
INTRODUCTION
Colon cancer is one of the major causes of cancer-related deaths in the world [1]. Colon cancer has an estimated incidence of over 1,000,000 new cases annually worldwide. Almost one of three patients with colon cancer dies from the disease. Colon cancer also more often affects people of well-developed countries in comparison to less developed countries [2]. Several epidemiological and laboratory studies have demonstrated the association of colon cancer with environmental factors such as western style dietary habits, low fiber intake, tobacco-smoking, high fat diet, low calcium/micronutrient intake and lack of physical activities [3]. However, there are also negative studies on fat, fiber and calcium in colon cancer development [4]. In addition, colon cancer is a heterogeneous disease. There is evidence that lifestyle factors like diet can modulate the course of this disease [5].
Constitutive activation of Nuclear Factor-κB (NF-κB) has been described in a great number of tumors including colon tumors. Human colon cancer cell lines and tumor samples as well as the nuclei of stromal macrophages in sporadic adenomatous polyps showed increased NF-κB activity [6, 7]. The inhibition of NF-κB has shown remarkable anti-tumor activity in preclinical and clinical studies [8]. NF-κB signaling has a pivotal role in cancer development and progression. NF-κB provides a mechanistic link between inflammation and cancer, and is a major factor controlling the ability of both pre-neoplastic and malignant cells to resist apoptosis-based tumor-surveillance mechanisms [9]. NF-κB might also regulate tumor angiogenesis and invasiveness, and the signaling pathways that adjust its activation provide attractive targets for new chemo-preventive and chemotherapeutic approaches [10]. A number of reports have shown that NF-κB is inhibited by apoptosis-inducing agents in human colon cancer cells [11, 12]. NF-κB acts as a cell survival factor through its regulatory role in the expression of an array of apoptotic (caspase-3 and Bax), antiapoptotic (Bcl-2 and IAP family), and cell proliferation genes (cyclooxygenase-2 and cyclins) [13]. Therefore, inhibition of NF-κB by chemotherapeutics is intended as a new strategy to eliminate cancerous cells through induction of apoptosis.
Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) induces apoptosis in cancer cells without toxicity to normal cells. It is well known that TRAIL binds to DRs, TRAIL-R1 (DR4) and TRAIL-R2 (DR5) expressed on cancer cell surface and activates apoptotic pathways. When DRs bind to their ligands, the death domains recruit the intracellular adaptor protein (Fas-associated death domain protein) which results in the activation of capase-8 that leads to the activation of downstream caspases, including capase-3, caspase-9 and Bax [14]. It is documented that the resistance of cancer cells is related to the activation of NF-κB and down-regulation of DRs; thus, down-regulation of NF-κB and up-regulation of DRs are implicated in the development of new anti-cancer treatment for chemo-resistant cancer cells. It has been demonstrated that up-regulation of DRs and inactivation of NF-κB are reciprocally associated in chemoresistance. Many chemotherapeutic agents or natural compounds such as garicinol, zerumbone, quercetin, snake venom toxin inhibit cancer cell growth via overexpression of death receptor associated with down-regulation of NF-κB [15–18]. In agreement with this notion, it is possible that BV could overcome resistance to TRAIL through inactivation of NF-κB and activation of DRs.
BV contains a variety of different peptides, including melittin (a major component of BV), apamin, adolapin, and mast cell degranulating peptide [19, 20]. BV has anti-cancer activity [21]. Melittin, a major polypeptide of BV, is thought to function as a lytic agent that has been used traditionally against chronic inflammation and cancer [22] and also used for the therapeutic agent of arthritis, rheumatism, atherosclerosis, and cancer in traditional medicine [23, 24]. Melittin inhibited the DNA-binding and transcriptional activity of NF-κB, an important transcriptional factor regulating inflammatory gene expression, through the suppression of IκB phosphorylation and the decrease in the translocation of the p50 and p65 subunits of NF-κB [25]. However, the anti-cancer effect of BV on colon cancer cells has not been studied yet. In this study, we investigated anti-cancer effects and possible mechanisms of BV on colon cancer cells. In particular, we determined the capacity of the BV induced apoptotic effect through activation of DRs and suppression of NF-κB.
RESULTS
Effect of BV on the growth of colon cancer cells
To evaluate the effect of BV on the growth of colon cancer cells, we analyzed cell viability using the MTT assay. BV (0–10 μg/ml) inhibited the growth of human colon cancer cells, and the IC50 values of HCT116 (Figure 1A) and SW480 (Figure 1B) were 8.2 and 7.8 μg/ml, respectively. We evaluated changes in the morphology of human colon cancer cells using phasecontrast microscopy. BV-treated SW480 cells for 48 h showed rounded morphology and tended to detach from substratum. However, BV-treated HCT116 cells for 48 h presented with membrane blebbing, cell shrinkage, cytoplasmic increasing density and irregularity in shape. (0–10 μg/ml) To evaluate the effect of BV on the colon epithelial normal cells, we treated FHC with BV (0–10 μg/ml) for 24 h, and BV showed no toxicity at all (Figure 1C). To further demonstrate the inhibitory effect of BV on cancer cell growth, we performed the soft agar colony formation assay. The number of colonies was concentration-dependently decreased by BV (Figure 1D), and the concentration of BV for inhibition of colony formation was similar to the cell growth inhibition. To determine whether the inhibition of cell growth by BV treatment was due to induction of apoptosis, we evaluated the changes in colon cancer cells by using DAPI staining followed by TUNEL assays, and then the double labeled cells were analyzed by fluorescence microscope. The cells were treated with BV (0–5 μg/ml) for 24 h. DAPI-stained TUNEL-positive cells were concentration-dependently increased and the highest concentration of BV (5 μg/ml) caused most of cells TUNEL-positive, and apoptosis rates were 91.35% in HCT116 cells (Figure 2A) and 87.23% in SW480 cells (Figure 2B). These results demonstrated that BV treatment strongly induced apoptosis in colon cancer cells.
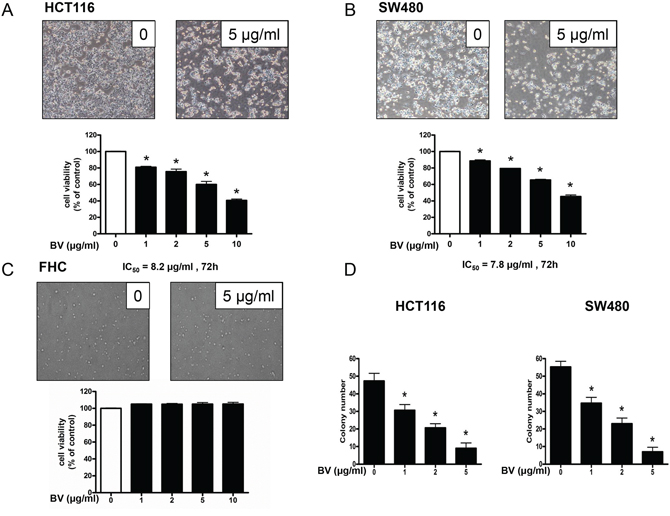
Figure 1: Effect of bee venom (BV) on the growth of colon cancer cells. A, B & C. Effect of BV on the growth of HCT116 and SW480 colon cancer cells and FHC colon epithelial normal cells. Cell viability was determined by MTT assay. D. Effect of BV on the colony formation in HCT116 and SW480 cells. Data was expressed as the mean ± S.D. of three experiments. *p < 0.05 indicates statistically significant differences from control group.
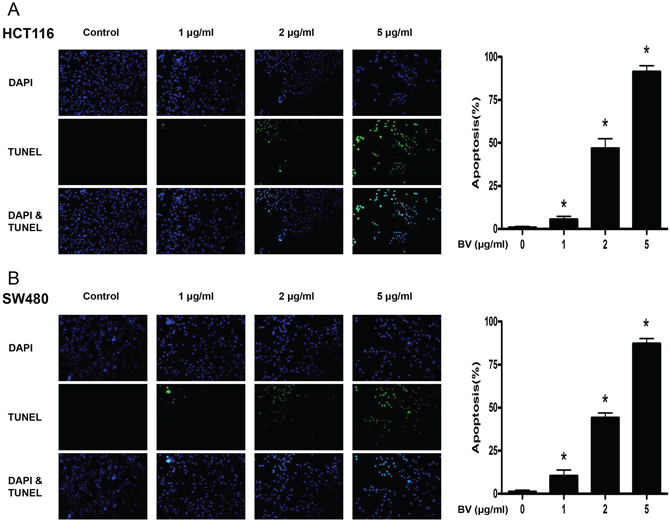
Figure 2: Effect of BV on apoptotic cell death. A. Apoptotic cell death of HCT116. B. Apoptotic cell death of SW480. Colon cancer cells were treated with BV (0–5 μg/ml) for 24 h, and then labeled with DAPI and TUNEL solution. Total number of cells in a given area was determined by using DAPI nuclear staining (fluorescent microscope). A green color in the fixed cells marks TUNEL-labeled cells. Apoptotic index was determined as the DAPI-stained TUNEL-positive cell number / total DAPI-stained cell number × 100%. Data was expressed as the mean ± S.D. of three experiments. *p < 0.05 indicates statistically significant differences from control cells.
Effect of BV on the expression of apoptosis regulatory proteins
To figure out the relationship between the induction of apoptosis and the expression of apoptosis regulatory protein by BV, the expression of apoptosis related intrinsic pathway (Figure 3A) and extrinsic pathway (Figure 3B) proteins was investigated. With the treatment of BV (0–5 μg/ml) in HCT116 and in SW480 colon cancer cells, we found that the expression of pro-apoptotic proteins such as Bax, cleaved caspase-3, cleaved caspase-8 and cleaved caspase-9 as well as the expression of DRs like DR3, DR4, DR5 and Fas was increased in a concentration dependent manner. However, the expression of anti-apoptotic protein Bcl-2 was decreased.
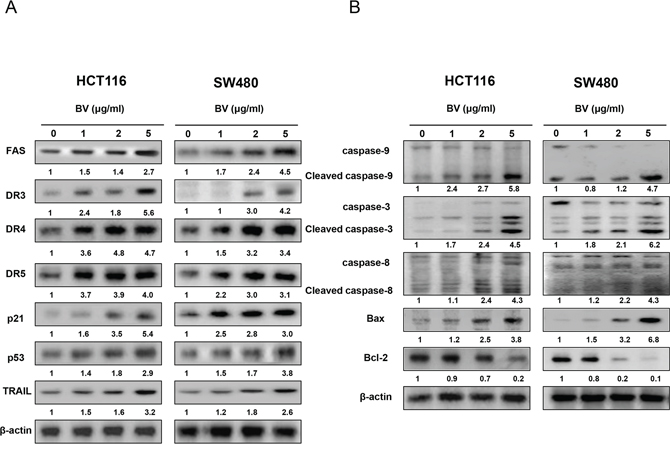
Figure 3: Effect of BV on the expression of apoptosis regulatory proteins. A. Expression of apoptosis regulatory proteins related intrinsic pathway was determined by Western blotting analysis with antibodies against capase-3, caspase-8, capase-9, Bax, Bcl-2 and β-actin (internal control). B. Extrinsic pathway was determined by Western blotting with antibodies against Fas, DR3, DR4, DR5, TRAIL, p21, p53 and β-actin (internal control). Values under Western band indicate the density of band. Each band is representative for three experiments.
Effect of BV on NF-κB activation
NF-κB plays a significant role in colon cancer cell growth. To investigate whether BV inactivates NF-κB, we performed EMSA for detecting DNA binding activity of NF-κB. We found that BV-untreated colon cancer cells showed highly constituted activation of NF-κB in both colon cancer cells. However, BV treatment concentration dependently inhibited DNA binding activity of NF-κB (Figure 4A). Agreed with the inhibition of NF-κB, cytosolic phosphorylation of IκB as well as the nucleus translocation of p50 and p65 was inhibited by BV treatment in both colon cancer cells (Figure 4B). The band of NF-κB was supershifted by p50 specific antibody in HCT116 colon cancer cells (Figure 4C).
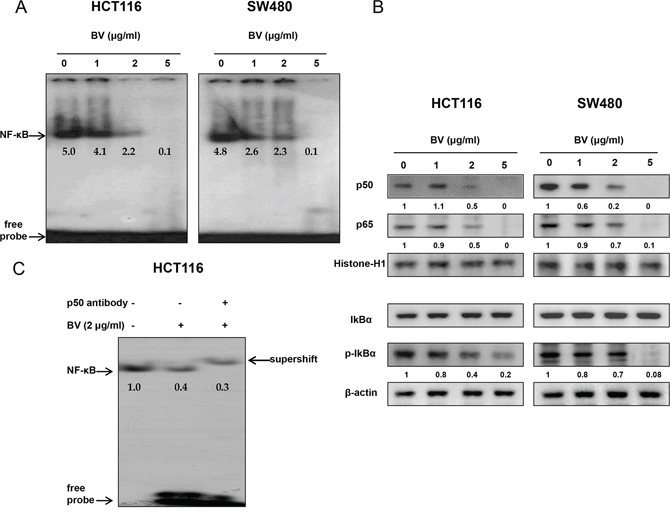
Figure 4: Effect of BV on NF-κB activation in colon cancer cells. A. Colon cancer cells were treated with BV (0–5 μg/ml) for 2 h, and then were lysed. Nuclear extract was incubated in binding reactions of 32p-end-labeled oligonucleotide containing the IκB sequence. The present EMSA results are representative for three experiments. B. Cytosolic proteins were used to determine expression of IκB, p-IκB and β-actin (internal control) and nuclear proteins were used to determine expression of p50, p65 and Histone H1 (internal control) in colon cancer cells. Values under Western band indicate the density of band. Each band is representative for three experiments. C. Supershift assay was performed on HCT116 cells, and a small volume of p50 antibody (1 μl) was added to the binding mix, and incubated at 37°C for 30 min before loading. The present results are representative for three experiments.
Reversed effect of DR4 siRNA, DR5 siRNA and TRAIL siRNA on BV-induced cell growth inhibition
To determine the effect of DR4, DR5 and their ligand TRAIL on the BV-induced apoptotic cell death, we performed siRNA transfection and MTT assay. Cells were transfected with DR4 siRNA, DR5 siRNA and TRAIL siRNA by a transfection agent for 24 h, and then were treated with BV (5 μg/ml) for another 24 h. Cell viability was determined by MTT assay. As a result, combination treatment with BV and DR4 or DR5 or TRAIL siRNA transfection partially abolished BV-induced cell growth inhibiton (Figure 5A). Besides, the expression of DR4 was significantly decreased by DR4 siRNA transfection and the expression of DR5 was significantly decreased by DR5 siRNA transfection (Figure 5B).
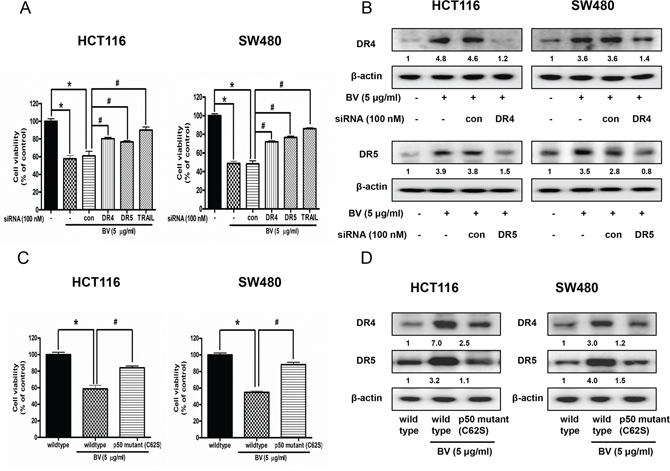
Figure 5: Effect of DR4 siRNA, DR5 siRNA and TRAIL siRNA transfection or p50 mutant plasmid (C62S) transfection on BV-treated colon cancer cell growth and expression of DR4 and DR5. A. Colon cancer cells were treated with non-targeting control siRNA and DR4, DR5, TRAIL siRNA (100 nM) for 24 h, and then were treated with BV (5 μg/ml) at 37°C for another 24 h. Cell viability was determined by MTT assay. Data was expressed as the mean ± S.D. of three experiments. *p < 0.05 indicates statistically significant differences from control cells. #p < 0.05 indicates significantly different from BV treated cells. B. Effect of DR4 siRNA transfection on the expression of DR4 and effect of DR5 siRNA transfection on the expression of DR5. Values under Western band indicate the density of band. Each band is representative for three experiments. C. Effect of p50 mutant plasmid (C62S) transfection on BV-treated colon cancer cell growth. Cell viability was determined by MTT assay. Data was expressed as the mean ± S.D. of three experiments. *p < 0.05 indicates statistically significant differences from control cells. #p < 0.05 indicates significantly different from BV treated cells. D. Effect of p50 mutant plasmid (C62S) transfection on the expression of DR4 and DR5. Cells were transfected by p50 mutant plasmid (C62S) for 24 h, and treated by BV (5 μg/ml) for another 24 h. Whole cell extracts were analyzed by Western blotting using DR4, DR5 and β-actin (internal control) antibodies. Values under Western band indicate the density of band. Each band is representative for three experiments.
Reversed effect of p50 mutant plasmid (C62S) on BV-induced cell growth inhibition and expression of DR4 and DR5
Colon cancer cells were transfected with p50 mutant plasmid (C62S) for 24 h, and then were treated with BV (5 μg/ml) for another 24 h. Cell viability was determined by MTT assay and expression of DR4 and DR5 was detected by Western blotting. As a result, p50 mutant plasmid (C62S) transfection partially abolished BV-induced cell growth inhibiton (Figure 5C), and BV-induced expression of DR4 and DR5 was partially prevented (Figure 5D).
Synergistic effect of BV and NF-κB inhibitor or p50 siRNA on the growth of human colon cancer cells and expression of DR4 and DR5
To further investigate whether NF-κB plays a critical role in BV-induced up-regulation of DR4 and DR5 as well as colon cancer cell growth inhibition, we pretreated the colon cancer cells with phenylarsine oxide (PAO), an NF-κB inhibitor (0.1 μM) for 1 h, and then these cells were treated with BV (1 μg/ml) for 24 h to assess cell growth, and DR4 and DR5 expression. We also knocked down p50 with p50 siRNA by a transfection agent for 24 h, and then treated the transfected cells with BV (5 μg/ml). Cell viability was determined by MTT assay. As a result, combination treatment with BV and p50 siRNA transfection augmented BV-induced cell growth inhibition (Figure 6A), and the expression of p50 translocation to nucleus was significantly decreased (Figure 6B). Besides, combination treatment with BV and NF-κB inhibitor (PAO) magnified BV-induced cell growth inhibtion (Figure 6C) and enhanced the BV-induced up-regulation of DR4 and DR5 (Figure 6D), suggesting that NF-κB pathway may be involved in BV-induced cell growth inhibition and up-regulation of DR4 and DR5.
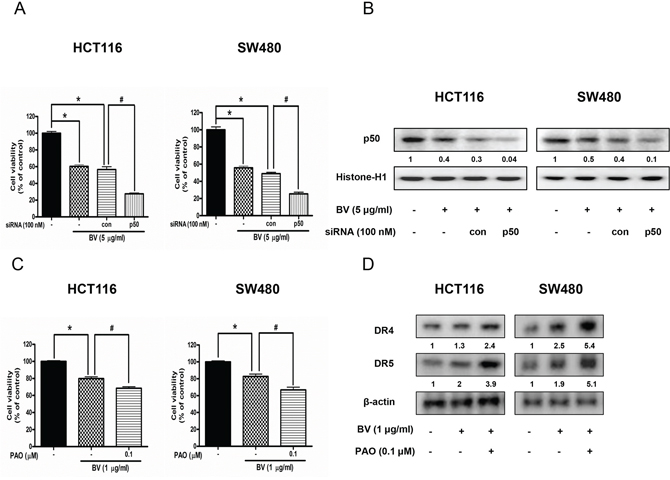
Figure 6: Effect of p50 siRNA transfection or NF-κB inhibitor (PAO) on BV-treated colon cancer cell growth and expression of DR4 and DR5. A. Effect of p50 knockdown with p50 siRNA transfection on the inhibitory effect of BV in colon cancer cell growth. Cell viability was determined by MTT assay. *p < 0.05 indicates statistically significant differences from control cells. #p < 0.05 indicates significantly different from BV treated cells. B. Effect of p50 siRNA transfection on the expression of p50 translocation to nucleus. Values under Western band indicate the density of band. Each band is representative for three experiments. C. Effect of NF-κB inhibitor (PAO) on BV-treated colon cancer cell growth. Cells were pretreated with NF-κB inhibitor, PAO (0.1 μM) for 1 h and then were treated with BV for 24 h. Cell viability was determined by MTT assay. *p < 0.05 indicates statistically significant differences from control cells. #p < 0.05 indicates significantly different from BV treated cells. D. Effect of NF-κB inhibitor (PAO) on the expression of DR4 and DR5. Cells were pretreated with PAO (0.1 μM) for 1 h and then were treated with BV for 24 h, and whole cell extracts were analyzed by Western blotting using DR4, DR5 and β-actin (internal control). Values under Western band indicate the density of band. Each band is representative for three experiments.
Reversed effect of p53 siRNA or p53 inhibitor on the growth of human colon cancer cells and expression of DR4 and DR5
To determine whether p53-dependent apoptosis pathway is involved in BV-induced cell death or not, the HCT116 and SW480 colon cancer cells were transfected with p53 siRNA by a transfection agent. The cells were transfected with 100 nM p53 siRNA for 24 h, and then treated with BV (2 μg/ml) for 24 h. Knock down of p53 partially abolished the cell growth inhibitory effect of BV in HCT116 and SW480 (Figure 7A). Expression of DR4 and DR5 by BV was suppressed by transfection with p53 siRNA both in HCT116 and SW480 cells (Figure 7B). To further investigate whether p53 plays an important role on BV-induced cell growth inhibition and up-regulation of DR4 and DR5, we pretreated HCT116 and SW480 colon cancer cells with 20 μM pifithrin-α, a p53 inhibitor for 1 h, and then these cells were treated with BV (2 μg/ml) for 24 h to assess cell viability and DR4 and DR5 expression. As a result, p53 inhibitor partially abolished BV-induced cell growth inhibition (Figure 7C) and suppressed the BV-induced up-regulation of DR4 and DR5 (Figure 7D), suggesting that p53-dependent apoptosis pathway may be involved in BV-induced cell growth inhibition and up-regulation of DR4 and DR5.
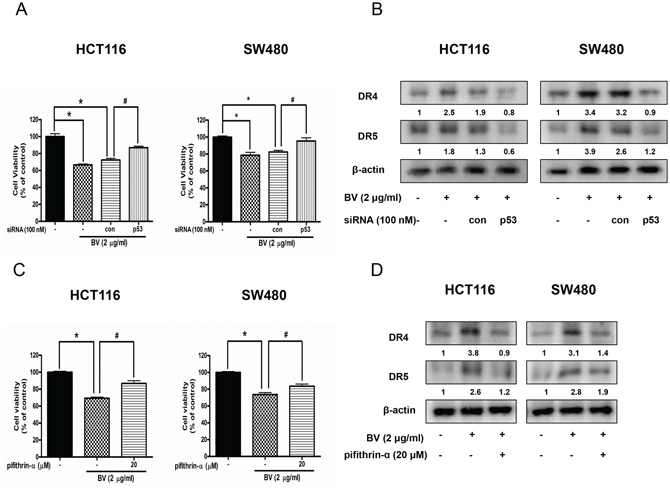
Figure 7: Effect of p53 siRNA transfection or p53 inhibitor (pifithrin-α) on BV-treated colon cancer cell growth and expression of DR4 and DR5. A. Colon cancer cells were treated with non-targeting control siRNA and p53 siRNA (100 nM) for 24 h, and then were treated with BV (5 μg/ml) at 37°C for another 24 h. Cell viability was determined by MTT assay. Data was expressed as the mean ± S.D. of three experiments. *p < 0.05 indicates statistically significant differences from control cells. #p < 0.05 indicates significantly different from BV treated cells. B. Effect of p53 siRNA transfection on the expression of DR4 and DR5. Whole cell extracts were analyzed by Western blotting with antibodies against DR4, DR5 and β-actin (internal control). Values under Western band indicate the density of band. Each band is representative for three experiments. C. Cells were pretreated with p53 inhibitor, pifithrin-α (20 μM) for 1 h and then were treated with BV for 24 h. Cell viability was determined by MTT assay. *p < 0.05 indicates significantly different from control cells. #p < 0.05 indicates significantly different from BV-treated cells. D. Effect of pifithrin-α on the expression of DR4 and DR5. Cells were pretreated with pifithrin-α (20 μM) for 1 h and then were treated with BV for 24 h, and whole cell extracts were analyzed by Western blotting using DR4, DR5 and β-actin (internal control). Values under Western band indicate the density of band. Each band is representative for three experiments.
Structure of melittin and molecular binding btween NF-κB p50 and melittin
The molecular binding between melittin, a major component of BV (Figure 8A) - p50 protein was assessed using pull-down assay. The interaction of melittin-Sepharose 6B beads with p50 was then detected by immunoblotting with p50 antibody. The results indicated that melittin bound with cell lysates containing p50 from SW480 cells and p50 recombinant protein (Figure 8B). To identify the binding site of melittin to p50, we performed computational docking experiments with melittin and NF-κB p50 subunit. Melittin bound somewhat near to the C-terminus of p50 where nuclear localization sequence (NLS) is located. Melittin bound to a pocket created by Tyr387, Val388, Pro390, Lys392, Thr424, Pro427, Lys428, Thr522, Ser524 and Phe525 of p50 (Figure 8C).
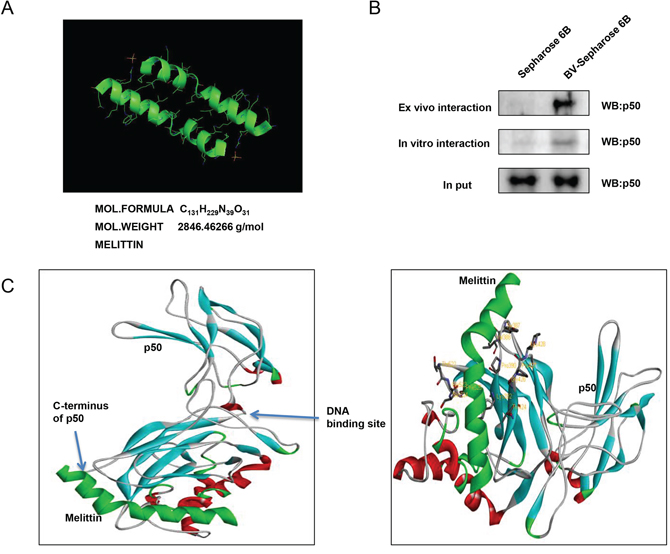
Figure 8: Structure of melittin, molecular binding between mellitin and NF-κB p50. A. Structure of mellitin. B. Pull down assay identifies the binding between mellitin and NF-κB p50. BV was conjugated with epoxy-activated Sepharose 6B. C. Docking model of mellitin with NF-κB p50 was determined as described in materials and methods.
Effect of BV on colon cancer tumor growth
To elucidate the anti-tumor effect of BV in vivo, the tumor growth on colon cancer cell xenograft bearing nude mice following BV treatments was investigated. In HCT116 xenograft studies, BV (1 mg/kg) was administrated intraperitoneally twice per week for 3 weeks to mice which have tumors ranging from 100 to 150 mm3. Tumor volume was measured twice a week, and all mice were sacrificed at the end of experiment when tumors were dissected and weighted. The inhibitory effect of BV on the growth of colon tumor was significant in xenograft model mice (Figure 9A). Tumor volume and weight were dose-dependently decreased (Figure 9B and 9C). The immunohistochemistry analysis of tumor section by hematoxylin and eosin staining and proliferation antibodies against PCNA staining revealed that BV (1 mg/kg) significantly suppressed tumor growth, and the expression level of DR4, DR5 and active caspase-3 was increased while the expression level of p50 was decreased in nude mice xenograft tissues (Figure 9D).
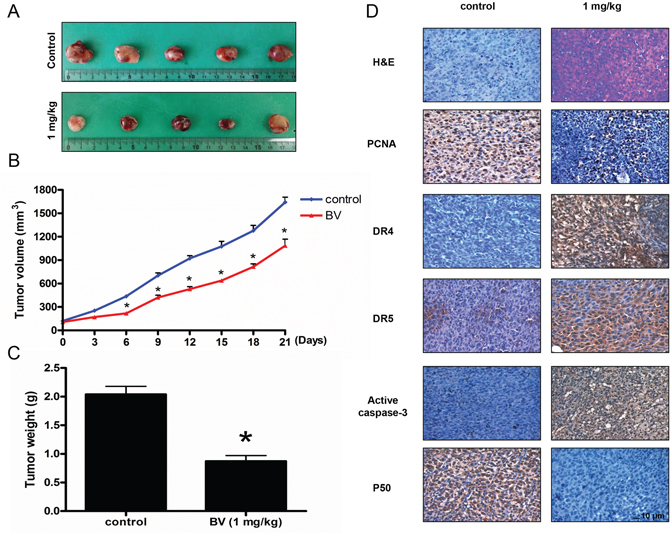
Figure 9: Effect of BV on the tumor growth, on the expression levels of proliferation, apoptosis regulatory proteins and NF-κB p50 subunit in immunohistochemistry. Growth inhibition (as assessed by tumor volume and weights) of subcutaneously transplanted HCT116 xenografts mice treated with BV (1 mg/kg) twice a week for three weeks. Xenografted mice (n = 6) were administrated intraperitoneally with BV (1 mg/kg). A. Figure represents the tumor photographs at day 21. B. Tumor volumes were measured twice a week for three weeks. C. Tumor weights were measured at study termination on Day 21. Mean ± S.D. estimates of tumor volume and weight from 6 mice in each treatment. D. Immunohistochemistry was used to determine expression levels of H&E, PCNA, DR4, DR5, active caspase-3 and p50 in nude mice xenograft tissues by the different treatments as described in materials and methods. All values represent mean ± S.D. from 5 animal tumor sections. *p < 0.05 indicates significantly different from the control group. Bar indicates 10 μm.
Effect of BV on the expression of apoptosis regulatory proteins and DNA binding activity of NF-κB in colon tumor tissues
To examine the relationship between colon tumor growth and apoptosis regulatory proteins as well as the DNA binding activity of NF-κB, we performed Western blotting and EMSA experiments. We found the expression of DR4, DR5, cleaved caspase-3, cleaved caspase-8 and Bax was increased, but the expression of Bcl-2 was decreased (Figure 10A). We also found the DNA binding activity of NF-κB was decreased in BV-treated colon tumor tissues (Figure 10B).
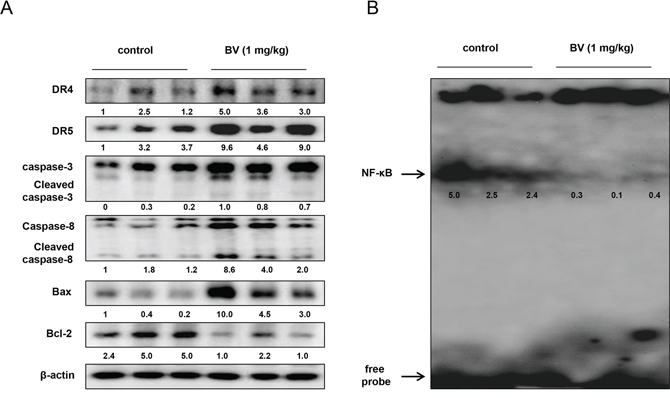
Figure 10: Effect of BV on the expression of apoptosis regulatory proteins and on the DNA binding activity of NF-κB in vivo. A. Expression of apoptosis regulatory proteins was detected by Western blotting using DR4, DR5, Bax, Bcl-2, caspase-3, caspase-8 and β-actin (internal control) in xenograft tumor samples (3 samples/each group) as described in materials and methods. B. DNA binding activity of NF-κB was determined by EMSA in nucleus extract from xenograft tumor samples (3 samples/each group) as described in materials and methods.
DISCUSSION
Although it has been previously reported that BV can induce apoptosis in many cancer cell lines such as lung cancer, hepatocellular carcinoma, breast cancer, prostate cancer and ovarian cancer cell lines [29–33], there is no study on the effect of BV on colon cancer cell lines. In this study, we demonstrated that BV inhibited cancer cell growth in HCT116 and SW480 colon cancer cells through activation of DR4 and DR5 and suppression of NF-κB. Our present findings showed that BV inhibited the growth of HCT116 and SW480 colon cancer cells in a concentration dependent manner (0–5 μg/ml). However, the clear mechanism has not been found yet. The possible molecular targets of BV and its component for inhibition of cancer cell growth are numerous and various [34]. Our previous study showed that BV and its major component melittin inhibited cancer cell growth in prostate carcinoma cells through induction of apoptotic cell death in vitro as well as in vivo xenograft model via activation of caspase pathway through suppression of constituted activation of NF-κB activity [32], BV and melittin induced apoptotic cell death in ovarian cancer cells through enhancement of DR3, DR4 and DR6 expression and inhibition of STAT3 pathway [33], BV inhibited cancer cell growth in NSCLC cells through the induction of apoptosis via increase of DR3 expression and inhibition of NF-κB pathway [35]. Similarly, we found that BV inhibited the growth of colon cancer cells through activation of DR4 and DR5 and inhibition of NF-κB pathway. These data suggest that differential signals are involved in the inhibitory effect of BV on cancer cell growth depending on cancer cell types.
It was reported that cancer cell growth inhibitory effect was correlated with the down-regulation of various cell proliferative genes regulated by NF-κB [36]. In agreement with this notion, we found that BV suppressed DNA binding activity of NF-κB. Moreover, the decrease of NF-κB DNA binding activity was associated with the inhibitory effect of BV on the IκB phosphorylation and nuclear translocation of p50 and p65 in colon cancer cells. The present data showed that BV also suppressed the expression of anti-apoptotic proteins like Bcl-2, while it increased the expression of pro-apoptotic proteins such as Bax, caspase-3, caspase-8 and caspase-9 which are regulated by NF-κB. Thus, BV may induce an alteration of expression of apoptosis and anti-apoptosis regulatory proteins to provide a favorable circumstance for the cancer cells to reach death status by down-regulation of NF-κB. Enhanced cell growth inhibition was found to occur with the combination treatment with NF-κB inhibitor PAO (phenylarsine oxide, 0.1 μM) or p50 siRNA trasfection and BV. It may be because BV binds to p50, and prevents its translocation to nucleus. P50 siRNA may work in other step (may inhibit its expression). Two different mechanisms could work synergistically or additively. These data suggest that inhibition of NF-κB contributed to the BV-induced inhibitory effect of colon cancer cell growth. Therefore, it is possible that reduced NF-κB activity may be correlated with the inhibition of colon cancer cell growth by BV treatment.
Recent studies on the signaling mechanisms of the DRs have revealed that members of the NF-κB and caspase families are key regulators of cell death. Expression of DRs leads to activation of caspase-8, which induces activation of effecter caspase like caspase-3 that can active caspase-9 to cause apoptotic cell death [37, 38]. NF-κB is also involved in growth arrest and/or apoptosis by regulating the expression of various target genes such as Cyclin D1, Bax, caspase-3, caspase-9, Bcl-2, IAP and survivin [39, 40]. These results may suggest that the inhibition of NF-κB correlates with the increased expression of DRs. Numerous studies have demonstrated that natural compounds-induced apoptosis in cancer cells may be correlated to the increase of DR expression. Garicinol, a polyisoprenylated benzophenone derivative, derived from dried rind of the fruit Garcinia indica can potentiate TRAIL-induced apoptotic cell death of human colon cancer cell through up-regulation of DR4 and DR5 [16]. Quercetin also enhanced TRAIL-mediated apoptosis in colon cancer cells by inducing the accumulation of death receptors in lipid rafts [17]. Zerumbone enhanced TRAIL-induced apoptosis through the induction of death receptors in human colon cancer cells [18]. In our previous study, we demonstrated that the snake venom toxin from Vipera lebetina turanica induces apoptosis of colon cancer cells through reactive oxygen species (ROS) and c-Jun N-terminal kinases (JNK) dependent death receptor (DR4 and DR5) expression [15], and (E)-2,4-Bis(p-hydroxyphenyl)-2-butenal inhibits colon cancer cell growth via suppression of NF-κB and induction of DR6 [41]. Similarly, our results showed that the expression of DR4 and DR5 was increased in both HCT116 and SW480 colon cancer cells. We also observed that BV induced the increasing expression of DR downstream apoptotic proteins such as cleaved caspase-3, cleaved caspase-8, cleaved caspase-9 and Bax but decreased expression of Bcl-2 in colon cancer cells. Transcriptional regulation of DR4 and DR5 is complex, and multiple potential binding sites of various transcription factors, including p53, are present in the upstream region of DR4 and DR5 [42]. Our data showed that the increasing expression of p53 and p21 are both induced by BV. Thus, the induction of DR4 and DR5 by BV treatment may be dependent on p53 in HCT116 and SW480 colon cancer cells. Besides, co-treatment with BV and p53 siRNA or pifithrin-α, a p53 inhibitor partially abolished BV-induced cell growth inhibition and activation of DR4 and DR5. These data suggest that BV-induced apoptotic cell death and inhibitory effect of cancer cell growth might be correlated with the activation of p53 dependent DR4 and DR5 expression.
Docking model and pull-down assay showed that melittin, the major component of BV, directly bound with NF-κB p50 subunit to inhibit the DNA binding activity of NF-κB. Melittin bound to the nuclear localization sequence (NLS: Tyr387, Val388, Pro390, Lys392, Thr424, Pro427, Lys 428, Thr522, Ser524 and Phe525), which was near to the C-terminus of p50. The binding site of p50 to melittin was not as the same as our previous finding which showed that melittin bound to the DNA binding site of p50 [20]. However, it is noteworthy that blocking NLS is important for the translocation of p50 to nucleus inhibiting NF-κB activity [43]. Importin α3 is shown to directly bind to NLS of NF-κB p50 and p65 proteins, and IκBα inhibits nuclear translocation of NF-κB by inhibiting the binding of p65 homodimers and p50/p65 heterodimers to importin α3 [44]. The inducible nuclear translocation of NF-κB in Jukat T lymphocytes is significantly inhibited by a cell-permeable3 peptide which could bind to the NLS of the NF-κB p50 subunit [45]. Therefore, the present data indicated that BV effectively inhibited colon cancer cell growth through inactivation of NF-κB by directly binding with p50 NF-κB subunit. Taken together, our results provide the mechanistic evidence that BV treatment results in induction of apoptosis of colon cancer cells through up-regulation of DR4 and DR5 as well as suppression of NF-κB. Therefore, our results suggest that BV could be applicable as an anti-colon cancer agent.
MATERIALS AND METHODS
Materials
Dried BV was purchased from You-Miel Bee Venom Ltd. (Hwasoon, Jeonnam, Korea). The composition of the BV was as previously described [26]. Caspase-3, caspase-8, caspase-9 antibodies were purchased from Cell Signaling Technology Inc. (Beverly, MA). DR3, DR4, DR5, Fas, TRAIL, IκBα, phospho-IκBα, p50, p65, p21, p53, Bcl-2, Bax, Histone-H1 and β-actin antibodies were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). The cell culture materials were obtained from Invitrogen (Carlsbad, CA), and other chemical reagents were from Sigma Chemical Co.
Cell culture
The HCT116 and SW480 colon cancer cell lines and FHC colon epithelial normal cells were obtained from American Type Culture Collection (ATCC). HCT116 was cultured in DMEM (Gibco, Life Technologies, Grand Island, NY) medium supplemented with 10% heat inactivated fetal bovine serum (FBS) and 100 units/ml penicillin, 100 μg/ml streptomycin (Invitrogen). SW480 was cultured in RPMI 1640 medium supplemented with 10% heat inactivated FBS,100 units/ml penicillin and 100 μg/ml streptomycin. FHC was cultured in DMEM: F12 (1:1) medium (ATCC) supplemented with 10% heat inactivated FBS, 25 mM HEPES, 100 units/ml penicillin and 100 μg/ml streptomycin. Cell cultures were then maintained in an incubator within a humidified atmosphere of 5% CO2 at 37°C.
Cell viability assay
Colon cancer cells HCT116 and SW480 were plated in 96-well plates for 24 h, and then were treated with BV (0–10 μg/ml) for 24 h. After treatment, cell viability was measured by MTT [3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide] assay (Sigma Aldrich, St. Louis, MO) according to the manufacturer’s instructions. Briefly, MTT (5 mg/ml) was added and plates were incubated at 37°C for 4 h before dimethyl sulfoxide (100 μl) was added to each well. Finally, the absorbance of each well was read at a wavelength of 540 nm using a plate reader.
Apoptosis evaluation
Colon cancer cells HCT116 and SW480 were cultured on 8-chamber slides. The cells were treated with BV (0–5 μg/ml) for 24 h, and then washed twice with phosphate buffered saline (PBS) and fixed by incubation in 4% paraformaldehyde in PBS for 1 h at room temperature. TdT-mediated dUTP nick and labeling TUNEL assays were performed by using the DeadEnd™ Fluorometric TUNEL System (Promega Corporation, Madison, USA) according to manufacturer’s instructions. Total number of cells in a given area was determined by using DAPI staining. The apoptotic index was determined as the number of TUNEL-positive stained cells divided by the total cell number counted × 100%.
Soft agar colony formation assay
Effect of BV on colony formation ability of the HCT116 and SW480 cells were assessed by soft agar colony formation assay. The assay was done in 6-well plates. In each well, 2 ml of 0.6% agar (in culture medium) was layered in the bottom followed by 3 ml of 0.3% agar as the top layer. Approximately 8 × 103 cells were the plated over the top layer. The cells were treated with BV (0–5 μg/ml) and maintained at 37°C in a humidified 5% CO2 atmosphere for 10 days. Then the colonies were counted under a microscope.
Western blot assay
Western blot assays were performed as previously described [26]. After a short washing in TBST, the membranes were immunoblotted with the following primary antibodies: caspase-3, caspase-8, caspase-9 (1:1000 dilutions; Cell Signaling, Beverly, MA) and p53, p21, DR3, DR4, DR5, Fas, TRAIL, Bax and Bcl-2 (1:1000 dilutions; Santa Cruz Biotechnology, Santa Cruz, CA). The blots were performed using specific antibodies followed by second antibodies and visualization by chemiluminescence (ECL) detection system.
Electrophoretic mobility shift assay and supershift assay
The DNA binding activity of NF-κB was determined using an electrophoretic mobility shift assay (EMSA) performed as according to the manufacturer’s recommendations (Promega). Supershift assay was performed on HCT116 cells, and a small volume of p50 antibody (1 μl) was added to the binding mix, and incubated at 37°C for 30 min before loading. The relative densities of the DNA–protein binding bands were scanned by densitometry using MyImage (SLB), and quantified by Labworks 4.0 software (UVP, Inc., Upland, CA).
Transfection of siRNA
2 × 105 cells were plated in 6-well plates and were transiently transfected with siRNA, using a mixture of siRNA and the WellFect-EX PLUS reagent in OPTI-MEM, according to the manufacturer’s specification (WelGENE, Seoul, Korea). The transfected cells were treated with BV (2 or 5 μg/ml) for 24 h and then used for detecting cell viability and protein expression.
Pull-down assay
Pull-down assay was performed as described elsewhere [27]. The proteins were then resolved by SDS-PAGE followed by immunoblotting with antibodies against p50 (1:1000 dilutions, Santa Cruz Biotechnology, Santa Cruz, CA).
Docking experiment
Docking studies between NF-κB p50 and melittin were performed using Autodock VINA [28] as described elsewhere [27].
Antitumor activity study in in vivo xenograft animal model
Eight-week-old male BALB/C nude mice were purchased from Orient-Bio (Gyunggi-do, Korea). The mice were maintained in accordance with the Korea Food and Drug Administration guidelines as well as the regulations for the care and use of laboratory animals of the animal ethics committee of Chungbuk National University (CBNU-278-11-01). The mice were held for 4 days after arriving before they were injected with cells. The last injection of BV was at the 18th day, and after 3 days the mice were euthanized at the 21th day. The mice were fed with mouse diet (S-11102 Altromin Moused Diet 1314 IRR) purchased from Samtako Inc. during the study. Human colon cancer cell line HCT116 cells were injected subcutaneous (1 × 107 tumor cells/0.1 ml PBS/animals) with a 27-gauge needle into the right lower flanks in carrier mice. After 14 days, when the tumors had reached an average volume of 100–150 mm3, the tumor-bearing nude mice were intraperitoneally (i.p.) injected with BV (1 mg/kg dissolved in PBS) twice per week for 3 weeks. The group untreated was designed as the control. The weight and tumor volume of the animals were monitored twice per week. The tumor volumes were measured with vernier calipers and calculated by the following formula: (A × B2)/2, where A is the larger and B is the smaller of the two dimensions. At the end of the experiment, the animals were sacrificed. The tumors were separated from the surrounding muscles and dermis, excised and weighed.
Immunohistochemistry
The animal tissues were fixed in 4% paraformaldehyde and cut into 5 μm sections using a freezing microtome (Thermo Scientific, Germany). The sections were stained with hematoxylin and eosin (H&E) for pathological examination. For immunohistological staining, tumor sections were incubated with primary antibody against PCNA, DR4, DR5, active caspase-3 and p50 (1:500, Abcam, Cambridge, UK). Immunohistochemistry experiment was performed as previously described [26].
Statistical analysis
All statistical analysis was performed with GraphPad Prism 4 software (Version 4.03, GraphPad Software, Inc., San Diego, CA, USA). Group differences in the Figures 5, 6, 7 were analyzed using two-way analysis of variance (ANOVA) followed by Tuckey’s test. Otherwise, data were analyzed by one-way ANOVA followed by Dunnett’s test. All values are presented as mean ± S.D. Significance was set at p < 0.05 for all tests.
ACKNOWLEDGMENTS AND FUNDING
This work was supported by the National Research Foundation of Korea [NRF] Grant funded by the Korea government (MSIP) (No. MRC, 2008-0062275).
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
Abbreviations
ATCC, American Type Culture Collection; Bax, Bcl-2-associated X protein; Bcl-2, B-cell leukemia/lymphoma-2; BV, bee venom; DAPI, 4; DMEM, Dulbecco’s modified Eagle’s medium; DR, death receptor; EMSA, electrophoretic mobility shift assay; FBS, fetal bovine serum; IAP, inhibitor of apoptosis protein; IHC, immunohistochemistry; JNK, c-Jun N-terminal kinases; MAPK, mitogen-activated protein kinase; NF-κB, nuclear factor κB; NLS, nuclear localization sequence; NSCLC, non-small cell lung cancer; PAO, phenylarsine oxide; PBS, phosphate buffered saline; PVDF, polyvinylidene fluoride; ROS, reactive oxygen species; SDS, sodium dodecyl sulfate; TRAIL, tumor necrosis factor-related apoptosis-inducing ligand; TUNEL, terminal deoxynucleotidyl transferase.
REFERENCES
1. Janakiram NB, Rao CV. The role of inflammation in colon cancer. Adv Exp Med Biol. 2014; 816:25–52.
2. Swiderska M, Choromanska B, Dabrowska E, Konarzewska-Duchnowska E, Choromanska K, Szczurko G, Mysliwiec P, Dadan J, Ladny JR, Zwierz K. The diagnostics of colorectal cancer. Contemp Oncol (Pozn). 2014; 18:1–6.
3. Johnson JJ, Mukhtar H. Curcumin for chemoprevention of colon cancer. Cancer Lett. 2007; 255:170–181.
4. Terry P, Giovannucci E, Michels KB, Bergkvist L, Hansen H, Holmberg L, Wolk A. Fruit, vegetables, dietary fiber, and risk of colorectal cancer. J Natl Cancer Inst. 2001; 93:525–533.
5. Fleet JC. New Mouse Models for Studying Dietary Prevention of Colorectal Cancer. Am J Physiol Gastrointest Liver Physiol. 2014.
6. Lee SY, Cho JS, Yuk DY, Moon DC, Jung JK, Yoo HS, Lee YM, Han SB, Oh KW, Hong JT. Obovatol enhances docetaxel-induced prostate and colon cancer cell death through inactivation of nuclear transcription factor-kappaB. J Pharmacol Sci. 2009; 111:124–136.
7. Lind DS, Hochwald SN, Malaty J, Rekkas S, Hebig P, Mishra G, Moldawer LL, Copeland EM 3rd, Mackay S. Nuclear factor-kappa B is upregulated in colorectal cancer. Surgery. 2001; 130:363–369.
8. Baldwin AS. Control of oncogenesis and cancer therapy resistance by the transcription factor NF-kappaB. J Clin Invest. 2001; 107:241–246.
9. Paul A, Das S, Das J, Samadder A, Bishayee K, Sadhukhan R, Khuda-Bukhsh AR. Diarylheptanoid-myricanone isolated from ethanolic extract of Myrica cerifera shows anticancer effects on HeLa and PC3 cell lines: signalling pathway and drug-DNA interaction. J Integr Med. 2013; 11:405–415.
10. Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006; 441:431–436.
11. Chen GG, Lee JF, Wang SH, Chan UP, Ip PC, Lau WY. Apoptosis induced by activation of peroxisome-proliferator activated receptor-gamma is associated with Bcl-2 and NF-kappaB in human colon cancer. Life Sci. 2002; 70:2631–2646.
12. Feinman R, Clarke KO, Harrison LE. Phenylbutyrate-induced apoptosis is associated with inactivation of NF-kappaB IN HT-29 colon cancer cells. Cancer Chemother Pharmacol. 2002; 49:27–34.
13. Pahl HL. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene. 1999; 18:6853–6866.
14. Sun SY. Understanding the Role of the Death Receptor 5/FADD/caspase-8 Death Signaling in Cancer Metastasis. Mol Cell Pharmacol. 2011; 3:31–34.
15. Park MH, Jo M, Won D, Song HS, Han SB, Song MJ, Hong JT. Snake venom toxin from Vipera lebetina turanica induces apoptosis of colon cancer cells via upregulation of ROS- and JNK-mediated death receptor expression. BMC Cancer. 2012; 12:228.
16. Prasad S, Ravindran J, Sung B, Pandey MK, Aggarwal BB. Garcinol potentiates TRAIL-induced apoptosis through modulation of death receptors and antiapoptotic proteins. Mol Cancer Ther. 2010; 9:856–868.
17. Psahoulia FH, Drosopoulos KG, Doubravska L, Andera L, Pintzas A. Quercetin enhances TRAIL-mediated apoptosis in colon cancer cells by inducing the accumulation of death receptors in lipid rafts. Mol Cancer Ther. 2007; 6:2591–2599.
18. Yodkeeree S, Sung B, Limtrakul P, Aggarwal BB. Zerumbone enhances TRAIL-induced apoptosis through the induction of death receptors in human colon cancer cells: Evidence for an essential role of reactive oxygen species. Cancer Res. 2009; 69:6581–6589.
19. Jentsch J, Mucke HW. Bee venom peptides XVIII. Peptide-m and mcd-peptide: Isolation and characterization. Int J Pept Protein Res. 1977; 9:78–79.
20. Park HJ, Lee SH, Son DJ, Oh KW, Kim KH, Song HS, Kim GJ, Oh GT, Yoon DY, Hong JT. Antiarthritic effect of bee venom: inhibition of inflammation mediator generation by suppression of NF-kappaB through interaction with the p50 subunit. Arthritis Rheum. 2004; 50:3504–3515.
21. Liu X, Chen D, Xie L, Zhang R. Effect of honey bee venom on proliferation of K1735M2 mouse melanoma cells in-vitro and growth of murine B16 melanomas in-vivo. J Pharm Pharmacol. 2002; 54:1083–1089.
22. Huh JE, Kang JW, Nam D, Baek YH, Choi DY, Park DS, Lee JD. Melittin suppresses VEGF-A-induced tumor growth by blocking VEGFR-2 and the COX-2-mediated MAPK signaling pathway. J Nat Prod. 2012; 75:1922–1929.
23. Terwilliger TC, Eisenberg D. The structure of melittin. I. Structure determination and partial refinement. J Biol Chem. 1982; 257:6010–6015.
24. Son DJ, Lee JW, Lee YH, Song HS, Lee CK, Hong JT. Therapeutic application of anti-arthritis, pain-releasing, and anti-cancer effects of bee venom and its constituent compounds. Pharmacol Ther. 2007; 115:246–270.
25. Park HJ, Son DJ, Lee CW, Choi MS, Lee US, Song HS, Lee JM, Hong JT. Melittin inhibits inflammatory target gene expression and mediator generation via interaction with IkappaB kinase. Biochem Pharmacol. 2007; 73:237–247.
26. Lee HL, Park SH, Kim TM, Jung YY, Park MH, Oh SH, Yun HS, Jun HO, Yoo HS, Han SB, Lee US, Yoon JH, Song MJ, Hong JT. Bee venom inhibits growth of human cervical tumors in mice. Oncotarget. 2015; 6:7280–7292. doi: 10.18632/oncotarget.3110.
27. Gu SM, Park MH, Hwang CJ, Song HS, Lee US, Han SB, Oh KW, Ham YW, Song MJ, Son DJ, Hong JT. Bee venom ameliorates lipopolysaccharide-induced memory loss by preventing NF-kappaB pathway. J Neuroinflammation. 2015; 12:124.
28. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010; 31:455–461.
29. Jang MH, Shin MC, Lim S, Han SM, Park HJ, Shin I, Lee JS, Kim KA, Kim EH, Kim CJ. Bee venom induces apoptosis and inhibits expression of cyclooxygenase-2 mRNA in human lung cancer cell line NCI-H1299. J Pharmacol Sci. 2003; 91:95–104.
30. Wang C, Chen T, Zhang N, Yang M, Li B, Lu X, Cao X, Ling C. Melittin, a major component of bee venom, sensitizes human hepatocellular carcinoma cells to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis by activating CaMKII-TAK1-JNK/p38 and inhibiting IkappaBalpha kinase-NFkappaB. J Biol Chem. 2009; 284:3804–3813.
31. Ip SW, Liao SS, Lin SY, Lin JP, Yang JS, Lin ML, Chen GW, Lu HF, Lin MW, Han SM, Chung JG. The role of mitochondria in bee venom-induced apoptosis in human breast cancer MCF7 cells. In Vivo. 2008; 22:237–245.
32. Park MH, Choi MS, Kwak DH, Oh KW, Yoon do Y, Han SB, Song HS, Song MJ, Hong JT. Anti-cancer effect of bee venom in prostate cancer cells through activation of caspase pathway via inactivation of NF-kappaB. Prostate. 2011; 71:801–812.
33. Jo M, Park MH, Kollipara PS, An BJ, Song HS, Han SB, Kim JH, Song MJ, Hong JT. Anti-cancer effect of bee venom toxin and melittin in ovarian cancer cells through induction of death receptors and inhibition of JAK2/STAT3 pathway. Toxicol Appl Pharmacol. 2012; 258:72–81.
34. Orsolic N. Bee venom in cancer therapy. Cancer Metastasis Rev. 2012; 31:173–194.
35. Choi KE, Hwang CJ, Gu SM, Park MH, Kim JH, Park JH, Ahn YJ, Kim JY, Song MJ, Song HS, Han SB, Hong JT. Cancer cell growth inhibitory effect of bee venom via increase of death receptor 3 expression and inactivation of NF-kappa B in NSCLC cells. Toxins (Basel). 2014; 6:2210–2228.
36. Barkett M, Gilmore TD. Control of apoptosis by Rel/NF-kappaB transcription factors. Oncogene. 1999; 18:6910–6924.
37. Elrod HA, Sun SY. Modulation of death receptors by cancer therapeutic agents. Cancer Biol Ther. 2008; 7:163–173.
38. Wen X, Lin ZQ, Liu B, Wei YQ. Caspase-mediated programmed cell death pathways as potential therapeutic targets in cancer. Cell Prolif. 2012; 45:217–224.
39. Suh J, Rabson AB. NF-kappaB activation in human prostate cancer: important mediator or epiphenomenon?. J Cell Biochem. 2004; 91:100–117.
40. Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, Kagnoff MF, Karin M. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004; 118:285–296.
41. Ban JO, Jung YS, Kim DH, Park KR, Yun HM, Lee NJ, Lee HP, Shim JH, Jeong HS, Lee YH, Ham YW, Han SB, Hong JT. (E)-2,4-Bis(p-hydroxyphenyl)-2-butenal inhibits tumor growth via suppression of NF-kappaB and induction of death receptor 6. Apoptosis. 2014; 19:165–178.
42. Gupta SC, Reuter S, Phromnoi K, Park B, Hema PS, Nair M, Aggarwal BB. Nimbolide sensitizes human colon cancer cells to TRAIL through reactive oxygen species- and ERK-dependent up-regulation of death receptors, p53, and Bax. J Biol Chem. 2011; 286:1134–1146.
43. Matthews JR, Watson E, Buckley S, Hay RT. Interaction of the C-terminal region of p105 with the nuclear localisation signal of p50 is required for inhibition of NF-kappa B DNA binding activity. Nucleic Acids Res. 1993; 21:4516–4523.
44. Fagerlund R, Kinnunen L, Kohler M, Julkunen I, Melen K. NF-{kappa}B is transported into the nucleus by importin {alpha}3 and importin {alpha}4. J Biol Chem. 2005; 280:15942–15951.
45. Torgerson TR, Colosia AD, Donahue JP, Lin YZ, Hawiger J. Regulation of NF-kappa B, AP-1, NFAT, and STAT1 nuclear import in T lymphocytes by noninvasive delivery of peptide carrying the nuclear localization sequence of NF-kappa B p50. J Immunol. 1998; 161:6084–6092.