foci whose size was more than 200 μm in diameter, and the total area of the liver section were measured with an image analyzer (Keyence, Osaka, Japan) (n=11-12). The proportion of hepatocytes positive for Ki-67, pErk, and p-c-Jun in GST-P positive foci (n=11-12), and the population of sinusoid lining cells positive for Ki-67 and pErk (n=5) were measured by counting at least 1,000 cells. The intensity score of Dusp1 immunostainings was evaluated using an image analyzer and associated software (Keyence), and was represented as a value relative to the Wt-control group (n=5 and 10 foci/rat, respectively).
Immunofluorescence staining for Cx32 and Cx26
The detailed methods for fluorescence immunohistochemistry employed in this study have been described previously [22]. Frozen sections were cut to 6 μm thickness and fixed in cold acetone and 10% buffered formalin. A polyclonal rabbit antibody against Cx32 (Thermo Fischer Scientific Inc., Waltham, MA) was used with biotin-conjugated anti-rabbit IgG and TRITC-labeled streptavidin (Thermo Fischer Scientific Inc.) to visualize the endogenous proteins using an image analyzer (Keyence). A monoclonal mouse antibody against Cx26 (Thermo Fischer Scientific Inc.) was used with biotin-conjugated anti-mouse IgG and FITC-labeled streptavidin (Thermo Fischer Scientific Inc.).
Triple immunofluorescence immunohistochemistry for pErk, Dusp1 and GST-P
Immunofluorescence staining for pErk, Dusp1 and GST-P was performed according to a modified protocol [39]. A monoclonal rabbit antibody for pErk (Cell Signaling Technology) was used with biotin-conjugated anti-rabbit IgG and FITC-labeled streptavidin (Thermo Fischer Scientific Inc.) in an immunoreaction enhancer solution, Can Get Signal (TOYOBO, Osaka, Japan), and then incubated at 95°c for 10 min in 10 mM citrate buffer (pH 6.0). Thereafter a polyclonal rabbit antibody against GST-P was used with biotin-conjugated anti-rabbit IgG and Cy5-labeled streptavidin (Thermo Fischer Scientific Inc.), and finally the section was incubated with an Alexa Fluor 350 conjugated rabbit anti-DUSP1 polyclonal antibody (Bioss Inc., Boston, MA). The nuclei were stained with Propidium Iodide (Vector Laboratories, Inc., Burlingame, CA).
Gap junction assay
This procedure was performed according to the method described in an earlier report [40]. Briefly, liver samples were obtained from Wt rats with each treatment (4 rats per group) and treated with Lucifer yellow (Sigma-Aldrich Corp., St. Louis, MO), a stain that can pass through the gap junction channel, and rhodamine-dextran (Sigma-Aldrich Corp.), which does not cross through the channel, to measure gap junction capability. Liver slices were cut to 5 mm-thick and 3 incisions of 1 mm depth were made, followed by the addition of a mixture of fluorescent dyes containing 0.05% Lucifer yellow and 0.05% rhodamine-dextran in PBS into the incisions. After 3 minutes, the slices were washed 3 times with PBS and frozen. Thereafter 7 μm thick frozen sections were made and spread of the dye was measured using an image analyzer (Keyence).
Western blotting
Liver tissues were homogenized with T-PER Tissue Protein Extraction Reagent (Thermo Fischer Scientific Inc.) and a protease inhibitor cocktail (Roche Diagnostic, Mannheim, Germany) on ice. Protein concentrations were determined by the Bradford method using a protein assay kit (Bio-Rad laboratories, Hercules, CA). Samples of 50 μg were mixed with SDS sample buffer, heated at 100°C for 10 min and then subjected to SDS-PAGE. The proteins were separated in 12% acrylamide gels and then transferred onto Hybond-ECL membranes (GE Healthcare UK Ltd., Buckinghamshire, UK). The antibodies used were against phosphorylated c-Raf (Ser338) (p-c-Raf), Erk 1/2, pErk, p38Mapk, phosphorylated p38Mapk (Thr180/Tyr182) (p-p38Mapk), pElk1 (Ser383), Sek1/Mkk4, phosphorylated Sek1/Mkk4 (Ser80) (pSek1/Mkk4), Sapk/Jnk, phosphorylated Sapk/Jnk (Thr183/Tyr185) (pSapk/Jnk), cyclin D1, Dusp1 (Cell Signaling Technology), c-Raf (BD bioscience, Franklin Lakes, NJ), and Cyp2e1 (Enzo Biochem Inc.). Equal protein loading was ascertained by western blotting with a β-actin antibody (Sigma-Aldrich corp.).
Quantitative RT-PCR
Total RNA was extracted using Isogen reagent (Nippon Gene Co. Ltd., Tokyo, Japan). One microgram of RNA was converted to cDNA with avian myeloblastosis virus reverse transcriptase (Takara, Otsu, Japan) in 20 μl reaction mixture. Aliquots of 2 μl of cDNA samples were subjected to quantitative PCR in a total volume of 25 μl using SYBR Premix ExTaqII (Takara) in a light cycler apparatus (Roche Diagnostic). Primers used for amplification of each mRNA were as follows:
Dusp1 forward, 5′-TGTAGCACCCCTCTCTACGA-3′; Dusp1 reverse, 5′-GACAATTGGCCGAGACGTTG-3′; c-Jun forward, 5′-TCATCCAGTCCAGCAATGGG-3′; c-Jun reverse, 5′-TATGCAGTTCAGCTAGGGCG-3′; c-Fos forward, 5′-ACCACGACCATGATGTTCTC-3′; c-Fos reverse, 5′-GACAGATCTGCACAAAAGTC-3′.
Microarray analysis
Gene expression analysis was performed using a Rat Oligo chip 20k (Toray Industries, Tokyo, Japan) according to the manufacturer’s instructions. Hepatic RNA expression of the 4 experimental groups (Tg-5%EtOH, Tg-Control, Wt-5%EtOH and Wt-Control) was compared.
Laser microdissection
The detailed methods employed in this study have been described previously [41]. Seven-micron thick frozen sections (6-8 sections) were mounted onto slides with films (Carl Zeiss, Oberkochen, Germany) and fixed in acetone for 10 min. The sections were treated with a polyclonal rabbit antibody against GST-P with RNaseOut (Thermo Fischer Scientific Inc.) for 25 min subsequently exposed to secondary antibody using the DAKO ENVISION™ System (DAKO Co., Tokyo, Japan) with RNaseOut and 2.5 μM EDTA for 25 min at 4°C and visualized with DAB after incubation for 5 min at room temperature. After immunostaining of the frozen tissue, laser microdissection was performed using the PALM MicroBeam system (Carl Zeiss) and 30 – 40 GST-P positive foci were collected into tubes. Thereafter, we quantified the mRNA level by quantitative RT-PCR as described above.
Statistical analysis
Differences in the quantitative data, expressed as mean ± SD, between groups were compared by one-way ANOVA and Dunnett’s post-hoc test using Graph Pad Prism 5 (GraphPad Software, Inc., La Jolla, CA).
ACKNOWLEDGMENTS
We thank Dr. Naoko Kaneko and Dr. Kazunobu Sawamoto (Depertment of Developmental and Regenerative Biology, Nagoya City University) for their helpful advice on laser microdissection technique.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
GRANT SUPPORT
This work was supported by Grant-in-Aid for Research in Nagoya City University to A.N.
REFERENCES
1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J and Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015; 65:87–108.
2. Nishikawa H and Osaki Y. Non-B, non-C hepatocellular carcinoma (Review). Int J Oncol. 2013; 43:1333–1342.
3. IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Personal habits and indoor combustions. Volume 100 E. A review of human carcinogens. In: Consumption of Alcoholic beverage. Lyon: IARC Monogr Eval Carcinog Risks Hum; vol 100E; 2012. pp373–499.
4. Donato F, Tagger A, Gelatti U, Parrinello G, Boffetta P, Albertini A, Decarli A, Trevisi P, Ribero ML, Martelli C, Porru S and Nardi G. Alcohol and hepatocellular carcinoma: the effect of lifetime intake and hepatitis virus infections in men and women. Am J Epidemiol. 2002; 155:323–331.
5. Tsutsumi M, Matsuda Y and Takada A. Role of ethanol-inducible cytochrome P-450 2E1 in the development of hepatocellular carcinoma by the chemical carcinogen, N-nitrosodimethylamine. Hepatology. 1993; 18:1483–1489.
6. Yamagiwa K, Higashi S and Mizumoto R. Effect of alcohol ingestion on carcinogenesis by synthetic estrogen and progestin in the rat liver. Jpn J Cancer Res. 1991; 82:771–778.
7. Kushida M, Wanibuchi H, Morimura K, Kinoshita A, Kang JS, Puatanachokchai R, Wei M, Funae Y and Fukushima S. Dose-dependence of promotion of 2-amino-3,8-dimethylimidazo[4,5-f]quinoxaline-induced rat hepatocarcinogenesis by ethanol: evidence for a threshold. Cancer Sci. 2005; 96:747–757.
8. Radike MJ, Stemmer KL and Bingham E. Effect of ethanol on vinyl chloride carcinogenesis. Environ Health Perspect. 1981; 41:59–62.
9. Mercer KE, Hennings L, Sharma N, Lai K, Cleves MA, Wynne RA, Badger TM and Ronis MJ. Alcohol consumption promotes diethylnitrosamine-induced hepatocarcinogenesis in male mice through activation of the Wnt/beta-catenin signaling pathway. Cancer Prev Res (Phila). 2014; 7:675–685.
10. Brandon-Warner E, Walling TL, Schrum LW and McKillop IH. Chronic ethanol feeding accelerates hepatocellular carcinoma progression in a sex-dependent manner in a mouse model of hepatocarcinogenesis. Alcohol Clin Exp Res. 2012; 36:641–653.
11. Griciute L, Castegnaro M and Bereziat JC. Influence of ethyl alcohol on carcinogenesis induced by volatile N-nitrosamines detected in alcoholic beverages. In; Relevance of N-nitroso Compounds in Human Cancer; Exposure and Mechanisms. Bartsch H, O’Neill IE, Schulte-Herman R editors. Lyon: IARC Sci Publ 1987; 84; pp264–5.
12. Holmberg B and Ekstrom T. The effects of long-term oral administration of ethanol on Sprague-Dawley rats—a condensed report. Toxicology. 1995; 96:133–145.
13. Yip-Schneider MT DC, McKillop IH, Wentz SC, Brandon-Warner E, Matos JM, Sandrasegaran K, Saxena R, Hennig ME, Wu H, Waters JA, Klein PJ, Froehlich JC, Schmidt CM. Alcohol induces liver neoplasia in a novel alcohol-preferring rat model. Alcohol Clin Exp Res 2011; Dec;35:2216–25.
14. Sosinsky GE and Nicholson BJ. Structural organization of gap junction channels. Biochim Biophys Acta. 2005; 1711:99–125.
15. Yamasaki H and Naus CC. Role of connexin genes in growth control. Carcinogenesis. 1996; 17:1199–1213.
16. Bruzzone R, White TW and Paul DL. Connections with connexins: the molecular basis of direct intercellular signaling. Eur J Biochem. 1996; 238:1–27.
17. Paul DL. Molecular cloning of cDNA for rat liver gap junction protein. J Cell Biol. 1986; 103:123–134.
18. Sagawa H, Naiki-Ito A, Kato H, Naiki T, Yamashita Y, Suzuki S, Sato S, Shiomi K, Kato A, Kuno T, Matsuo Y, Kimura M, et al. Connexin 32 and luteolin play protective roles in non-alcoholic steatohepatitis development and its related hepatocarcinogenesis in rats. Carcinogenesis. 2015; 36:1539–1549.
19. Krutovskikh VA, Oyamada M and Yamasaki H. Sequential changes of gap-junctional intercellular communications during multistage rat liver carcinogenesis: direct measurement of communication in vivo. Carcinogenesis. 1991; 12:1701–1706.
20. Nakashima Y, Ono T, Yamanoi A, El-Assal ON, Kohno H and Nagasue N. Expression of gap junction protein connexin32 in chronic hepatitis, liver cirrhosis, and hepatocellular carcinoma. J Gastroenterol. 2004; 39:763–768.
21. Yamaoka K, Nouchi T, Kohashi T, Marumo F and Sato C. Expression of gap junction protein connexin 32 in chronic liver diseases. Liver. 2000; 20:104–107.
22. Naiki-Ito A, Kato H, Asamoto M, Naiki T and Shirai T. Age-dependent carcinogenic susceptibility in rat liver is related to potential of gap junctional intercellular communication. Toxicol Pathol. 2012; 40:715–721.
23. Asamoto M, Hokaiwado N, Murasaki T and Shirai T. Connexin 32 dominant-negative mutant transgenic rats are resistant to hepatic damage by chemicals. Hepatology. 2004; 40:205–210.
24. Hokaiwado N, Asamoto M, Futakuchi M, Ogawa K, Takahashi S and Shirai T. Both early and late stages of hepatocarcinogenesis are enhanced in Cx32 dominant negative mutant transgenic rats with disrupted gap junctional intercellular communication. J Membr Biol. 2007; 218:101–106.
25. Eferl R and Wagner EF. AP-1: a double-edged sword in tumorigenesis. Nat Rev Cancer. 2003; 3:859–868.
26. Sinclair J, Lambrecht L and Smith EL. Hepatic alcohol dehydrogenase activity in chick hepatocytes towards the major alcohols present in commercial alcoholic beverages: comparison with activities in rat and human liver. Comp Biochem Physiol B. 1990; 96:677–682.
27. Besnard A, Galan-Rodriguez B, Vanhoutte P and Caboche J. Elk-1 a transcription factor with multiple facets in the brain. Front Neurosci. 2011; 5:35.
28. Fowler SL, Akins M, Zhou H, Figeys D and Bennett SA. The liver connexin32 interactome is a novel plasma membrane-mitochondrial signaling nexus. J Proteome Res. 2013; 12:2597–2610.
29. King TJ, Gurley KE, Prunty J, Shin JL, Kemp CJ and Lampe PD. Deficiency in the gap junction protein connexin32 alters p27Kip1 tumor suppression and MAPK activation in a tissue-specific manner. Oncogene. 2005; 24:1718–1726.
30. Koffler L, Roshong S, Kyu Park I, Cesen-Cummings K, Thompson DC, Dwyer-Nield LD, Rice P, Mamay C, Malkinson AM and Ruch RJ. Growth inhibition in G(1) and altered expression of cyclin D1 and p27(kip-1) after forced connexin expression in lung and liver carcinoma cells. J Cell Biochem. 2000; 79:347–354.
31. Tsujita E, Taketomi A, Gion T, Kuroda Y, Endo K, Watanabe A, Nakashima H, Aishima S, Kohnoe S and Maehara Y. Suppressed MKP-1 is an independent predictor of outcome in patients with hepatocellular carcinoma. Oncology. 2005; 69:342–347.
32. Calvisi DF, Pinna F, Pellegrino R, Sanna V, Sini M, Daino L, Simile MM, De Miglio MR, Frau M, Tomasi ML, Seddaiu MA, Muroni MR, et al. Ras-driven proliferation and apoptosis signaling during rat liver carcinogenesis is under genetic control. Int J Cancer. 2008; 123:2057–2064.
33. Lawan A, Zhang L, Gatzke F, Min K, Jurczak MJ, Al-Mutairi M, Richter P, Camporez JP, Couvillon A, Pesta D, Roth Flach RJ, Shulman GI, et al. Hepatic mitogen-activated protein kinase phosphatase 1 selectively regulates glucose metabolism and energy homeostasis. Mol Cell Biol. 2015; 35:26–40.
34. Tomasi ML, Ramani K, Lopitz-Otsoa F, Rodriguez MS, Li TW, Ko K, Yang H, Bardag-Gorce F, Iglesias-Ara A, Feo F, Pascale MR, Mato JM, et al. S-adenosylmethionine regulates dual-specificity mitogen-activated protein kinase phosphatase expression in mouse and human hepatocytes. Hepatology. 2010; 51:2152–2161.
35. Yamaji S, Droggiti A, Lu SC, Martinez-Chantar ML, Warner A and Varela-Rey M. S-Adenosylmethionine regulates connexins sub-types expressed by hepatocytes. Eur J Cell Biol. 2011; 90:312–322.
36. Lu SC and Mato JM. Role of methionine adenosyltransferase and S-adenosylmethionine in alcohol-associated liver cancer. Alcohol. 2005; 35:227–234.
37. Pascale RM, Simile MM, De Miglio MR, Nufris A, Daino L, Seddaiu MA, Rao PM, Rajalakshmi S, Sarma DS and Feo F. Chemoprevention by S-adenosyl-L-methionine of rat liver carcinogenesis initiated by 1,2-dimethylhydrazine and promoted by orotic acid. Carcinogenesis. 1995; 16:427–430.
38. Lu SC, Ramani K, Ou X, Lin M, Yu V, Ko K, Park R, Bottiglieri T, Tsukamoto H, Kanel G, French SW, Mato JM, et al. S-adenosylmethionine in the chemoprevention and treatment of hepatocellular carcinoma in a rat model. Hepatology. 2009; 50:462–471.
39. Ikeda K, Suzuki T, Tate G and Mitsuya T. Multiple immunoenzyme labeling using heat treatment combined with the polymer method: an analysis of the appropriate inactivation conditions of primary antibodies. Acta Histochem. 2011; 113:117–124.
40. Sai K, Kanno J, Hasegawa R, Trosko JE and Inoue T. Prevention of the down-regulation of gap junctional intercellular communication by green tea in the liver of mice fed pentachlorophenol. Carcinogenesis. 2000; 21:1671–1676.
41. Suzuki S, Asamoto M, Tsujimura K and Shirai T. Specific differences in gene expression profile revealed by cDNA microarray analysis of glutathione S-transferase placental form (GST-P) immunohistochemically positive rat liver foci and surrounding tissue. Carcinogenesis. 2004; 25:439–443.