
INTRODUCTION
Ligand-induced activation of the NK cell activating receptor NKG2D has been demonstrated to be significant in controlling tumor growth in experimental animal models [1, 2]. NKG2D, an NK cell group 2 member D activating receptor, is also expressed by most NKT cells, subsets of gamma-delta T cells, all human CD8+ T cells, and activated mouse CD8+ T cells as co-stimulatory receptors [1, 3–7]. Ectopic expression of NKG2D ligands results in effective tumor rejection mediated by NK cells, and in some cases primed cytotoxic T cells [5, 6]. Neutralization of NKG2D was shown to increase the incidence of carcinogen-induced tumor formation [8]. In the spontaneous prostate tumor model TRAMP (transgenic adenocarcinoma of the mouse prostate), mice deficient in NKG2D exhibited accelerated tumor progression in comparison to their NKG2Dwt counterparts [9]. Enforced expression of membrane-bound NKG2D ligands on TRAMP tumors prevented disease progression through sustained NKG2D signaling in effector cells [10]. These studies have collectively demonstrated the importance of NKG2D signaling in active tumor immune surveillance.
The most prevalently expressed NKG2D ligands in human malignancies are the MHC class I-related molecules A and B (MICA/B, collectively termed MIC) [11–14]. However, human cancer cells frequently evade NKG2D signaling through protease or exosome-mediated shedding of MIC to produce the immune suppressive soluble MIC (sMIC). Clinically, levels of sMIC correlated with disease stage in many malignancies [10, 15–19]. Mechanistically, sMIC shed from tumor cells has been shown to down-modulate surface NKG2D expression on NK cells and effector NKT and T cells [17, 20, 21]. Very recently, we have further shown that sMIC perturbs NK cell homeostatic maintenance in tumor-bearing hosts and facilitates the expansion of arginase I+ myeloid suppressor cells in the tumor microenvironment [10, 22]. These studies suggest that sMIC may be a valid target for immunotherapy to re-instate endogenous NK and effector T cell anti-tumor immune responses. Our recent study has provided proof for this concept [14].
The common gamma-chain cytokine interleukin-15 (IL-15) is considered as one of the most promising cytokines for cancer immunotherapy [23–27]. IL-15 not only is important for maintaining NK cell homeostasis [28–33], but also can upregulate NKG2D expression on NK and T cells and prime NKG2D signaling pathways [34–37]. IL-15 has also been shown to be important for maintaining CD8+ memory T cell populations [38–42]. The IL-15 superagonist ALT-803, a complex of the mutant IL-15 (IL-15N72D) and the IL-15RαSu/Fc [43, 44], was shown to confer at least 25-fold higher bioactivity in vivo and extended half-life compared to native IL-15 [45]. Pre-clinical studies have demonstrated that a single dose of ALT-803 was able to eliminate well-established primary myeloma cells in the bone marrow and to further reject tumor re-challenge due to expansion of CD44hi memory CD8+ T cells [45]. These pre-clinical studies have signified the cancer therapeutic potential of ALT-803 and have led to the current clinical trials for treating various human malignancies [46]. However, due to the facts that mice do not express human MIC and the human onco-immune dynamics of NKG2D ligand shedding and tumor progression have not been described in these mouse models, the impact of tumor-derived immune suppressive sMIC on the therapeutic potential of ALT-803 remains unknown.
To overcome the limitation that mice do not express human MIC, we have developed syngeneic transplantable tumor models in which sMIC-overexpressing mouse tumor cell lines were implanted into the sMIC-tolerant transgenic mouse [10]. Using this transplantable system, we tested the hypothesis that ALT-803 and a sMIC-neutralizing antibody can generate a cooperative therapeutic anti-tumor effect. We demonstrate that combinatory therapy of an antibody targeting sMIC and ALT-803 significantly enhanced the survival of mice bearing sMIC+ tumors in comparison with monotherapy. Mechanistically, we show that combined therapy cooperatively enhanced the homeostatic maintenance and functional potential of NK cells and memory CD8+ T cells. Combinatory therapy also heightened the potential of CD4+ T cells to produce IFN-γ and cooperatively eliminated myeloid derived suppressor cells (MDSCs) in tumor infiltrates. We also demonstrate in vitro that ALT-803 and a sMIC-neutralizing antibody cooperatively enhanced the activation of STAT5 signaling pathways in effector cells. Our findings provide the rationale for a translational approach whereby combinatory therapy of an antibody targeting tumor-derived sMIC and ALT-803 can cooperatively enhance innate and adaptive anti-tumor responses.
RESULTS
ALT-803 and sMIC-neutralizing antibody combined therapy inhibits tumor growth and prolongs survival of animals bearing sMIC+ tumors
Tumor shedding of sMIC is a human-specific mechanism of tumor immunoevasion. To test the hypothesis that targeting sMIC can enhance the therapeutic potential of IL-15 superagonist ALT-803 in a pre-clinical model, we developed multiple transplantable syngeneic tumor models by: 1) overexpressing human soluble MICB in transplantable mouse tumor cell lines, and 2) inoculating tumor lines secreting sMICB into the MICB transgenic mouse. As membrane-bound MIC can stimulate anti-tumor immunity [10], in order to eliminate experimental variation, we chose to develop these tumor models using the soluble form of MICB instead of membrane-bound MIC. Since mice do not express homologs of the human MIC ligand family, we utilized MICB transgenic mice as hosts to eliminate the effect of autoantibodies against the human sMICB. The MICB transgenic mice were produced by using the minimal rat probasin (rPb) promoter to direct expression of the transgene encoding the native form of MICB to the prostate epithelium. These mice have a similar phenotype as wild type B6 animals; however, they do not generate immune responses to syngeneic tumors expressing human MIC [10].
We implanted the murine mouse prostate tumor cell line RM9 and melanoma cell line B16F10 that were engineered to express human sMICB (designated as RM9-sMICB and B16-sMICB respectively) subcutaneously into cohorts of syngeneic MICB transgenic mice. When tumors reached approximately 75–100 mm3 in volume, mice were randomized into four therapeutic groups (n = 8–10 per group, Figure 1a). Although monotherapy with the sMIC-neutralizing antibody B10G5 and ALT-803 elicited survival benefits in comparison to control treatment, combined therapy further significantly prolonged survival in comparison to monotherapy in two independent tumor transplants (p < 0.05 and p < 0.0001 respectively, Figure 1b and 1d). Using linear regression analyses, we compared tumor growth rate prior to animals in the control group (or any treatment group) reaching the survival endpoint. B10G5 or ALT-803 monotherapy significantly reduced tumor growth rate by 35% and 51%, respectively, in comparison to control IgG treatment, whereas combined therapy further reduced tumor growth rate by 60% (Figure 1c). We observed similar trends in mice bearing B16-sMICB tumors (Figure 1d). Monotherapy with either B10G5 or ALT-803 reduced the average tumor burden by about 50%, whereas combined therapy further reduced tumor burden by more than 70% (p < 0.001) (Figure 1e). The enhanced therapeutic effect of combined therapy was further confirmed by the significant reduction in final B16-sMICB tumor weight in comparison to monotherapy in repeated experiments in which all animals were euthanized at day 10 of therapy (p < 0.01) (Figure 1f). Similar trend in survival benefit and tumor growth rate with combined therapy in comparison to monotherapy was presented with treatment up to 55 days where majority (>90%) of animals reached defined survival end point (data not shown). These data clearly demonstrate that monotherapy of ALT-803 or the sMIC-neutralizing antibodyB10G5 has respective therapeutic effects; however combined therapy elicited significantly enhanced therapeutic effects.
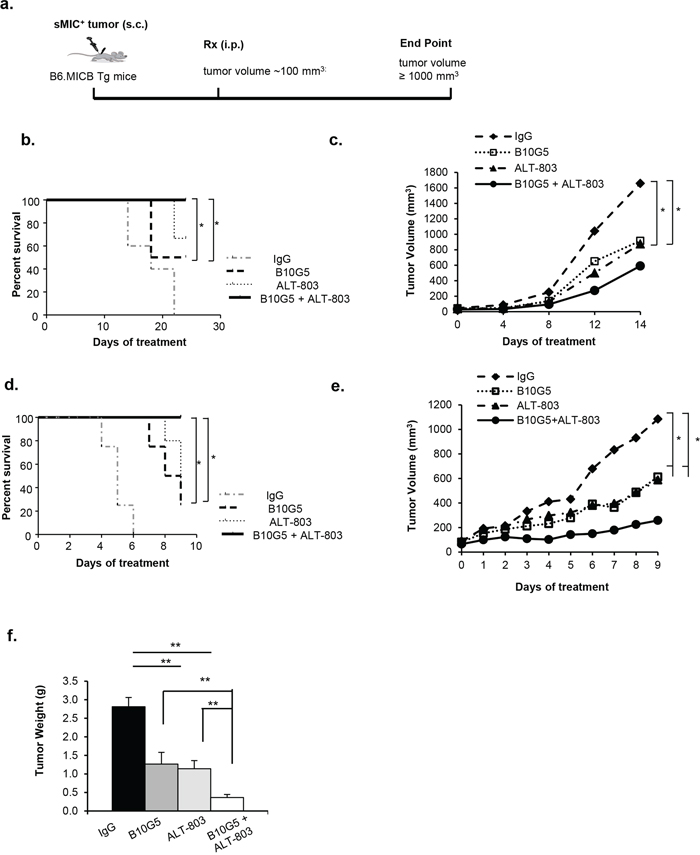
Figure 1: Combined therapy inhibits tumor growth and prolongs survival of animals bearing sMIC+ tumors. a. Depiction of treatment scheme. B6/MICB mice received right flank subcutaneous injection of 4 × 106 RM9-sMICB or B16F10-sMICB cells were treated i.p. with: 1) control mIgG (n = 10), 2) B10G5 (n = 9), 3) IL-15 superagonist ALT-803 (n = 8), and 4) a combination of B10G5 and ALT-803 (n = 10). b. Survival curve of mice bearing RM9-sMICB tumors with different therapy. c. Growth curve of RM9-sMICB tumors under different therapies. d. Survival curve of mice bearing B16-sMICB tumors with different therapy. e. and f. Growth curve of B16-sMICB tumors during different therapies. (f) Total weights of B16-sMICB tumors when animals were sacrificed at day 10 of therapy in repeated experiments. n = 8–10 per experimental group. *p < 0.05. **p < 0.01.
ALT-803 and sMIC-neutralizing antibody combined therapy markedly enhances NK cell number and function
We sought to understand mechanisms associated with the enhanced efficacy of combined therapy. Tumor-derived soluble MIC not only impairs NK cell function by downmodulating surface NKG2D expression but also perturbs peripheral NK cell maintenance [10, 21]. We first investigated the therapeutic effect of monotherapy and combined therapy on NK cells. As representatively shown with the B16-sMICB tumor model, in response to monotherapy with B10G5 or ALT-803, not only the number of NK cells (CD3−NK1.1+) but also the level of NKG2D expression, measured by the percentage of NKG2D+ NK cells and the mean fluorescence intensity (MFI) of the NKG2D expression on NKG2D+ NK cells, in the spleen and tumor-draining inguinal lymph nodes (dLN) was markedly increased in comparison to the control treatment group (Figure 2a–2c and Supplementary Figure S1a). However, combined therapy resulted in further significant increase in the number of NK cells and the level of NKG2D expression on cells in the spleen and dLN compared to monotherapy (Figure 2a–2c; Supplementary Figure S1a). Moreover, significantly increased activation of NK cells as evaluated by surface CD25 expression, was also observed in response to therapy, either by monotherapy of B10G5 or ALT-803 (Figure 2d and 2e; Supplementary Figure S1b). A further significant increase in surface CD25 expression on NK cells was elicited with combination therapy in comparison to monotherapy (Figure 2d and 2e; Supplementary Figure S1b), either with B10G5 or ALT-803, indicating enhanced NK cell activation with combination treatment. No significant change in other NK cell surface receptor expression, such as NKp46, Ly49A, Ly49C/I/F/H, NKG2A/C/E, was observed with combined therapy in comparison to monotherapy (data not shown).
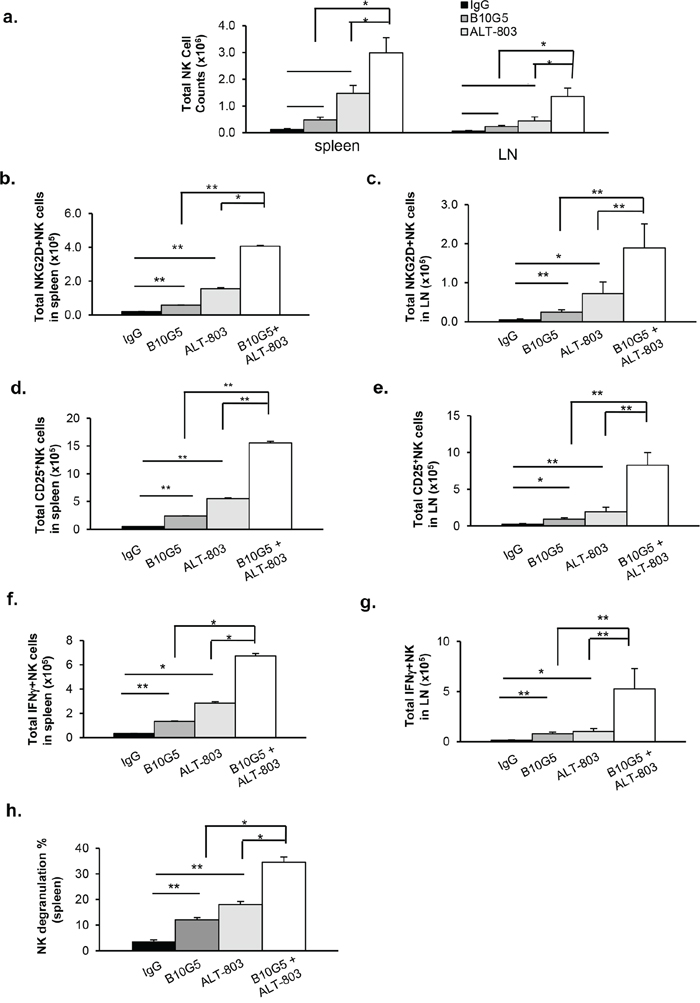
Figure 2: ALT-803 and sMIC-neutralizing antibody combined therapy markedly enhances NK cell homeostatic maintenance and function in sMIC-B16 tumor bearing mice. a. Therapy significantly increased total number of NK (CD3−NK1.1+) cells in the spleen or tumor-draining lymph nodes (dLN). b, c. Therapy significantly increased total number of NKG2D+ NK cells in the spleen and dLN. d, e. Therapy significantly increased NK cell activation shown by increased population of CD25+NK cells. f-i. Therapy significantly enhanced NK cell function shown by IFNγ production in response to PMA/Ionomycin in vitro re-stimulation. f and g, summary data of total IFNγ+NK cells. h. Summary of splenic NK cell cytotoxic function shown by CD107a degranulation when co-cultured with target RMA-S-RAE-1β cells (4:1 ratio). In all criteria, combined therapy elicited significantly better response than monotherapy. Similar results were obtained from sMIC-RM9 tumor bearing mice. *p < 0.05. **p < 0.01. Groups with statistically non-significances were not marked.
We further tested functional potential of NK cells from these mice by responses to ex vivo re-stimulation. NK cell IFNγ production in response to ex vivo PMA/I stimulation or degranulation shown by CD107a expression in response to NKG2D ligand-positive RMA-S-RAE-1β stimulation was significantly increased with monotherapy of B10G5 or ALT-803 (Figure 2f–2h). When B10G5 and ALT-803 were combined, therapy resulted in a further significant increase in the number of IFNγ-producing NK cells in response to PMA/I stimulation and the level of NK cell degranulation in response to RMA-S-RAE1β stimulation (Figure 2f–2h, Supplementary Figure 1c and 1d). These data clearly demonstrate that, in comparison to monotherapy, combined therapy significantly enhanced innate NK cell anti-tumor potential by further restoring homeostatic maintenance and function.
Combined therapy enhances functional potential of CD8 T cells
We next sought to determine the effects of combined therapy on adaptive immunity, primarily CD8+ T cells, as ALT-803 has been demonstrated to enhance CD8+ populations in vivo [48]. As shown in Figure 3a, in mice bearing B16-sMICB tumors, monotherapy of B10G5 or ALT-803 increased splenic CD8 numbers by 75% and 860%, respectively, compared to control treatment. With combined treatment, splenic CD8 numbers further increased significantly compared to monotherapy with B10G5 (p < 0.005) or ALT-803 (p = 0.05) (Figure 3a). The frequency of NKG2D+CD8+ T cells was significantly increased in both the spleen and dLN with monotherapy of B10G5 or ALT-803 compared to control IgG therapy (Figure 3b and 3c, Supplementary Figure S2a). With combined treatment, splenic NKG2D+CD8+ T cell populations and the intensity of NKG2D expression (measured by MFI) on NKG2D+CD8+ T cell were further markedly increased in comparison to monotherapy of B10G5 (p < 0.05) or ALT-803 (p < 0.01) (Figure 3b and 3c; Supplementary Figure S2a). Since NKG2D is only expressed by activated CD8+ T cells in mice [49], these data indicate a cooperation of B10G5 and ALT-803 in activating CD8+ T cells. This conclusion was further confirmed by examination of CD25+ CD8+ T cells populations (Figure 3d and 3e; Supplementary Figure S2b).
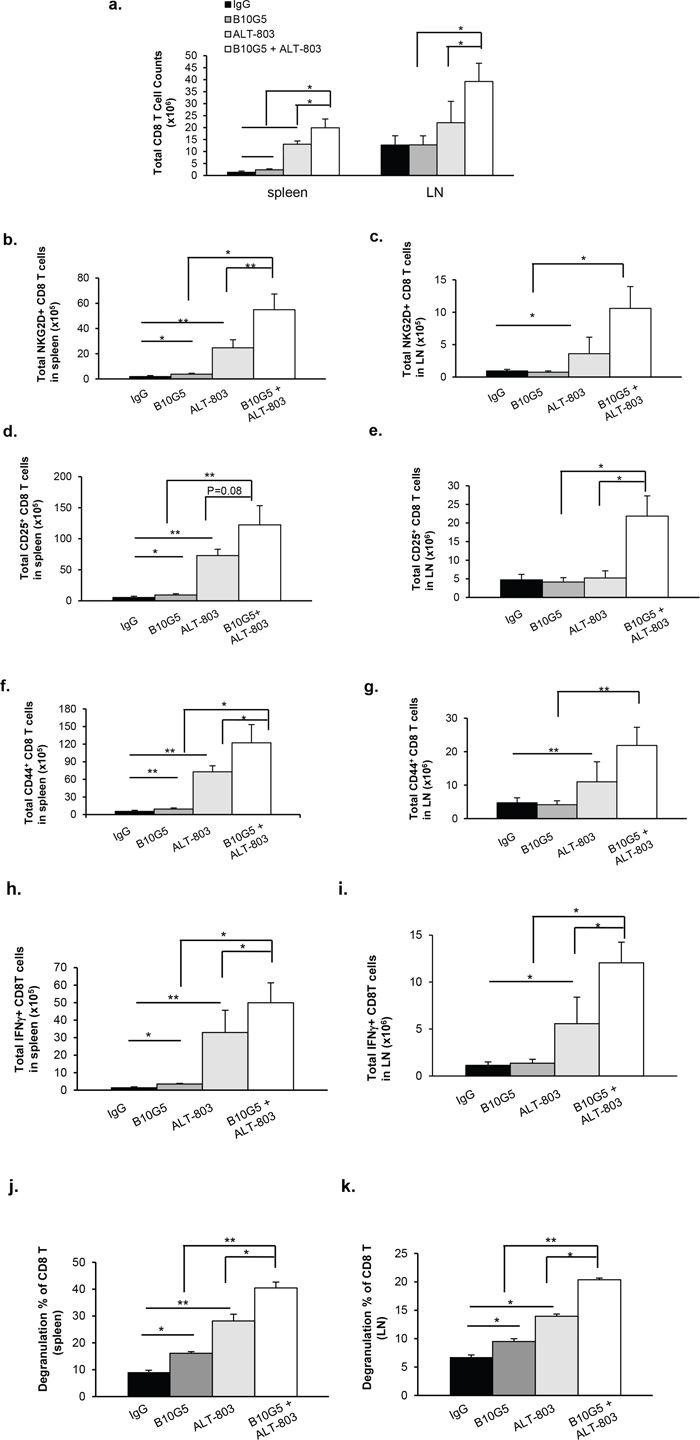
Figure 3: Combined therapy remarkably enhanced functional potential of CD8 T cells in sMICB-B16 tumor bearing mice. a. Combined therapy significantly increased total number of CD8 T cells in the spleen and lymph node. b, c. Combined therapy remarkably increased NKG2D+CD8 T cell populations in the spleen and dLN. d, e. Combined therapy significantly increased the number CD25+ CD8 T cells in the spleen and dLN, whereas monotherapy only elicited limited effect. f, g. Combined therapy significantly increased CD44+ CD8 T cell population, whereas monotherapy only elicited limited effect. h, i. Combined therapy significantly increased CD8 T cell anti-tumor potential shown by IFNγ-production in response to in vitro PMA/Ionomycin re-stimulation; whereas monotherapy only elicited limited effect. Similar results were obtained from sMIC-RM9 tumor bearing mice. j, k. Combined therapy significantly increased CD8 T cell cytotoxic potential shown by CD107a degranulation in response to tumor peptide antigen gp100 stimulation. Data were obtained at day 10 since treatment initiation. *p < 0.05. **p < 0.01. Groups with statistically non-significances were not marked.
Previous studies have shown that ALT-803 can increase CD44hi memory CD8+ T cell populations in tumor-bearing hosts [45]. Consistent with these studies, monotherapy with ALT-803 increased the population of memory-like CD44hi CD8+ T cells in both the spleen and dLN. B10G5 alone resulted in a significant increase in CD44hi CD8+ T cells in the spleen, but not in the dLN. Intriguingly, a remarkable increase in the population of CD44hi CD8+ T cells in both spleen and dLN was elicited as a result of the combined therapy (Figure 3f and 3g; Supplementary Figure S2c). These data demonstrate the cooperative effect of ALT-803 and B10G5 in generating or likelihood maintaining memory CD8+ T cells in the tumor-bearing host.
IL-15 has been shown to revive CD8+ T cells from an anergic state in tumor hosts [50–52]. sMIC has been shown to impair NKG2D expression and thus the function of CD8+ T cells [20]. These findings prompted us to evaluate whether there is a combined therapeutic effect on CD8+ T cell function. Consistent with other studies, ALT-803 therapy significantly increased the frequency and total number of splenic IFN-γ-producing CD8+ T cells in response to PMA and ionomycin re-stimulation in vitro (Figure 3h and 3i; Supplementary Figure S2d). To a lesser extent than ALT-803, B10G5 therapy also significantly increased the number of splenic IFN-γ-producing and CD8+ T cells. With the combined therapy of ALT-803 and B10G5, CD8+ T cells elicited a markedly enhanced response to re-stimulation in comparison to monotherapy (Figure 3h and 3i, Supplementary Figure S2d). Moreover, with combined therapy, CD8 T cells exhibited greater cytotoxic responses shown by increased CD107a degranulation with melanoma peptide antigen gp100 stimulation (Figure 3j and 3k, Supplementary Figure S2e). Together, these data have suggested that combined therapy of B10G5 and ALT-803 cooperatively not only can improve the generally machinery responsiveness of CD8 T cells in sMIC+ tumor-bearing mice, but also can enhance antigen-specific effector CD8 T cell responses.
Combination therapy of B10G5 and ALT-803 heightens CD4+ T cell anti-tumor potential
IL-15 has been shown to be important in driving TCR-dependent expansion of CD4+ T cells and maintaining memory function [53–56]. Thus, we sought to determine our combined therapeutic effect on CD4+ T cells. ALT-803 monotherapy had a trend of reduced total CD4 T cell population in the spleen (p = 0.049), however, combined therapy did not significantly impact total CD4 numbers in the spleen (Figure 4a). Neither monotherapy with B10G5 or ALT-803 nor combined therapy had a significant impact on total CD4+ T cell numbers in tumor-draining LNs (Figure 4a). Monotherapy of B10G5 or ALT-803 had differential impact on potentiating CD4+ T cell to Th1 responses in the spleen and dLN. As shown by IFNγ production in response to in vitro PMA/ionomycin re-stimulation, B10G5 therapy resulted in a better Th1 response in the spleen, whereas ALT-803 therapy remarkably potentiated CD4+ T cells to Th1 responses in the spleen and dLN (Figure 4b–4d).
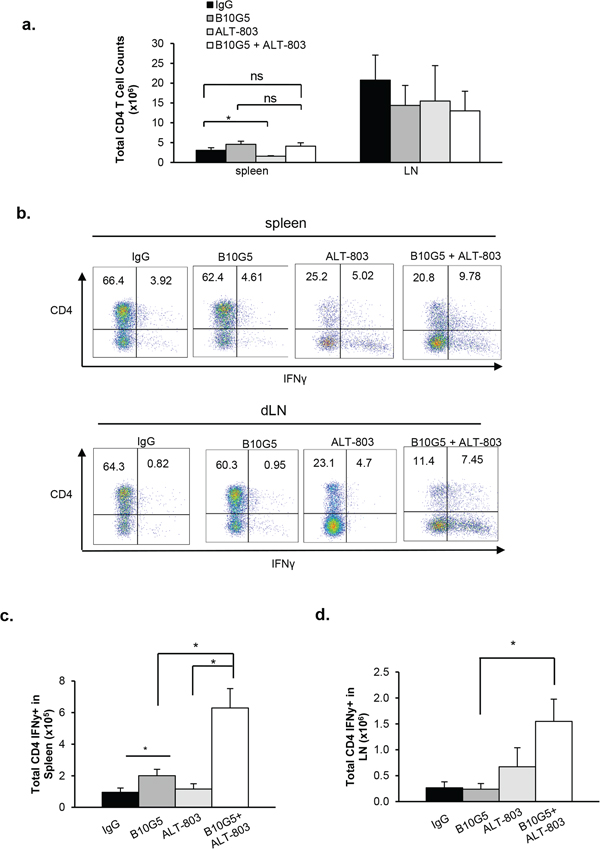
Figure 4: Combinatory therapy of B10G5 and ALT-803 significantly heightens CD4 T cell anti-tumor potential. a. Total number of CD4 T cells remains unchanged in the spleen and dLN in response to therapy, whether monotherapy or combined therapy. b. Representative dot plots demonstrating the impact of therapy on the frequency of IFNγ-producing CD4 T cells in the spleen and dLN in response to in vitro PMA/Ionomycin re-stimulation. c. Summary data showing the total number of IFNγ-producing CD4 T cells in response to therapy. Data were obtained at day 10 since treatment initiation. Similar results were obtained from sMIC-RM9 tumor bearing mice. *p < 0.05.
B10G5 and ALT-803 reduce MDSC populations in TILs
We have recently shown that tumor-derived sMIC can facilitate the expansion of immune suppressive MDSCs, generally defined as CD11b+Gr-1+ cells [22]. Consistent with our previous findings, neutralizing sMIC significantly reduced the population of MDSCs in RM9-sMICB tumor infiltrates. ALT-803 monotherapy did not significantly impact MDSC population in tumor infiltrates. However, the combination therapy of B10G5 and ALT-803 significantly reduced the MDSC population in tumor infiltrates with a trend of further reduction in comparison to B10G5 monotherapy (Figure 5). We also observed a similar trend toward decreased MDSC frequency in TILs isolated from mice bearing B16-sMICB tumors (data not shown). These results demonstrate that B10G5 can enhance the therapeutic efficacy of ALT-803 through cooperatively eliminating MDSC-mediated immune suppression.
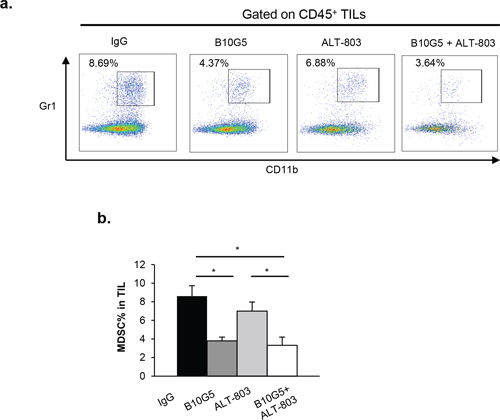
Figure 5: Combination therapy of B10G5 and ALT-803 reduces MDSC population in TILs. a. Representative dot plots showing percentage of MDSC, defined by CD11b+Gr-1+, in tumor-infiltrating lymphocytes (TILs) in RM9-sMICB tumors on day 14 of therapy. b. Quantification of MDSC percentages in TILs. One grams of tumor from different treatment group were digested to single cell suspension and used for quantifying MDSC percentage. Similar results were obtained from sMIC-B16 tumors. *p < 0.05.
Combined therapy cooperatively enhances STAT5 signaling
To further understand the potential cellular mechanisms whereby combined therapy of B10G5 and ALT-803 enhances NK functionality, purified mouse splenic NK cells were co-cultured with the murine prostate cancer cell line RM9 overexpressing sMICB (RM9-sMICB) in the presence of IgG, B10G5, ALT-803, or the combination of ALT-803 and B10G5. We analyzed the activation status of AKT and STAT5, the two essential molecules for cell survival and effector function. As shown in Figure 6a, the levels of AKT and STAT5 phosphorylation were both increased in the presence of ALT-803 or B10G5. However, the combination of ALT-803 and B10G5 only cooperatively further enhanced STAT5 phosphorylation at a significant level (Figure 6b). Given that STAT5 is essential for NK cell survival and maintaining memory CD8 T cells function, these data suggest that, at the cellular level, the combined therapy of ALT-803 and B10G5 cooperatively enhances NK cell survival and presumably CD8+ T cell memory function. Given that AKT is critical for cellular survival and NK cell effector function, our data also suggest that monotherapy of ALT-803 or B10G5 can respectively enhance cellular survival and NK cell functional potential although with a lesser magnitude than the combined therapy.
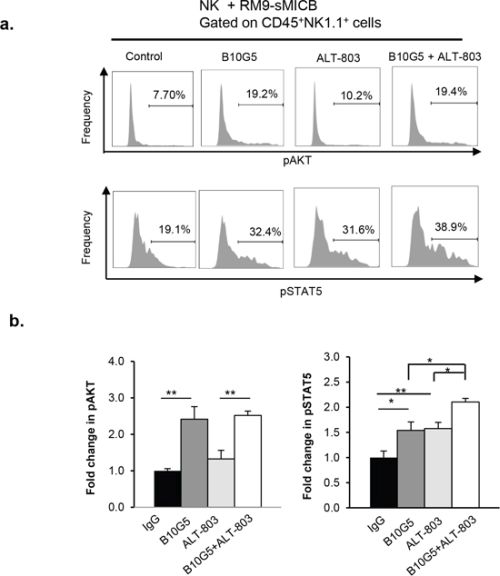
Figure 6: Combined therapy cooperatively sustains STAT5 activation in NK cells in the presence of tumor-derived soluble MIC. NK cells derived from spleens of B6/Rag1−/− mice were co-cultured in a 1:1 ratio with sMICB-RM9 cells in the presence of control mIgG, B10G5 (10 μg/ml), ALT-803 (71 ng/ml), or combination of B10G5 and ALT-803. At 36 h of co-culture, live cells were gated for CD45+NK1.1+ and analyzed for AKT and STAT5 phosphorylation with intracellular staining and flow cytometry analyses. No IL-2 was present in the co-culture. a. Histograms representatively show the phosphorylation status of AKT and STAT5 in NK cells in various culture conditions. b. Summary of data from five replicates of four independent experiments. *p < 0.05. **p < 0.01.
Depletion of NK cells eliminates the cooperative effect of B10G5 and ALT-803
As both sMIC and IL-15 pose significant effect (negative and positive respectively) on NK cell homeostasis and function, we thus addressed the impact of NK cells in the combined therapeutic effect. As shown in Figure 7, depletion of NK cells abolished the cooperative therapeutic effect of B10G5 and ALT-803, although monotherapy or combined therapy respectively exhibited significant inhibition in tumor growth and prolonged survival. Given that B10G5 or ALT-803 therapy alone has been shown to enhance cytotoxic CD8 T cell function (Figures 3 and 4), the respective therapy effect under the depletion of NK cells is reasonably anticipated.
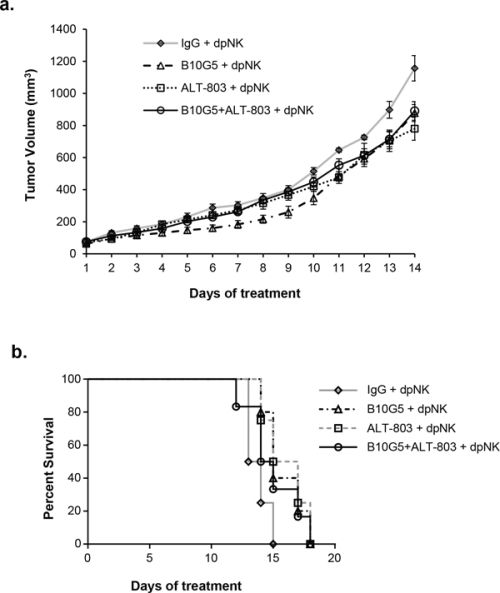
Figure 7: NK depletion impairs the cooperative therapeutic effect of B10G5 and ALT-803. NK cells were depleted by anti-NK1.1 antibody during therapy. a. Tumor growth curve. b. Kaplan-Meier survive curve. Tumor volume of 1000 mm3 was defined as survival endpoint. dpNK, depletion of NK cells.
DISCUSSION
In the current study, we demonstrated for the first time that the combination therapy of the IL-15 superagonist ALT-803 with a monoclonal antibody B10G5 targeting soluble MIC confers a significant survival benefit in comparison to monotherapy in sMIC+ tumor hosts. We showed that the combination therapy not only decreased the rate of tumor growth but also prolonged survival of animals bearing primary tumors in two independent sMIC+ syngeneic tumor models. Mechanistically, we further demonstrated that combined therapy cooperatively enhanced homeostasis and function of NK cells, functional potential of effector CD8+ and CD4+ T cells, and the expansion or sustainability of memory CD8 T cells. We further demonstrated in vitro that, in the presence of tumor-derived sMIC, ALT-803 and an antibody neutralizing sMIC cooperatively enhanced STAT5 signaling pathways, which are critical for maintenance of NK cell homeostasis, memory CD8+ T cells, and responses of effector CD8+ T cells in vivo [57–60]. Finally, we demonstrated that NK cells played a significant role in generating the cooperative therapeutic effect.
Human tumor-shed soluble MIC was considered as a viable target for immunotherapy, rationalized by its immune suppressive effect on NK cells and effector T cells and its ability to facilitate the expansion of MDSCs [10, 14, 22]. We have previously shown that MIC neutralization can decrease lung micrometastases of sMIC+ tumors and restore NK cell populations and proliferation [10]. In this study, we demonstrated that ALT-803 administration further increased the functional potential of NK, CD4+, and CD8+ cells in combination with a sMIC-neutralizing antibody. Our findings are consistent with published pre-clinical studies demonstrating the immune stimulatory effect of ALT-803. Xu et al. have shown that in models of murine multiple myeloma, ALT-803 therapy induced expansion of memory CD8+ T cells with upregulated NKG2D expression and increased secretion of IFN-γ; in addition, the effect of ALT-803 was shown to be independent of antigen-specific stimulation [45]. Gomes-Giacoia et al. showed that combination therapy of ALT-803 and intravesical Bacillus Calmette-Guerin (BCG) in a rat bladder cancer model resulted in an enhanced therapeutic effect compared to monotherapy. Gomes-Giacoia et al. further demonstrated that the enhanced combinatory therapeutic effect is associated with increased serum and urinary cytokine levels (IL-1α, IL-1β, and RANTES), increased NKG2D+ NK populations in the spleen and peripheral blood, and increased NK infiltration into the bladder [61]. These published studies strongly support the underlying mechanisms associated with the enhanced therapeutic effect of ALT-803 and a sMIC-neutralizing antibody as demonstrated in our current study.
Our data demonstrate that combined therapy not only increased NKG2D expression on CD8+ T cells but greatly increased the population of CD44hi memory CD8+ T cell subset. While the role of IL-15 and ALT-803 in generating and maintaining CD8+ memory populations has been well established [38, 45, 62–64], the understanding of NKG2D in memory CD8+ T cell homeostasis is limited. Studies by Andre et al. utilizing transgenic mice constitutively expressing the human NKG2D ligand MICA demonstrated that functional NKG2D was dispensable for the generation of memory CD8+ responses but necessary for effector functions of reactivated memory cells [65]. Other studies by Zloza et al. demonstrated that NKG2D was important for expansion and maintaining function of memory cells upon reactivation [66]. Zloza et al also demonstrated that IL-15 was crucial for rescue of memory resulting in enhanced cytokine secretion and cytolysis [66]. These limited investigations have suggested that NKG2D is critical for sustaining the immunity of activated memory CD8+ T cells. Provided that IL-15 has been well-demonstrated in upregulating NKG2D expression and priming NKG2D signaling pathways, it is reasonable to conclude that the increase in NKG2D function to enhance both effector and memory CD8+ T cell function confers a critical mechanism for the cooperative combinatory therapeutic effect.
We show that the combined therapy also cooperatively potentiated CD4 T cells to Th1 responses represented by a significant increase in IFN-γ production in response to re-stimulation, whereas monotherapy only elicited a limited and inconsistent impact in the spleen and dLN. Our observations are consistent with published studies demonstrating that NKG2D+CD4+ T cells are prone to Th1 cytokine secretion upon co-triggering of IL-15 and NKG2D [67]. Moreover, it has been shown that IL-15 requires IL-12 to cooperatively stimulate Th1 cytokine production by CD4+ T cells [68]. In this sense, because activated dendritic cells (DC) in the dLN are the major in vivo source for IL-12, it is thus anticipated that ALT-803 may promote Th1 responses by CD4+ T cells in the dLN as we have observed. NKG2D is normally absent in CD4+ T cells but is expressed by a rare population of effector CD4+ T cells in tumor hosts or virally infected individuals [67, 69]. It is an intriguing question how NKG2D signaling alone may regulate potentiation of CD4+ T cells to a Th1 response and warrants further investigation.
Tumor-associated MDSCs have been shown to be highly immune suppressive through negatively regulating the function of multiple lymphocyte subsets [70, 71]. MDSCs can inhibit the function of CD8+ and dendritic cells though arginase activity and induce the anergy of NK cells through membrane-bound TGFβ1 [72–75]. MDSCs can expand dramatically during tumor progression by tumor-secreted growth factors and cytokines [76, 77]. We have recently shown that tumor-shed sMIC can directly facilitate the expansion of MDSCs through upregulating STAT3 pathways [22]. IL-15 or its agonists have not been shown to play a direct role in regulating MDSC expansion or activity, thus it is anticipated that ALT-803 therapy may only have marginal effect in the population of MDSCs in tumor infiltrates. With B10G5 neutralizing sMIC, we show in this study a significant decrease in the population of MDSCs in tumor infiltrates than ALT-803 treatment alone. As tumor-derived growth factors have been shown to favor the expansion of MDSCs, it is possible that the reduction of MDSC in the tumor infiltrated with the combined therapy is the result of smaller tumor burden. However, since we have shown that neutralizing sMIC reduces the number of MDSC in tumor infiltrates [22], it is also conceivable that the reduction of MDSC may have alleviated its immune suppressive effects on both NK and CD8 T cells and thus cooperatively inhibited tumor growth.
IL-15 can activate several signaling cascades, including the PI3K/AKT/mTOR and STAT5 pathways in effector T and NK cells [78, 79], but the enhancement of signaling in the context of MIC neutralization has not been previously studied. STAT5 has been shown to be essential for NK maturation, peripheral maintenance, and function [57]. Phosphorylation of STAT5 correlates with levels of IL-15 trans-presentation [80]. However, recent studies have shown that IL-15-primed NK cells also require intact PI3K/AKT/mTOR signaling for optimal cytotoxicity, cytokine secretion, and proliferation [81]. These studies suggest that STAT5 and AKT are the two most essential pathways in maintaining the homeostasis and function of NK and memory CD8+ T cells. We show that the combination therapy cooperatively enhanced activation of STAT5 pathways and that monotherapy respectively enhanced activation of AKT pathways in NK cells. Although we did not extend our analyses to other cell types, one could anticipate similar outcomes in CD8+ T cells. However, it is interesting that the combined therapy only demonstrated limited impact on activating AKT pathways at cellular levels, presumably due to a feedback regulatory mechanism to control normal cellular homeostatic balances.
In summary, given the global immune suppressive nature of human tumor-derived sMIC and the cancer therapeutic potential of the IL-15 superagonist complex ALT-803, which is currently in clinical trials to treat advanced malignancies, our findings provide the rationale for combination therapy of an antibody targeting sMIC and ALT-803 to improve clinical outcomes.
MATERIALS AND METHODS
Mice and cell lines
Mice were bred and housed under specific pathogen-free conditions in the animal facility at the Medical University of South Carolina in accordance with institutional guidelines with approved IACUC protocols. All mice used in this study were male rPB-MICB mice on the B6 background as previously described [10]. Transgenic progeny were identified by PCR analysis of DNA extracted from tail biopsies using the forward primer specific for rPB (5′-ACAAGTGCATTTAGCCTCTCCAG TA-3′) and the reverse primer specific for MICB (5′-TGTGTCTTGGTCTTCATGGC-3′).
sMIC-expressing RM9-sMICB and B16F10-sMICB cell lines were developed by transduction with a IRES-GFP retroviral vector containing the construct for recombinant soluble MICB, as described previously [47]. sMIC+ cells were selected by puromycin and further by flow cytometry sorting for GFP-positive cells.
ALT-803 and B10G5 antibody
Generation and characterization of ALT-803 was previously described [48]. Briefly, ALT-803 is a complex consisted of IL-15 with the N72D mutation and the sushi-domain of soluble IL-15Ra fused to the human Fc region of IgG1. Generation of the anti-MIC mAb B10G5 was also previously described [10]. B10G5 is a non-blocking sMIC-neutralizing monoclonal antibody, which neutralizes free sMIC but does not block the interaction of MIC with the receptor NKG2D [14].
Tumor inoculation and in vivo experiments
RM9-sMICB and B16F10-sMICB cells were implanted subcutaneously into cohorts of syngeneic B6/MICB male mice (n = 8–10 per group) (4 × 105 cells/mouse) at ages 8–10 weeks old. When tumor volume reached approximately 75–100 mm3, animals were randomized into four therapy groups (n = 8–10 per group): 1) control mouse IgG (3.0 mg/kg BW); 2) anti-MIC mAb B10G5 (3.0 mg/kg BW); 3) ALT-803 (0.2 mg/kg BW); and 4) B10G5 and ALT-803. All therapies were given via I.P. routine twice weekly. In some studies, mice received anti-NK1.1 (PK136, BioXcell) antibody twice weekly via I.P. to deplete NK cell during therapy. Tumor volume of 1000 mm3 was defined as survival endpoint. After euthanization, spleens, tumor draining lymph nodes and tumors were harvested for analyses.
Ex vivo cytokine re-stimulation and cytotoxicity assay
Single-cell suspensions of splenocytes and tumor draining lymph nodes were stimulated at 37˚C for 6 hr with 50 ng/ml phorbol myristate acetate (PMA) and 500 ng/ml ionomycin. Cells were harvested and analyzed by flow cytometry for intracellular IFN-γ. For cytotoxicity assay, we use CD107a expression as a marker for NK and CD8 T cells degranulation in response to re-stimulation. Specifically, for CD8 T cell function, single cell suspension of bulked splenocytes or tumor draining lymph nodes from B16-sMICB mice was stimulated with 1 μg/ml of melanoma peptide antigen gp100 overnight with addition of PE-labeled anti-CD107a antibody in the culture. For NK cell cytotoxicity assay, NK cells isolated from the spleens were incubated with MHC-I-deficient NKG2D ligand RAE-1β expression RAM-S-RAE-1β cells at 4:1 ratio for 5 h with addition of PE-labeled anti-CD107a antibody in the culture. 1 μM Golgi Plug was added to the culture 4 h before flow cytometry assay.
In vitro co-culture assay
NK cells were generated from spleens of Rag1−/− mice as described elsewhere. Briefly, single cell suspensions of splenocytes from Rag1−/− mice were seeded at 37ºC for 2 h to remove adherent cells. Non-adherent cells were collected and cultured in 1000U/ml of IL-2 for 3–4 days before being harvested for experiments. Cells were > 95% CD3−NK1.1+ at harvest (data not shown). NK cells were co-cultured with RM9-sMICB cells at 1:1 ratio for up to 36 hours in the presence of control mIgG (10 μg/ml), anti-MICB mAb B10G5 (10 μg/ml), ALT-803 (71 ng/ml), or a combination of B10G5 and ALT-803. Of note, no IL-2 was present in the co-culture. Suspension cells were harvested for analyzing phosphorylation status of pAKT and pSTAT5 in NK cells using flow cytometry by intracellular staining. NK cells were identified by surface CD45 and NK1.1 double positive staining.
Flow cytometry analysis
Single-cell suspensions were incubated on ice for 30 minutes with a combination of antibodies specific to cell surface markers for identification lymphocyte subsets. These antibodies are: APC- or PE-Cy7-conjugated anti-NK1.1 (clone PK136), APC-Cy7-conjugated anti-CD3ε (clone 145–2C11), FITC-conjugated anti-CD8a (clone 53–6.7), Alexa Fluor 700-conjugated anti-CD4 (clone RM4–5), PE-conjugated anti-NKG2D (clone CX5), PE-conjugated anti-CD44 (clone IM7), PECy7-conjugated anti-CD25 (clone PC61), FITC-conjugated anti-CD11b (clone M1/70), and/or PE-conjugated anti-Gr1 (clone 1A8) (BD Biosciences). For NK cell receptors, fluorochrome-conjugated NKp46, Ly49A, Ly49C/I/F/H, NKG2A/C/E, CD16 were all from eBiosciences. For intracellular staining, cells were stained with surface markers followed by fixation and permeabilization with BD Perm/Fix kits and antibodies specific to intracellular molecules. Fluorochrome-conjugated antibodies specific to IFN-γ, phospho-AKT (pS473), and phospho-STAT5 (pY694) were all from BD Biosciences. Cells were analyzed using the BD Fortessa. Data were analyzed using the FlowJo software (Tree Star).
Statistics
All statistical data were expressed as mean ± SEM. Difference between means of populations was compared by standard Student’s t test using One-way ANOVA. Survival was determined via Kaplan-Meier analysis with comparison of curves using the Mantel-Haenszel log-rank test. A P value of 0.05 or less was considered significant. GraphPad Prism software was used for all analyses.
ACKNOWLEDGMENTS
This work was supported by NIH-NCI grant 1R01CA149405 and A. David Mazzone - Prostate Cancer Foundation Challenge Award (to J.Wu) and in part supported by the Flow Cytometry Core Facility Shared Resource, Hollings Cancer Center, Medical University of South Carolina (P30 CA138313). We thank Dr. James Cook for critical reading of the manuscript.
CONFLICTS OF INTEREST
F. Basher and J. Wu declare no conflict of interest. Hing C. Wong and Emily K. Jeng are the employees and stockholders of Altor biosciences Corporation who developed ALT-803.
REFERENCES
1. Nausch N, Cerwenka A. NKG2D ligands in tumor immunity. Oncogene 2008;27:5944–58.
2. Gasser S, Orsulic S, Brown EJ, Raulet DH. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature 2005;436:1186–90.
3. Long EO. Versatile signaling through NKG2D. Nat Immunol 2002;3:1119–20.
4. Wu J, Groh V, Spies T. T cell antigen receptor engagement and specificity in the recognition of stress-inducible MHC class I-related chains by human epithelial gamma delta T cells. J Immunol 2002;169:1236–40.
5. Diefenbach A, Jensen ER, Jamieson AM, Raulet DH. Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature 2001;413:165–71.
6. Cerwenka A, Baron JL, Lanier LL. Ectopic expression of retinoic acid early inducible-1 gene (RAE-1) permits natural killer cell-mediated rejection of a MHC class I-bearing tumor in vivo. Proc Natl Acad Sci U S A 2001;98:11521–6.
7. Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, et al. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 1999;285:727–9.
8. Smyth MJ, Swann J, Cretney E, Zerafa N, Yokoyama WM, Hayakawa Y. NKG2D function protects the host from tumor initiation. J Exp Med 2005;202:583–8.
9. Guerra N, Tan YX, Joncker NT, Choy A, Gallardo F, Xiong N, et al. NKG2D-deficient mice are defective in tumor surveillance in models of spontaneous malignancy. Immunity 2008;28:571–80.
10. Liu G, Lu S, Wang X, Page ST, Higano CS, Plymate SR, et al. Perturbation of NK cell peripheral homeostasis accelerates prostate carcinoma metastasis. J Clin Invest 2013;123:4410–22.
11. Groh V, Bahram S, Bauer S, Herman A, Beauchamp M, Spies T. Cell stress-regulated human major histocompatibility complex class I gene expressed in gastrointestinal epithelium. Proc Natl Acad Sci U S A 1996;93:12445–50.
12. Groh V, Steinle A, Bauer S, Spies T. Recognition of stress-induced MHC molecules by intestinal epithelial gammadelta T cells. Science 1998;279:1737–40.
13. Pende D, Rivera P, Marcenaro S, Chang CC, Biassoni R, Conte R, et al. Major histocompatibility complex class I-related chain A and UL16-binding protein expression on tumor cell lines of different histotypes: analysis of tumor susceptibility to NKG2D-dependent natural killer cell cytotoxicity. Cancer Res 2002;62:6178–86.
14. Lu S, Zhang J, Liu D, Li G, Staveley-O’Carroll KF, Li Z, et al. Nonblocking Monoclonal Antibody Targeting Soluble MIC Revamps Endogenous Innate and Adaptive Antitumor Responses and Eliminates Primary and Metastatic Tumors. Clin Cancer Res 2015.
15. Raffaghello L, Prigione I, Airoldi I, Camoriano M, Levreri I, Gambini C, et al. Downregulation and/or release of NKG2D ligands as immune evasion strategy of human neuroblastoma. Neoplasia 2004;6:558–68.
16. Coudert JD, Zimmer J, Tomasello E, Cebecauer M, Colonna M, Vivier E, et al. Altered NKG2D function in NK cells induced by chronic exposure to NKG2D ligand-expressing tumor cells. Blood 2005;106:1711–7.
17. Oppenheim DE, Roberts SJ, Clarke SL, Filler R, Lewis JM, Tigelaar RE, et al. Sustained localized expression of ligand for the activating NKG2D receptor impairs natural cytotoxicity in vivo and reduces tumor immunosurveillance. Nat Immunol 2005;6:928–37.
18. Holdenrieder S, Stieber P, Peterfi A, Nagel D, Steinle A, Salih HR. Soluble MICB in malignant diseases: analysis of diagnostic significance and correlation with soluble MICA. Cancer Immunol Immunother 2006;55:1584–9.
19. Holdenrieder S, Stieber P, Peterfi A, Nagel D, Steinle A, Salih HR. Soluble MICA in malignant diseases. Int J Cancer 2006;118:684–7.
20. Groh V, Wu J, Yee C, Spies T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature 2002;419:734–8.
21. Wu JD, Higgins LM, Steinle A, Cosman D, Haugk K, Plymate SR. Prevalent expression of the immunostimulatory MHC class I chain-related molecule is counteracted by shedding in prostate cancer. J Clin Invest 2004;114:560–8.
22. Xiao G, Wang X, Sheng J, Lu S, Yu X, Wu JD. Soluble NKG2D ligand promotes MDSC expansion and skews macrophage to the alternatively activated phenotype. J Hematol Oncol 2015;8:13.
23. Waldmann TA. The biology of interleukin-2 and interleukin-15: implications for cancer therapy and vaccine design. Nat Rev Immunol 2006;6:595–601.
24. Fehniger TA, Cooper MA, Caligiuri MA. Interleukin-2 and interleukin-15: immunotherapy for cancer. Cytokine Growth Factor Rev 2002;13:169–83.
25. Koka R, Burkett PR, Chien M, Chai S, Chan F, Lodolce JP, et al. Interleukin (IL)-15R[alpha]-deficient natural killer cells survive in normal but not IL-15R[alpha]-deficient mice. J Exp Med 2003;197:977–84.
26. Cheever MA. Twelve immunotherapy drugs that could cure cancers. Immunol Rev 2008;222:357–68.
27. Wu J. NKG2D Ligands in Cancer Immunotherapy: Target or Not? Austin J Clin Immunol 2014;1:2.
28. Ranson T, Vosshenrich CA, Corcuff E, Richard O, Muller W, Di Santo JP. IL-15 is an essential mediator of peripheral NK-cell homeostasis. Blood 2003;101:4887–93.
29. Keppel MP, Yang L, Cooper MA. Murine NK cell intrinsic cytokine-induced memory-like responses are maintained following homeostatic proliferation. J Immunol 2013;190:4754–62.
30. Zhao YM, French AR. Two-compartment model of NK cell proliferation: insights from population response to IL-15 stimulation. J Immunol 2012;188:2981–90.
31. Kennedy MK, Glaccum M, Brown SN, Butz EA, Viney JL, Embers M, et al. Reversible defects in natural killer and memory CD8 T cell lineages in interleukin 15-deficient mice. J Exp Med 2000;191:771–80.
32. Vosshenrich CA, Ranson T, Samson SI, Corcuff E, Colucci F, Rosmaraki EE, et al. Roles for common cytokine receptor gamma-chain-dependent cytokines in the generation, differentiation, and maturation of NK cell precursors and peripheral NK cells in vivo. J Immunol 2005;174:1213–21.
33. Elpek KG, Rubinstein MP, Bellemare-Pelletier A, Goldrath AW, Turley SJ. Mature natural killer cells with phenotypic and functional alterations accumulate upon sustained stimulation with IL-15/IL-15Ralpha complexes. Proc Natl Acad Sci U S A 2010;107:21647–52.
34. Meresse B, Chen Z, Ciszewski C, Tretiakova M, Bhagat G, Krausz TN, et al. Coordinated induction by IL15 of a TCR-independent NKG2D signaling pathway converts CTL into lymphokine-activated killer cells in celiac disease. Immunity 2004;21:357–66.
35. Tang F, Sally B, Ciszewski C, Abadie V, Curran SA, Groh V, et al. Interleukin 15 primes natural killer cells to kill via NKG2D and cPLA2 and this pathway is active in psoriatic arthritis. PLoS One 2013;8:e76292.
36. Roberts AI, Lee L, Schwarz E, Groh V, Spies T, Ebert EC, et al. NKG2D receptors induced by IL-15 costimulate CD28-negative effector CTL in the tissue microenvironment. J Immunol 2001;167:5527–30.
37. Horng T, Bezbradica JS, Medzhitov R. NKG2D signaling is coupled to the interleukin 15 receptor signaling pathway. Nat Immunol 2007;8:1345–52.
38. Weng NP, Liu K, Catalfamo M, Li Y, Henkart PA. IL-15 is a growth factor and an activator of CD8 memory T cells. Ann N Y Acad Sci 2002;975:46–56.
39. Zhang X, Sun S, Hwang I, Tough DF, Sprent J. Potent and selective stimulation of memory-phenotype CD8+ T cells in vivo by IL-15. Immunity 1998;8:591–9.
40. Kanegane H, Tosato G. Activation of naive and memory T cells by interleukin-15. Blood 1996;88:230–5.
41. Alpdogan O, van den Brink MR. IL-7 and IL-15: therapeutic cytokines for immunodeficiency. Trends Immunol 2005;26:56–64.
42. Mueller YM, Petrovas C, Bojczuk PM, Dimitriou ID, Beer B, Silvera P, et al. Interleukin-15 increases effector memory CD8+ t cells and NK Cells in simian immunodeficiency virus-infected macaques. J Virol 2005;79:4877–85.
43. Zhu X, Marcus WD, Xu W, Lee HI, Han K, Egan JO, et al. Novel human interleukin-15 agonists. J Immunol 2009;183:3598–607.
44. Wong HC, Jeng EK, Rhode PR. The IL-15-based superagonist ALT-803 promotes the antigen-independent conversion of memory CD8 T cells into innate-like effector cells with antitumor activity. Oncoimmunology 2013;2:e26442.
45. Xu W, Jones M, Liu B, Zhu X, Johnson CB, Edwards AC, et al. Efficacy and mechanism-of-action of a novel superagonist interleukin-15: interleukin-15 receptor alphaSu/Fc fusion complex in syngeneic murine models of multiple myeloma. Cancer Res 2013;73:3075–86.
46. AltorBioscienceCorporation. A Study of ALT-803 in Patients With Relapsed or Refractory Multiple Myeloma.
47. Wu JD, Atteridge CL, Wang X, Seya T, Plymate SR. Obstructing shedding of the immunostimulatory MHC class I chain-related gene B prevents tumor formation. Clin Cancer Res 2009;15:632–40.
48. Han KP, Zhu X, Liu B, Jeng E, Kong L, Yovandich JL, et al. IL-15:IL-15 receptor alpha superagonist complex: high-level co-expression in recombinant mammalian cells, purification and characterization. Cytokine 2011;56:804–10.
49. Ehrlich LI, Ogasawara K, Hamerman JA, Takaki R, Zingoni A, Allison JP, et al. Engagement of NKG2D by cognate ligand or antibody alone is insufficient to mediate costimulation of human and mouse CD8+ T cells. J Immunol 2005;174:1922–31.
50. King JW, Thomas S, Corsi F, Gao L, Dina R, Gillmore R, et al. IL15 can reverse the unresponsiveness of Wilms’ tumor antigen-specific CTL in patients with prostate cancer. Clin Cancer Res 2009;15:1145–54.
51. Oelert T, Papatriantafyllou M, Pougialis G, Hammerling GJ, Arnold B, Schuler T. Irradiation and IL-15 promote loss of CD8 T-cell tolerance in response to lymphopenia. Blood 2010;115:2196–202.
52. Teague RM, Sather BD, Sacks JA, Huang MZ, Dossett ML, Morimoto J, et al. Interleukin-15 rescues tolerant CD8+ T cells for use in adoptive immunotherapy of established tumors. Nat Med 2006;12:335–41.
53. Van Belle TL, Dooms H, Boonefaes T, Wei XQ, Leclercq G, Grooten J. IL-15 augments TCR-induced CD4+ T cell expansion in vitro by inhibiting the suppressive function of CD25 High CD4+ T cells. PLoS One 2012;7:e45299.
54. Zhu X, Wang M, Mavi P, Rayapudi M, Pandey AK, Kaul A, et al. Interleukin-15 expression is increased in human eosinophilic esophagitis and mediates pathogenesis in mice. Gastroenterology 2010;139:182–93 e7.
55. Ruckert R, Brandt K, Ernst M, Marienfeld K, Csernok E, Metzler C, et al. Interleukin-15 stimulates macrophages to activate CD4+ T cells: a role in the pathogenesis of rheumatoid arthritis? Immunology 2009;126:63–73.
56. Purton JF, Tan JT, Rubinstein MP, Kim DM, Sprent J, Surh CD. Antiviral CD4+ memory T cells are IL-15 dependent. J Exp Med 2007;204:951–61.
57. Eckelhart E, Warsch W, Zebedin E, Simma O, Stoiber D, Kolbe T, et al. A novel Ncr1-Cre mouse reveals the essential role of STAT5 for NK-cell survival and development. Blood 2011;117:1565–73.
58. Mitchell DM, Williams MA. Disparate roles for STAT5 in primary and secondary CTL responses. J Immunol 2013;190:3390–8.
59. Tripathi P, Kurtulus S, Wojciechowski S, Sholl A, Hoebe K, Morris SC, et al. STAT5 is critical to maintain effector CD8+ T cell responses. J Immunol 2010;185:2116–24.
60. Hand TW, Cui W, Jung YW, Sefik E, Joshi NS, Chandele A, et al. Differential effects of STAT5 and PI3K/AKT signaling on effector and memory CD8 T-cell survival. Proc Natl Acad Sci U S A 2010;107:16601–6.
61. Gomes-Giacoia E, Miyake M, Goodison S, Sriharan A, Zhang G, You L, et al. Intravesical ALT-803 and BCG treatment reduces tumor burden in a carcinogen induced bladder cancer rat model; a role for cytokine production and NK cell expansion. PLoS One 2014;9:e96705.
62. Tanchot C, Lemonnier FA, Perarnau B, Freitas AA, Rocha B. Differential requirements for survival and proliferation of CD8 naive or memory T cells. Science 1997;276:2057–62.
63. Judge AD, Zhang X, Fujii H, Surh CD, Sprent J. Interleukin 15 controls both proliferation and survival of a subset of memory-phenotype CD8(+) T cells. J Exp Med 2002;196:935–46.
64. Schluns KS, Williams K, Ma A, Zheng XX, Lefrancois L. Cutting edge: requirement for IL-15 in the generation of primary and memory antigen-specific CD8 T cells. J Immunol 2002;168:4827–31.
65. Andre MC, Sigurdardottir D, Kuttruff S, Pommerl B, Handgretinger R, Rammensee HG, et al. Impaired tumor rejection by memory CD8 T cells in mice with NKG2D dysfunction. Int J Cancer 2012;131:1601–10.
66. Zloza A, Kohlhapp FJ, Lyons GE, Schenkel JM, Moore TV, Lacek AT, et al. NKG2D signaling on CD8(+) T cells represses T-bet and rescues CD4-unhelped CD8(+) T cell memory recall but not effector responses. Nat Med 2012;18:422–8.
67. Romero AI, Chaput N, Poirier-Colame V, Rusakiewicz S, Jacquelot N, Chaba K, et al. Regulation of CD4(+)NKG2D(+) Th1 cells in patients with metastatic melanoma treated with sorafenib: role of IL-15Ralpha and NKG2D triggering. Cancer Res 2014;74:68–80.
68. Niedbala W, Wei X, Liew FY. IL-15 induces type 1 and type 2 CD4+ and CD8+ T cells proliferation but is unable to drive cytokine production in the absence of TCR activation or IL-12 / IL-4 stimulation in vitro. Eur J Immunol 2002;32:341–7.
69. Jensen H, Folkersen L, Skov S. Regulation and gene expression profiling of NKG2D positive human cytomegalovirus-primed CD4+ T-cells. PLoS One 2012;7:e41577.
70. Lindau D, Gielen P, Kroesen M, Wesseling P, Adema GJ. The immunosuppressive tumour network: myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology 2013;138:105–15.
71. Khaled YS, Ammori BJ, Elkord E. Myeloid-derived suppressor cells in cancer: recent progress and prospects. Immunol Cell Biol 2013;91:493–502.
72. Schouppe E, Van Overmeire E, Laoui D, Keirsse J, Van Ginderachter JA. Modulation of CD8(+) T-cell activation events by monocytic and granulocytic myeloid-derived suppressor cells. Immunobiology 2013;218:1385–91.
73. Rodriguez PC, Ochoa AC. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: mechanisms and therapeutic perspectives. Immunol Rev 2008;222:180–91.
74. Ostrand-Rosenberg S, Sinha P, Beury DW, Clements VK. Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Semin Cancer Biol 2012;22:275–81.
75. Li H, Han Y, Guo Q, Zhang M, Cao X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta 1. J Immunol 2009;182:240–9.
76. Serafini P, De Santo C, Marigo I, Cingarlini S, Dolcetti L, Gallina G, et al. Derangement of immune responses by myeloid suppressor cells. Cancer Immunol Immunother 2004;53:64–72.
77. Serafini P, Borrello I, Bronte V. Myeloid suppressor cells in cancer: recruitment, phenotype, properties, and mechanisms of immune suppression. Semin Cancer Biol 2006;16:53–65.
78. Ma A, Koka R, Burkett P. Diverse functions of IL-2, IL-15, and IL-7 in lymphoid homeostasis. Annu Rev Immunol 2006;24:657–79.
79. Kovanen PE, Leonard WJ. Cytokines and immunodeficiency diseases: critical roles of the gamma(c)-dependent cytokines interleukins 2, 4, 7, 9, 15, and 21, and their signaling pathways. Immunol Rev 2004;202:67–83.
80. Lee GA, Liou YH, Wang SW, Ko KL, Jiang ST, Liao NS. Different NK cell developmental events require different levels of IL-15 trans-presentation. J Immunol 2011;187:1212–21.
81. Nandagopal N, Ali AK, Komal AK, Lee SH. The Critical Role of IL-15-PI3K-mTOR Pathway in Natural Killer Cell Effector Functions. Front Immunol 2014;5:187.