
INTRODUCTION
Small cell carcinoma of the ovary, hypercalcemic type (SCCOHT) is the most common undifferentiated ovarian malignancy diagnosed in women under age 40, with a mean diagnosis age of 23.9 years [1]. It is an extremely aggressive tumor, with long-term survival rates of early stage diagnoses at 33% [2]. We and others recently discovered that SCCOHT is in fact a monogenic disease, where almost all cases are attributable to germline and/or somatic deleterious mutations in a single gene, SMARCA4, which is a key component of the SWI/SNF chromatin remodeling complexes [3-5]. These complexes utilize the energy of ATP hydrolysis to mobilize nucleosomes and remodel chromatin and have been found to promote transcriptional activation by inducing changes in DNA methylation patterns [6, 7].
Although SCCOHT is classified as a miscellaneous ovarian tumor by the World Health Organization [8], our previous publications describe its genetic and histological similarity to rhabdoid tumors [5, 9]. Rhabdoid tumors are pediatric soft tissue tumors that can manifest as either atypical teratoid/rhabdoid tumors (ATRTs) in the brain, or extra-cranial malignant rhabdoid tumors (MRTs) that most often develop in the kidney, but can arise in other tissues [10]. In 98% of cases, they are caused by inactivating mutations in SMARCB1 (also known as SNF5/INI1/BAF47), another component of the SWI/SNF complex [6]. The other 2% are caused by deleterious mutations in SMARCA4 (also known as BRG1), and when this is the case, the patients have been reported to have a worse prognosis [11]. While ATRTs and MRTs by definition arise in different tissues, gene expression profiling of ATRTs and MRTs has shown that they are molecularly similar tumors [10]. These tumors were originally thought to be variants of primitive neuroectodermal tumors (PNET) and Wilms tumor respectively, but were reclassified once genetic testing showed that they were distinct entities [9]. Likewise, our recent publications [5, 9] describe phenotypic and genetic similarities between SCCOHT, MRT, and ATRT, and propose that SCCOHT should be part of the rhabdoid tumor family and therefore be renamed MRT of the ovary (MRTO). Further similarities between MRT and SCCOHT include that hypercalcemia has been seen in both tumor types, with an incidence of approximately 30% in SCCOHT and 26% in patients with MRT [12]. Often, this serum hypercalcemia is due to increased parathyroid hormone production by the tumor, but it is still unclear whether the mechanism of hypercalcemia in these patients is related to the SWI/SNF mutations [12].
In contrast to SCCOHT, ovarian high grade serous carcinoma (HGSC) is the most common sub-type of ovarian cancer, accounting for 80-85% of all ovarian cancers, with a median age of diagnosis of 60 years. While the cell of origin of SCCOHT is still unknown, much work has been done on the characterization of HGSC; we therefore added HGSC to our comparison of SCCOHT and ATRT in an attempt to distinguish SCCOHT as a molecularly distinct entity from this common ovarian tumor.
In the present study, we further explore the landscape of somatic genetic and epigenetic alterations in SCCOHT and compare it to that of ATRTs and HGSC. Since SMARCA4 and SMARCB1 are two main components of the SWF/SNF complexes and are thought to modify DNA methylation in a similar manner, we hypothesized that they would show similarity in their global methylation profile. To our knowledge, no study has previously investigated the genome-wide methylation pattern of SCCOHT in comparison with other tumor types.
RESULTS
Somatic genomic alterations reveal SCCOHTs, like ATRTs, have “simple” genomes
To characterize the landscape of somatic alterations in SCCOHT, we performed whole exome sequencing (WES) on 14 formalin-fixed paraffin-embedded (FFPE) SCCOHT tumors and their matched normal tissues, and compared their overall genomic alterations with 14 ATRTs from our lab and 14 ovarian high-grade serous carcinomas (HGSCs) from TCGA.
In line with previous studies, our WES analysis revealed SMARCA4 and SMARCB1 to be the only frequently mutated genes in SCCOHT and ATRTs, respectively (Figure 1, Table S1) [3-5, 13]. In contrast, HGSCs frequently showed mutations in TP53, consistent with the data published by TCGA [14]. This gene was somatically mutated in one ATRT (ATRT_8) and was not mutated in any SCCOHTs. Furthermore, no SCCOHTs had mutations in any of the eight other genes recurrently mutated in HGSC (BRCA1, BRCA2, RB1, NF1, FAT3, CSMD3, GABRA6, and CDK12) [14].
Consistent with the above results, Fisher’s least significant difference (LSD) test revealed that the average number of somatic protein-coding mutations was significantly lower in SCCOHT (8.5 mutations) and ATRTs (4 mutations) than in HGSCs (74 mutations, p-values <0.0001 for both SCCOHT vs HGSC and for ATRT vs HGSC) (Figure S1). Moreover, our analysis also revealed fewer mutations per megabase (Mb) in the coding regions in SCCOHT and ATRTs (0.28 and 0.14 mutations/Mb, respectively) than in HGSCs (2.8 mutations/Mb) (Figure 1).
To detect and compare recurrent somatic allelic imbalance (AI) in tumors, we analyzed WES data from each tumor type using ExomeAI (see methods) [15]. This analysis revealed remarkable genome-wide differences in the AI patterns of SCCOHT and ATRTs compared to those in HGSC (Figure 2). We found that SCCOHTs and ATRTs have very “simple” genomes and the only recurrent AI aberration identified was on chr19p surrounding SMARCA4 (57% of cases), and chr22q surrounding SMARCB1 (100% of cases), respectively (Figures 1 and 2). Regardless of the way in which the gene was altered, however, almost all SCCOHT and ATRT samples previously showed loss of the respective SMARCA4 or SMARCB1 protein (Table S1). In contrast to the quiescent SCCOHT and ATRT genomes, several abnormalities that included chromosomal arms or entire chromosomes were observed across the genome of HGSCs (Figure 2).
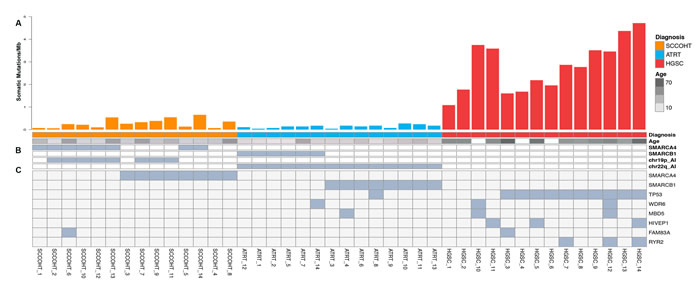
Figure 1: Results from genomic analysis of SCCOHT, ATRT, and HGSC. A. Histograms show protein-coding somatic mutation rates for each sample in SCCOHT (orange), ATRT (blue) and HGSC (red). Age range of each patient is indicated below the histogram (age annotation was not available for HGSC_10, HGSC_4 and HGSC_6). B. Germline mutations identified in SMARCA4 and SMARCB1. C. Somatic mutations (substitutions and indels) and allelic imbalance (chr19p and chr22q for SCCOHT and ATRT, respectively). SCCOHT_4 and SCCOHT_8 had two somatic mutations each. The mutation in SCCOHT_3 was homozygous.
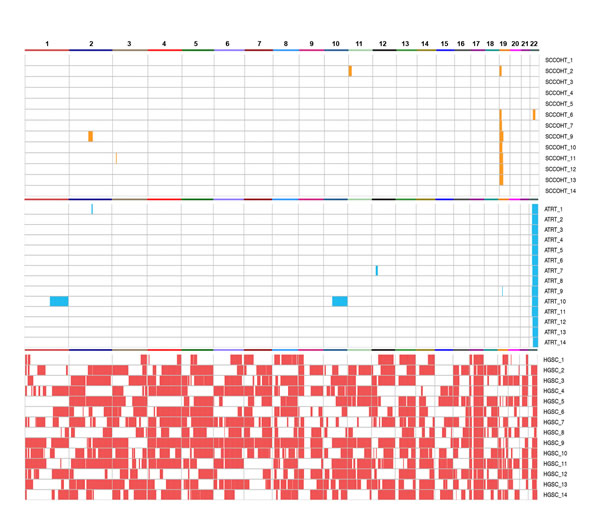
Figure 2: Genome-wide analysis of AI in SCCOHT (orange), ATRT (blue), and HGSC (red). Recurrent AI was observed only on chr19p and chr22q in SCCOHT and ATRTs, respectively, while many AI aberrations were detected across the genome of HGSCs.
The global DNA methylation patterns of SCCOHTs and ATRTs are strongly correlated
DNA methylation is a major regulator of gene expression and its alteration is frequently reported in tumorigenesis [16]. It is known that SWI/SNF complexes are involved in the establishment of DNA methylation patterns [7]. In order to characterize the genome-wide DNA methylation pattern of SCCOHT and to examine its similarity to ATRT and HGSC, we performed multidimensional scaling (MDS) analysis of methylation data using the 10,000 most variable CpGs. We found that the methylation pattern of SCCOHT was distinct from that of HGSC (Figure 3A). Similarly, ATRTs were clustered apart from other brain tumors, including glioblastomas, embryonal tumors with multilayered rosettes (ETMRs), and primitive neuroectodermal tumors (PNETs), although some degree of heterogeneity was present within the ATRT group. The latter is in line with a recent study that demonstrating that up to three subgroups may be detected in ATRTs [17].
We next quantified the degree to which methylation profiles of SCCOHT and ATRT are similar by applying a model-based analysis, which estimates the methylation effects between diagnostic groups, using a segment-wise approach (see methods). The analysis showed that SCCOHT has a higher Pearson correlation with ATRT (r = 0.93) than with HGSC (r = 0.78) (Figure 3B). Similar correlations (though of lesser magnitude) were obtained when applying a CpG-wise approach (Figure S3). Not surprisingly, all pairs of correlations are highly significant with p-values < 10e-16 (alternative hypothesis: true correlation is not equal to 0). This suggests that similar mechanisms, likely linked to chromatin remodeling by the SWI/SNF complexes, might contribute to similar methylation alterations in both groups (SCCOHT and ATRT).
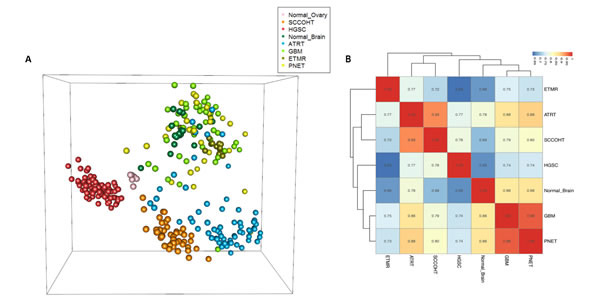
Figure 3: Methylation analyses of SCCOHT, ATRT, and HGSC compared to other samples. A. Multidimensional scaling (MDS) analysis of methylation data. Methylation values of 10,000 most variable CpGs are projected into three dimensions. Each sphere represents a sample, with colors representing diagnostic groups as indicated in the legend. B. Hierarchical clustering of the methylation effect of diagnostic groups. The values used for clustering are segment-wise estimations of methylation group effects, as estimated by the model-based method (see methods). Pairwise correlation of the values (r) are shown. All pairs of correlations are highly significant (p-value < 10e-16). HGSC: High grade serous carcinoma; SCCOHT: Small cell carcinoma of the ovary, hypercalcemic type; ATRT: Atypical teratoid/rhabdoid tumors; GBM: Glioblastoma; ETMR: Embryonal tumor with multilayered rosettes; PNET: Primitive neuroectodermal tumor.
DISCUSSION
Understanding SCCOHT on a molecular level is crucial for determining the best treatment with which to combat the disease, as tissue of origin of a tumor is not always the best indicator of successful therapy.
Here we compared the genomic and epigenomic landscapes of SCCOHT, ATRT, and HGSC using WES and methylation analyses. Until now, several papers have discussed the similarities between SCCOHT and ATRT on genetic and histological levels [5, 9, 18], but this is the first study in which a comprehensive comparison has been made between these tumor types on genomic and epigenomic levels.
Using WES analysis, we were able to show the remarkable genomic similarities between SCCOHT and ATRT (Figures 1, 2, S2). Interestingly, while all ATRTs had LOH on chr22q (where SMARCB1 is located) in addition to one SMARCB1 mutation, only 8 of 14 SCCOHTs showed chr19p loss (where SMARCA4 is located), with the remaining samples having only point mutations or small indels (see more details in Table S1). It is notable that one SCCOHT sample had only one mutation with no chr19p LOH. This sample did show loss of the SMARCA4 protein, but we were unable to find the second mutation by sequencing.
In addition to DNA sequencing analysis, analysis of the methylation data showed that SCCOHTs are considerably more similar to ATRTs than they are to HGSC (Figure 3). These genomic and epigenomic findings are mirrored by the clinical observations. The clinical presentation, histological appearance, and overall prognosis of SCCOHT differ from that of HGSC, in addition to loss of SMARCA4 in SCCOHT [19].
Perhaps the most valuable use of the results from our genomic and epigenomic analyses done here will be in forming new therapeutic strategies. Many case reports and studies of small cohorts of SCCOHT have described therapies combining surgical resection and adjuvant chemotherapy with many conflicting results. For SCCOHT, as with many cancers, stage at diagnosis seems to be the biggest factor in determining survival, with sometimes only surgery and radiotherapy needed for stage I patients [20]. For newly diagnosed SCCOHT, treatment is usually akin to that for the more common small cell lung cancer and ovarian germ cell tumors, where in addition to surgery, platinum-based therapy is used [21]. Other ovarian cancers, including HGSC, are treated similarly, by surgery with or without adjuvant chemotherapy, including platinums alone or in combination, depending on the stage at diagnosis.
While most patients do not survive a diagnosis of SCCOHT, intensive chemotherapy protocols have been occasionally found to result in durable responses, but mostly in early stage disease [22, 23]. For advanced stage disease, multiple combinations of almost all chemotherapy agents have been tried but with little success, as it has increasingly been shown that multi-modal therapy is required to effectively treat these patients [24]. This is similar to primary treatment for MRTs and ATRTs, where surgery has been shown to be beneficial with adjuvant chemotherapy and radiation therapy [25-27]. Additionally, high dose chemotherapy with stem cell rescue has been reported to be promising and has been incorporated in the primary treatment of ATRT [28, 29]. Similarly, some cases of SCCOHT have been treated with high-dose chemotherapy and stem cell rescue [30, 31].
Aside from current protocols, trials and in vitro studies in ATRT patients have resulted in some success. Due to the extensive research that has been done on the effects of SMARCB1 loss, many studies have tested the effect of targeting overexpressed proteins in ATRT. Alisertib (MLN8237), an Aurora K inhibitor, has shown early evidence of remarkable activity in the treatment of ATRT patients [32], while CDK, MEK, and EZH2 inhibitors have been shown to be effective in restricting tumor cell growth in rhabdoid tumor cell line and xenograft-based models [33-36]. In SCCOHT, on the other hand, no clinical trials have been conducted recently. While several recent papers show that SMARCA2, the ATPase counterpart of SMARCA4, is overexpressed in SMARCA4-deficient tumors and may be a good therapeutic target [37, 38], in the BIN-67 SCCOHT cell line, SMARCA2 is expressed at the mRNA level, but its protein is not detectable [5]. While BIN-67 is one of only two SCCOHT cell lines, it is indeed representative of SCCOHT tumors in vivo, as it was recently shown that loss of both SMARCA2 and SMARCA4 proteins is specific to SCCOHT [39]. Studies have been conducted in which other drugs and oncolytic viruses were tested in in vitro and in vivo models of SCCOHT [40,41], but thus far none have reached clinical trials.
Taken together, it is clear that not only is SCCOHT significantly more similar to ATRT than to HGSC on genetic and epigenetic levels, but that therapies used to treat ATRT seem to result in a better response in SCCOHT patients than those used to treat other types of ovarian cancer. Our methylation analysis results show that similar pathways may be dysregulated in SCCOHT and ATRT downstream of SMARCA4 or SMARCB1 loss. These results together with similarities between SCCOHT and MRT/ATRT on a histological and clinical level, demonstrate that management with multimodal therapy, including stem cell transplant, should be considered in the primary treatment of these devastating ovarian tumors. Furthermore, despite their distinct tissues of origin and ages at diagnosis, our data suggests that the loss of distinct SWI/SNF subunits leads to convergent biological features in ATRT and SCCOHT. This suggests an instructive role of the loss of subunits of this chromatin remodeling complex in shaping convergent cancer formation and dependencies.
materials and METHODS
Patients and Tumor samples
To characterize the landscape of somatic alterations in SCCOHT, we performed WES on 14 formalin-fixed paraffin-embedded tumors and their matched normal tissues [42]. The mutation status of all 14 SCCOHT samples was previously published and all samples were previously seen by a gynecological pathologist to evaluate the histological appearance of SCCOHT [5]. To confirm the nature of SCCOHT tumors by loss of SMARCA4, immunohistochemistry on these tumors was done as previously described [5]. We then expanded the analysis by including 14 rhabdoid tumor samples (11 Brain, 1 kidney, 1 bladder, and 1 soft-tissue), carrying somatic or germline mutations in SMARCB1, from a previous study by Lee and colleagues [43], and 14 publically available HGSC samples from the TCGA consortium [44]. DNA methylation profiling was carried out on DNA extracted from tumor and normal ovarian tissue (45 SCCOHT and 6 normal ovary), and 110 previously published [45, 46] and 61 unpublished samples of tumor and normal brain tissue: 65 ATRTs, 31 PNETs, 12 ETMRs, 46 GBMs, and 17 normal brain samples. We also included methylation data from 12 TCGA HGSCs (Batch 409, Level 1 data downloaded from the TCGA data portal, https://tcga-data.nci.nih.gov/tcga/) and 80 HGSCs from a recent study by Patch et al. [47].
Whole Exome Sequencing
Whole-exome library preparation, exon capture and sequencing were performed using our standard protocols at the McGill University and Génome Québec Innovation Centre as previously described by Witkowski et al. [5]. Briefly, DNA samples were extracted and visualized on an agarose gel. The Agilent SureSelect V4 and the Illumina Nextera Rapid-Capture Exome kits were used for whole exome capture as previously described [5]. All libraries were sequenced on either Illumina HiSeq 2000 or 2500 sequencer with paired-end 100-bp reads.
Somatic genomic alterations analysis
Bioinformatics analysis of exome sequencing data was performed using our WES pipeline as previously described (Figure S2) [48, 49]. In brief, alignment of sequenced reads to the reference genome (hg19) was performed using BWA (v. 0.5.9) [50]. Subsequently, the Genome Analysis Toolkit (GATK) was used to do local realignment of reads around small insertions and deletions (indels) and to get coverage of consensus coding sequence (CCDS) bases (Table S1) [51]. A mean coverage of 76X (SCCOHT), 71X (ATRT) and 144X (HGSC) was obtained for all CCDS exons in each tumor type (for HGSCs, we also performed the analysis with 80X coverage which led to similar results; data not shown). Potential somatic substitutions, single nucleotide variants (SNVs) and indels, were called using Mutect (see https://confluence.broadinstitute.org/display/CGATools/MuTect for method) and IndelLocator (see https://confluence.broadinstitute.org/display/CGATools/Indelocator for methods) on the basis of BWA alignments and were then annotated with ANNOVAR [52]. To remove common variants and false positive calls, candidate somatic mutations were subjected to several filtering steps and eliminated if they fulfilled any one of the following criteria: (i) genomic position of variant covered by <10X, (ii) <5 reads support the alternative variant, (iii) variant has allelic ratio <5% for SNVs or <15% for indels, (iv) variant has allele frequency > 0.001 in our non-cancer (~1000 exomes sequenced previously in our center) or ExAC databases, or (v) variant seen as homozygote in ExAC database. Finally, only the most likely damaging variants (nonsense, canonical splice-site, and missense mutations, and coding indels) were considered for further analysis.
To compare mutation rates of the three diagnostic groups (SCCOHT, ATRT, and HGSC), we used Fisher’s least significant difference (LSD) test [53] with an alpha level of 0.05 and Bonferroni-adjusted P-values using the R package ‘agricolae’ (http://CRAN.R-project.org/package=agricolae).
AI analysis was performed on WES data using ExomeAI [15]. In brief, ExomeAI detects AI events across all samples by investigating the B allele frequency (BAF) profile of exomes. To do this, all heterozygous variants (BAF values of 0.05 to 0.95) are extracted from VCF files, and AI segments are called and summarized across all samples. In samples with low quality and quantity (e.g. FFPE tumors), AI-based methods produce more reliable calls, as inadequate genome-wide coverage consistency across tumor and matched normal samples can lead to false positive copy number calls [15].
DNA Methylation analysis
Raw methylation intensities of 314 samples were imported and inspected for quality using the Bioconductor package Minfi [54]. Functional normalization was applied on all samples, where the first n (here n = 2) principal components of the internal control probes are used to adjust intensities for technical variations and batch effects [53, 55]. CpG sites were annotated using the IlluminaHumanMethylation450kanno.ilmn12.hg19 Bioconductor package, which is based on hg19, Illumina’s version 1.2 annotations. Known SNP sites and CpGs on sex chromosomes were removed from further analysis. Similar to that described by Jaffe et al [56], we used a model-based method to estimate the methylation effect of all diagnostic groups, at both CpG and segment levels. Methylation effects of all diagnostic groups were estimated in comparison with normal ovary. For all model-based analyses, we used normalized beta values, as described above. More details of the method will be described elsewhere (J.N. et al, unpublished data). For MDS analysis, the most variable CpGs (n = 10,000) were used to calculate Euclidean distance between samples. Classical Multidimensional Scaling transformation was then applied to project the distances into 3 dimensions for visualization.
ACKNOWLEDGMENTS
We wish to acknowledge the contribution of the high-throughput and Sanger sequencing platforms of the McGill University and Genome Québec Innovation Centre (Montreal, Canada).
FUNDING
JM is a recipient of a Canada Research Chair. SF received a postdoctoral Fellowship from the Network of Applied Medical Genetics. MH received funding from IZKF Münster (Ha3/019/15). LW received funding from the Fonds de recherche du Québec – Santé. WDF received funds from the Canadian Cancer Society Research Institute and the Small Cell Ovarian Cancer Foundation.
CONFLICTs OF INTERESTS
None.
REFERENCES
1. Young RH, Oliva E, Scully RE. Small cell carcinoma of the ovary, hypercalcemic type. A clinicopathological analysis of 150 cases. The American journal of surgical pathology. 1994; 18: 1102-1116.
2. Estel R, Hackethal A, Kalder M, Munstedt K. Small cell carcinoma of the ovary of the hypercalcaemic type: an analysis of clinical and prognostic aspects of a rare disease on the basis of cases published in the literature. Arch gynecol obstet. 2011; 284: 1277-1282.
3. Jelinic P, Mueller JJ, Olvera N, Dao F, Scott SN, Shah R, Gao J, Schultz N, Gonen M, Soslow RA, Berger MF, Levine DA. Recurrent SMARCA4 mutations in small cell carcinoma of the ovary. Nat Genet. 2014; 46: 424-426.
4. Ramos P, Karnezis AN, Craig DW, Sekulic A, Russell ML, Hendricks WP, Corneveaux JJ, Barrett MT, Shumansky K, Yang Y, Shah SP, Prentice LM, Marra MA, et al. Small cell carcinoma of the ovary, hypercalcemic type, displays frequent inactivating germline and somatic mutations in SMARCA4. Nat genet. 2014; 46: 427-429.
5. Witkowski L, Carrot-Zhang J, Albrecht S, Fahiminiya S, Hamel N, Tomiak E, Grynspan D, Saloustros E, Nadaf J, Rivera B, Gilpin C, Castellsague E, Silva-Smith R, et al. Germline and somatic SMARCA4 mutations characterize small cell carcinoma of the ovary, hypercalcemic type. Nat Genet. 2014; 46: 438-443.
6. Witkowski L, Foulkes WD. In Brief: Picturing the complex world of chromatin remodelling families. J Pathol. 2015. doi: 10.1002/path.4585.
7. Banine F, Bartlett C, Gunawardena R, Muchardt C, Yaniv M, Knudsen ES, Weissman BE, Sherman LS. SWI/SNF Chromatin-Remodeling Factors Induce Changes in DNA Methylation to Promote Transcriptional Activation. Cancer Res. 2005; 65: 3542-3547.
8. Pathology and Genetics of Tumours of the Breast and Female Genital Organs. In World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of the Breast and Female Genital Organs, 3rd. Tavassoli FA, Devilee P, (eds). IARC Press: Lyon, 2003.
9. Foulkes WD, Clarke BA, Hasselblatt M, Majewski J, Albrecht S, McCluggage WG. No small surprise - small cell carcinoma of the ovary, hypercalcaemic type, is a malignant rhabdoid tumour. J Pathol. 2014; 233: 209-214.
10. Brennan B, Stiller C, Bourdeaut F. Extracranial rhabdoid tumours: what we have learned so far and future directions. Lancet Oncol. 2013; 14: e329-e336.
11. Hasselblatt M, Nagel I, Oyen F, Bartelheim K, Russell RB, Schuller U, Junckerstorff R, Rosenblum M, Alassiri AH, Rossi S, Schmid I, Gottardo NG, Toledano H, et al. SMARCA4-mutated atypical teratoid/rhabdoid tumors are associated with inherited germline alterations and poor prognosis. Acta neuropathologica. 2014; 128: 453-456.
12. Amar AM, Tomlinson G, Green D, Breslow NE, de Alarcon PA. Clinical Presentation of Rhabdoid Tumors of the Kidney. J Pediatr Hematol Oncol. 2001; 23: 105-108.
13. Lee RS, Stewart C, Carter SL, Ambrogio L, Cibulskis K, Sougnez C, Lawrence MS, Auclair D, Mora J, Golub TR, Biegel JA, Getz G, Roberts CWM. A remarkably simple genome underlies highly malignant pediatric rhabdoid cancers. J Clin Invest. 2012; 122: 2983-2988.
14. Integrated genomic analyses of ovarian carcinoma. Nature. 2011; 474: 609-615.
15. Nadaf J, Majewski J, Fahiminiya S. ExomeAI: detection of recurrent allelic imbalance in tumors using whole-exome sequencing data. Bioinformatics. 2015; 31: 429-431.
16. Kulis M, Esteller M. DNA methylation and cancer. Adv Genet. 2010; 70: 27-56.
17. Torchia J, Picard D, Lafay-Cousin L, Hawkins CE, Kim S-K, Letourneau L, Ra Y-S, Ho KC, Chan TSY, Sin-Chan P, Dunham CP, Yip S, Ng H-k, et al. Molecular subgroups of atypical teratoid rhabdoid tumours in children: an integrated genomic and clinicopathological analysis. Lancet Oncol. 16: 569-582.
18. Rabinovich A, Witkowski L, Shaco-Levi R, Meirovitz M, Hasselblatt M, Foulkes WD. Primary rhabdoid tumor of the ovary: When large cells become small cells. Gynecol Oncol Rep. 2015; 12: 64-66.
19. Berek JS, Thomas GM, Ozols RF. Ovarian Cancer. In Holland-Frei Cancer Medicine, 5th. Bast R, Kufe D, Pollock R, (eds). BC Decker: Hamilton, Ontario, 2000.
20. Callegaro-Filho D, Burke TW, Eifel PJ, Ramirez PT, Euscher EE, Schmeler KM. Radiotherapy for recurrent small cell carcinoma of the ovary: A case report and review of the literature. Gynecol Oncol Rep. 2015; 11: 23-25.
21. Reed N, Millan D, Verheijen R, Castiglione M. Non-epithelial ovarian cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2010; 21: v31-v36.
22. Senekjian EK, Weiser PA, Talerman A, Herbst AL. Vinblastine, cisplatin, cyclophosphamide, bleomycin, doxorubicin, and etoposide in the treatment of small cell carcinoma of the ovary. Cancer. 1989; 64: 1183-1187.
23. Wallbillich JJ, Nick AM, Ramirez PT, Watkins JL, Euscher ED, Schmeler KM. Vinblastine, cisplatin, cyclophosphamide, bleomycin, doxorubicin, and etoposide (VPCBAE) in the management of three patients with small-cell carcinoma of the ovary. Gynecol Oncol Rep. 2012; 2: 58-60.
24. Harrison ML, Hoskins P, du Bois A, Quinn M, Rustin GJS, Ledermann JA, Baron-Hay S, Friedlander ML. Small cell of the ovary, hypercalcemic type—Analysis of combined experience and recommendation for management. A GCIG study. 2006; 100: 233-238.
25. Lau CSM, Mahendraraj K, Chamberlain RS. Atypical teratoid rhabdoid tumors: a population-based clinical outcomes study involving 174 patients from the Surveillance, Epidemiology, and End Results database (1973–2010). 2015; 7: 301-309.
26. Brennan B, Stiller C, Bourdeaut F. Extracranial rhabdoid tumours: what we have learned so far and future directions. 14: e329-e336.
27. Uwineza A, Gill H, Buckley P, Owens C, Capra M, O’Sullivan C, McDermott M, Brett F, Farrell M, Pears J, O’Sullivan MJ. Rhabdoid tumor: the Irish experience 1986–2013. 207: 398-402.
28. Hilden JM, Meerbaum S, Burger P, Finlay J, Janss A, Scheithauer BW, Walter AW, Rorke LB, Biegel JA. Central Nervous System Atypical Teratoid/Rhabdoid Tumor: Results of Therapy in Children Enrolled in a Registry. 2004; 22: 2877-2884.
29. Gardner SL, Asgharzadeh S, Green A, Horn B, McCowage G, Finlay J. Intensive induction chemotherapy followed by high dose chemotherapy with autologous hematopoietic progenitor cell rescue in young children newly diagnosed with central nervous system atypical teratoid rhabdoid tumors. 2008; 51: 235-240.
30. Distelmaier F, Calaminus G, Harms D, Sträter R, Kordes U, Fleischhack G, Göbel U, Schneider DT. Ovarian small cell carcinoma of the hypercalcemic type in children and adolescents. Cancer. 2006; 107: 2298-2306.
31. Pressey JG, Kelly DR, Hawthorne HT. Successful treatment of preadolescents with small cell carcinoma of the ovary hypercalcemic type. J Pediatr Hematol Oncol. 2013; 35: 566-569.
32. Wetmore C, Bendel A, Gajjar A. Activity of Alisertib (MLN8237) as Single Agent in Recurrent Atypical Teratoid Rhabdoid Tumor (AT/RT) in Four Children: A Single Patient Treatment Plan Pilot Study. Cancer Genet. 207: 453.
33. Unland R, Borchardt C, Clemens D, Kool M, Dirksen U, Frühwald MC. Analysis of the antiproliferative effects of 3-deazaneoplanocin A in combination with standard anticancer agents in rhabdoid tumor cell lines. Anticancer Drugs. 2015; 26: 301-311.
34. Smith ME, Cimica V, Chinni S, Jana S, Koba W, Yang Z, Fine E, Zagzag D, Montagna C, Kalpana GV. Therapeutically targeting cyclin D1 in primary tumors arising from loss of Ini1. Proc Natl Acad Sci U S A. 2011; 108: 319-324.
35. Knutson SK, Warholic NM, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, Porter Scott M, Chesworth R, Moyer MP, Copeland RA, Richon VM, Pollock RM, Kuntz KW, et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A. 2013; 110: 7922-7927.
36. Weingart MF, Roth JJ, Hutt-Cabezas M, Busse TM, Kaur H, Price A, Maynard R, Rubens J, Taylor I, Mao X-g, Xu J, Kuwahara Y, Allen SJ, et al. Disrupting LIN28 in atypical teratoid rhabdoid tumors reveals the importance of the mitogen activated protein kinase pathway as a therapeutic target. Oncotarget. 2015; 6: 3165-3177. doi: 10.18632/oncotarget.3078.
37. Hoffman GR, Rahal R, Buxton F, Xiang K, McAllister G, Frias E, Bagdasarian L, Huber J, Lindeman A, Chen D, Romero R, Ramadan N, Phadke T, et al. Functional epigenetics approach identifies BRM/SMARCA2 as a critical synthetic lethal target in BRG1-deficient cancers. Proc Natl Acad Sci U S A. 2014; 111: 3128-3133.
38. Oike T, Ogiwara H, Tominaga Y, Ito K, Ando O, Tsuta K, Mizukami T, Shimada Y, Isomura H, Komachi M, Furuta K, Watanabe S-I, Nakano T, et al. A Synthetic Lethality–Based Strategy to Treat Cancers Harboring a Genetic Deficiency in the Chromatin Remodeling Factor BRG1. Cancer Res. 2013; 73: 5508-5518.
39. Karnezis AN, Wang Y, Ramos P, Hendricks WPD, Oliva E, D’Angelo E, Prat J, Nucci MR, Nielsen TO, Chow C, Leung S, Kommoss F, Kommoss S, et al. Dual loss of the SWI/SNF complex ATPases SMARCA4/BRG1 and SMARCA2/BRM is highly sensitive and specific for small cell carcinoma of the ovary, hypercalcemic type. 2015. doi: 10.1002/path.4633.
40. Gamwell L, Gambaro K, Merziotis M, Crane C, Arcand S, Bourada V, Davis C, Squire J, Huntsman D, Tonin P, Vanderhyden B. Small cell ovarian carcinoma: genomic stability and responsiveness to therapeutics. Orphanet J Rare Dis. 2013; 8: 33.
41. Otte A, Rauprich F, Hillemanns P, Park-Simon T-W, Ohe Jvd, Hass R. In vitro and in vivo therapeutic approach for a small cell carcinoma of the ovary hypercalcaemic type using a SCCOHT-1 cellular model. Orphanet J Rare Dis. 2014; 9: 126-126.
42. Witkowski L, Carrot-Zhang J, Albrecht S, Fahiminiya S, Hamel N, Tomiak E, Grynspan D, Saloustros E, Nadaf J, Rivera B, Gilpin C, Castellsague E, Silva-Smith R, et al. Germline and somatic SMARCA4 mutations characterize small cell carcinoma of the ovary, hypercalcemic type. Nature genetics. 2014; 46: 438-443.
43. Lee RS, Stewart C, Carter SL, Ambrogio L, Cibulskis K, Sougnez C, Lawrence MS, Auclair D, Mora J, Golub TR, Biegel JA, Getz G, Roberts CW. A remarkably simple genome underlies highly malignant pediatric rhabdoid cancers. J Clin Invest. 2012; 122: 2983-2988.
44. Network TCGAR. Integrated genomic analyses of ovarian carcinoma. Nature. 2011; 474: 609-615.
45. Kleinman CL, Gerges N, Papillon-Cavanagh S, Sin-Chan P, Pramatarova A, Quang DA, Adoue V, Busche S, Caron M, Djambazian H, Bemmo A, Fontebasso AM, Spence T, et al. Fusion of TTYH1 with the C19MC microRNA cluster drives expression of a brain-specific DNMT3B isoform in the embryonal brain tumor ETMR. Nat genet. 2014; 46: 39-44.
46. Fontebasso AM, Papillon-Cavanagh S, Schwartzentruber J, Nikbakht H, Gerges N, Fiset PO, Bechet D, Faury D, De Jay N, Ramkissoon LA, Corcoran A, Jones DT, Sturm D, et al. Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nat genet. 2014; 46: 462-466.
47. Patch AM, Christie EL, Etemadmoghadam D, Garsed DW, George J, Fereday S, Nones K, Cowin P, Alsop K, Bailey PJ, Kassahn KS, Newell F, Quinn MC, et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature. 2015; 521: 489-494.
48. Fahiminiya S, Majewski J, Mort J, Moffatt P, Glorieux FH, Rauch F. Mutations in WNT1 are a cause of osteogenesis imperfecta. J Med Genet. 2013; 50:345-8.
49. Fahiminiya S, Al-Jallad H, Majewski J, Palomo T, Moffatt P, Roschger P, Klaushofer K, Glorieux FH, Rauch F. A Polyadenylation Site Variant Causes Transcript-Specific BMP1 Deficiency and Frequent Fractures in Children. Hum mol genet. 2015; 24:516-24.
50. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 25: 1754-1760.
51. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010; 20: 1297-1303.
52. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010; 38: e164.
53. Steel RGD. Citation Classic - Principles and Procedures of Statistics. Curr Contents. 1977: 20-20.
54. Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, Irizarry RA. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014; 30: 1363-1369.
55. Fortin JP, Labbe A, Lemire M, Zanke BW, Hudson TJ, Fertig EJ, Greenwood CM, Hansen KD. Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome biol. 2014; 15: 503.
56. Jaffe AE, Murakami P, Lee H, Leek JT, Fallin MD, Feinberg AP, Irizarry RA. Bump hunting to identify differentially methylated regions in epigenetic epidemiology studies. Int J Epidemiol. 2012; 41: 200-209.