
Introduction
EGFR kinase inhibitors have become routine treatment for patients with EGFR-mutant lung cancer [1-4]. However, resistance will ultimately emerge, thereby limiting the overall efficacy of such treatment. Resistance may emerge due to EGFR second site mutations, mainly the T790M-gatekeeper mutation, which prevents binding of the typical quinazoline-based compounds [5-8], due to genome alterations such as amplification of MET [9-15] that activate phosphatidyl-inositide-3 kinase (PI3K) signaling, or by processes changing cellular differentiation [16]. Both the T790M mutation and MET amplification may exist in a subclone present at the time of therapy; thus, a proportion of cases of resistance are likely to occur due to clonal selection of such resistant subclones under therapy [17, 18].
It was recently proposed that high-dose pulses of kinase inhibitors lead to enhanced target suppression and eradication of tumor cells more effectively by more potent induction of apoptosis [19]. As a consequence, intermittent high-dose schedules were shown to enhance efficacy in ERBB2-amplified breast cancer [20] as well as in BRAF-mutant melanomas [21]. In a cancer evolution modeling approach an intermittent scheduling of erlotinib could prevent the appearance of resistance despite the presence of EGFRT790M positive subclones ab initio [22, 23].
Since the duration of target suppression is likely to affect the efficacy of a given compound in addition to the magnitude of target inhibition [24, 25] we sought to determine, whether enhanced trough levels or peak plasma levels might be more relevant to offer enhanced therapeutic efficacy.
Results
High dose pulses of erlotinib potently inhibit tumorcell growth of EGFR-mutant NSCLC cell lines in vivo and in vitro
In line with a recent report [19] 20 minutes of treatment with 10µM of erlotinib potently suppressed tumor cell growth of the erlotinib sensitive cell lines HCC827 and PC9 (both carry EGFR exon 19 deletion mutations) as effectively as a continuous 72 hours exposure with 0.1µM. As expected, any such treatment had minor effect on the resistant cell lines H1975 (EGFR L858R and T790M) and HCC827GR (EGFR exon 19 deletion, MET-amplified) (Figure S1), thus confirming that efficacy was due to on-target activity of the compound. A pulse of 10µM of erlotinib reduced pAkt and pErk1/2 levels in the sensitive cell lines, despite re-establishment of pEGFR signaling (Figure 1A, upper panels). Continuous exposure to erlotinib inhibited pEGFR, pAkt and pErk1/2 levels at a concentration of 0.1µM. Again, downstream signaling was only slightly reduced in the two resistant control cell lines (Figure 1A, lower panels). Phosphorylated levels of MET were reduced in HCC827, PC9 and H1975 cells by high concentrations of erlotinib, but not in the MET-amplified cell line HCC827GR (Figure S2). Induction of apoptosis (Figure 1A, 1B) was also similar in the sensitive cells treated with a high-dose pulse of 10µM for 20 minutes and those treated with 0.1µM, but not in the resistant ones.
Given the high efficacy of high-dose treatment of erlotinib in EGFR-mutant tumor cells in vitro, we tested whether pulsatile high doses of erlotinib enhanced tumor control in vivo. Tumor shrinkage occurred more rapidly and was of greater magnitude in tumor xenografts of PC9 and HCC827 cells treated with intermittent high doses of erlotinib (200mg/kg every other day, “intermittent_2day”) than in those treated daily with 30mg/kg (Figure 1C). Treatment with either 15mg/kg daily or 200mg/kg every fourth day (“intermittent_4day”) were less effective, although this effect was more pronounced in PC9 xenografts (Figure S3). Again, H1975 was unresponsive to therapy using any of these schedules (Figure 1C and Figure S3, right panels).
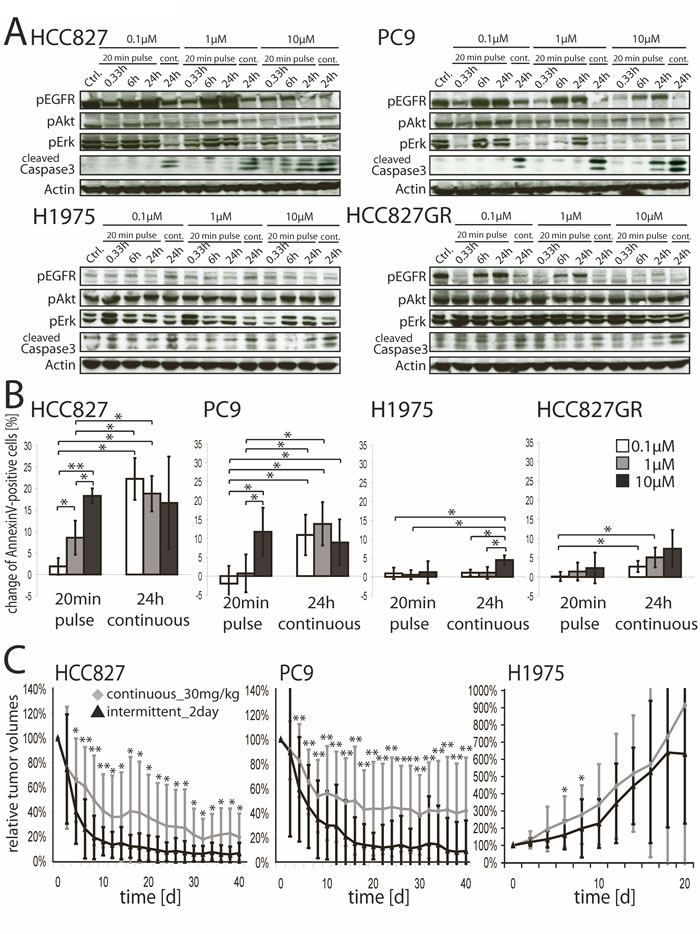
Figure 1: A. Western Blot analyses of HCC827, PC9, H1975 and HCC827GR cells treated with 0.1µM, 1µM or 10µM of erlotinib or DMSO for 20 minutes or continuously till preparation of lysates. Whole-cell lysates were analyzed for expression levels of the indicated proteins by western blotting. B. AnnexinV flow cytometry of HCC827, PC9, H1975 and HCC827GR treated with 0.1µM, 1µM, 10µMerlotinib or DMSO for 20 minutes or continuously. FACS-analysis was done 24 hours after initial exposure to erlotinib and read-out was normalized to DMSO-control. Change of Annexin V/PI-double positive cells ±SD are shown. *p < 0.05, **p < 0.001. C. shows relative tumor volumes of xenografts ±SD (HCC827, PC9 and H1975). Xenograft harboring mice were treated with 30mg/kg erlotinib daily or 200mg/kg erlotinib every 2nd day p.o. and tumor volumes were measured every 2nd day.*p < 0.05, **p < 0.001.
Toxicity as well as pharmacokinetic aspects of high-dose pulse treatment
Alignment of murine and human EGFR revealed only one different amino acid in the kinase domain (Y > F at 771), which is unlikely to affect binding of erlotinib (Figure S4A). Thus, erlotinib is expected to inhibit EGFR signaling in all murine tissues, in which EGFR is expressed. We found that mice treated with 100mg/kg erlotinib daily lost about 20% of weight. The intermittent_2day schedule led to an initial weight loss of 10%, too, but mice typically recovered within 20 days. Mice treated with 50mg/kg daily and the intermittent_4day schedule initially showed a slight reduction of weight of about 5%, but recovered within 10 days. Mice treated with 15mg/kg or 30mg/kg daily and the control group did not exhibit significant weight loss (Figure 2A). Both the 50mg/kg and 100mg/kg daily schedules were highly toxic: severe diarrhea occurred in 88% and 73% of mice, respectively. By contrast, diarrhea occurred in 49% of the mice in the intermittend_2day and 20% of the intermittend_4day groups (Figure S4B). In the 100mg/kg daily group significantly more mice died during therapy than in the other groups (Figure S4C). Rash occurred in 38% of mice in the 100mg/kg daily cohort, whereas rash was rare in the mice treated with the other schedules (Figure S4D). There was no case of diarrhea or rash in the control group. Thus, intermittent high-dose treatment of EGFR-mutant tumors with erlotinib enhanced tumor control with limited toxicity.
Estimates for erlotinib peak plasma concentrations after a single dose of 30mg/kg or 200mg/kg of erlotinib were 6.5µmol/l and 11.7µmol/l, respectively. The area under the curve (AUC) showed linear increase with the dose (3.84µmol*h vs. 24.45µmol*h (p < 0.001)). Thus, erlotinib clearance was independent on the dose (2.72l/h vs. 3.05l/h (p = 0.45)); however, the apparent absorption rate constant was much higher for the low dose (0.36/h vs. 0.08/h (p < 0.001)), suggesting some saturation of absorbtion of erlotinib in the intestine (Figure 2B). The concentrations of erlotinib in tumor lysates of HCC827, PC9 and H1975 xenografts peaked after 6 hours. In mice treated with 30mg/kg of erlotinib the peak concentrations reached about 100ng of erlotinib/µg of protein and declined after 12 hours. Treatment with 200mg/kg led to peak tumor tissue concentrations of 200-250ng/µg. In muscle tissue concentrations were lower, suggesting enrichment in tumor tissue (Figure 2C). The peak tumor concentrations of the active metabolite OSI-420 were about 10ng/µg for 30mg/kg erlotinib and declined completely within 24 hours, and reached 30 to 50ng/µg in tumor tissue and about 20ng/µg in muscle tissue for the 200mg/kg dose (Figure S5). We finally assessed the pharmacodynamic effects of high-dose treatment in tumors explanted from treated mice by pEGFR-immunohistochemistry. In the sensitive HCC827- and PC9-xenografts both 30mg/kg and 200mg/kg of erlotinib reduced pEGFR compared to untreated controls and the resistant H1975-xenografts. However single dosing of 200mg/kg of erlotinib reduced pEGFR much stronger than 30mg/kg (Figure 2D).
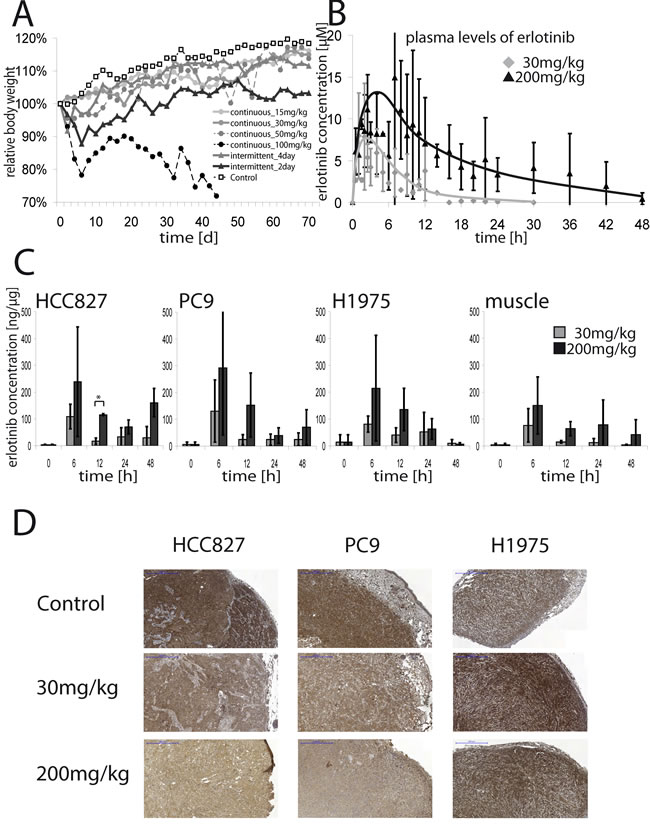
Figure 2: A. Relative body weight of mice treated witherlotinib15mg/kg daily, 30mg/kg daily, 50mg/kg daily, 100mg/kg daily, 200mg/kg every 4th day, 200mg/kg every 2nd day or vehicle detergent alone. Shown is the mean weight of mice, set relative to the weight at the beginning of the therapy. In B. mean plasma concentrations of erlotinib ±SD in mice are shown. Non-tumor harboring mice were treated orally with a single dose of either 30mg/kg or 200mg/kg and blood samples were taken from the tail-vein. Plasma-concentrations were determined by liquid chromatography tandem mass spectrometry. C. Mean erlotinib concentrations ±SD in tumor lysates (HCC827, PC9, H1975) or lysates of muscle tissue of mice treated with a single dose of either 30mg/kg or 200mg/kg erlotinib are shown. Lysates were prepared from untreated mice or 6, 12, 24 or 48 hours after administration of erlotinib. Erlotinib concentrations in the supernatant were assessed by mass spectrometry and set relative to the protein-amount of the lysate.*p < 0.05. D. Representative IHC-stainings for pEGFR of tumors (HCC827, PC9, H1975) of mice either untreated or treated with a single dose of 30mg/kg or 200mg/kg erlotinib. Tumors were resected 12 hours after treatment. 5x magnification, blue scale bar indicates 500µm.
In vivo pharmacodynamic assessments by 18F-FLT-PET
We have recently shown in mice and in patients that erlotinib induces early cell cycle arrest in EGFR-mutant tumors that precedes induction of apoptosis and that can be monitored in vivo using 18F-FLT-PET [26, 27]. We therefore determined, whether the dynamics of induction of cell cycle arrest and tumor shrinkage might also be similar in the 30mg/kg daily and the intermittend_2day schedules. While in H1975 xenografts uptake of 18F-FLT was not reduced by erlotinib treatment (Figure 3A, lower panel, Figure 3B, right panel, Figure S6 and S7), the decrease in relative FLT-uptake was similar in both the continuous 30mg/kg and the intermittent_2day schedules in HCC827 and PC9 xenografts (Figure 3A, upper panel, Figure 3B left panel, Figure 3C and Figure S6 and S7). In PC9 tumors the intermittent_4day showed a similar decline in FLT-uptake at days 1, 6 and 8; however 18F-FLT-uptake increased again at days 20 and 27, but not in the continuous_30mg/kg and intermittent_2day group (p < 0.05) (Figure 3C). This observation corroborates the notion that both high trough and peak levels of erlotinib are relevant for cell cycle arrest [28, 29] and tumor shrinkage.
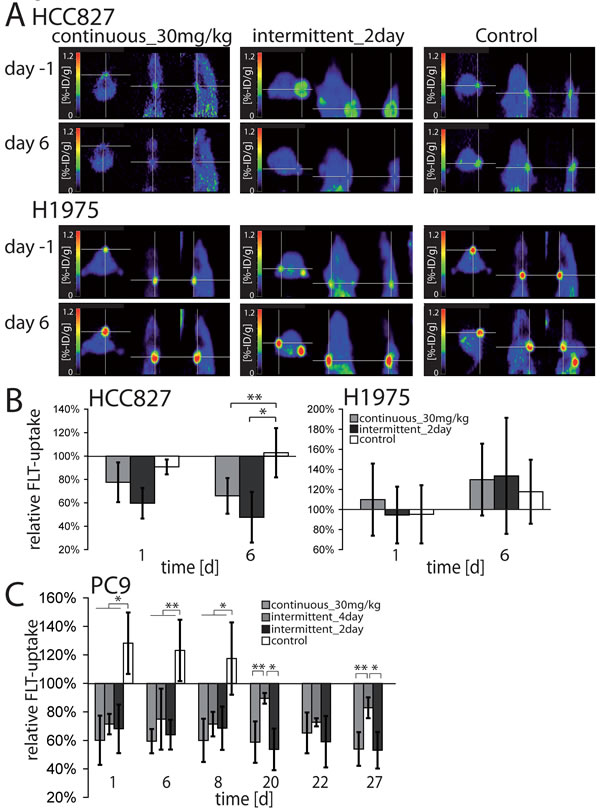
Figure 3: In A. representative 18F-FLT-images of mice harboring HCC827 or H1975 xenografts treated with 30mg/kg erlotinib daily, 200mg/kg erlotinib every 2nd day or vehicle are shown. 18F-FLT-PET measurements were performed the day before start of therapy and at day 6 of therapy. The cross hairs indicate tumor positions. B. Change in relative 18F-FLT-uptake of HCC827- and H1975-xenografts. Mice were treated with either 30mg/kg erlotinib daily or 200mg/kg erlotinib every 2nd day or vehicle. 18F-FLT-PET-imaging was performed the day before stat of therapy (day -1), day 1 and 6 after start of therapy. All values were set relative to day -1. Error bars indicate ±SD, *p < 0.05, **p < 0.001. C. Change in relative 18FFLT-uptake of PC9 xenografts. Mice were treated with either 30mg/kg erlotinib daily,200mg/kg erlotinib every 4th day, 200mg/kg erlotinib every 2nd day (up to day 27) or vehicle (up to day 8). 18F-FLT-PET- imaging was performed at day -1, day 1, 6, 8, 20, 22 and27 after start of therapy. Treatment days of the intermittent_4day schedule were: day 0, 4, 8, 12, 16, 20, 24, 28. All values were set relative to day -1. Error bars indicate ±SD.*p < 0.05, **p < 0.001.
Intermittent high-dose erlotinib treatment improves progression-free survival of mice bearing EGFR-mutant xenografts.
High doses of erlotinib improved tumor shrinkage by enhanced target signaling suppression. Efficacy of EGFR inhibitors is ultimately limited by the emergence of resistance. We therefore tested the hypothesis that intermittent high dose treatment of EGFR-mutant tumors might also enhance the duration of response. Mice engrafted with HCC827 or PC9 cells were therefore treated with the most effective, but still tolerable schedules for up to 400 days: 30mg/kg erlotinib daily or 200mg/kg erlotinib every other day. Median follow-up time for HCC827 was 280 (continuous_30mg/kg) and 400 (intermittent_2day) days and for PC9 102 and 160 days, respectively (Figure S9). All tumor volumes of these mice are individually shown in Figure 4A. After initial response several tumors restarted to grow. However, in the case of HCC827 xenografts treated with 200mg/kg every second day no resistance emerged, whereas 40% of the tumors treated with 30mg/kg daily became resistant (p < 0.05). PC9-xenografts showed acquired resistance in both therapy groups; however, recurrence occurred later (Figure 4A, right) and progression-free survival was longer in the group receiving intermittent high-dose treatment (p < 0.05) (Figure 4B).
Sequencing of tumors explanted at the time of resistance revealed the T790M resistance mutation in PC9 xenografts (Figure S10). These results show that more pronounced target suppression not only leads to higher magnitude, but also greater duration of response.
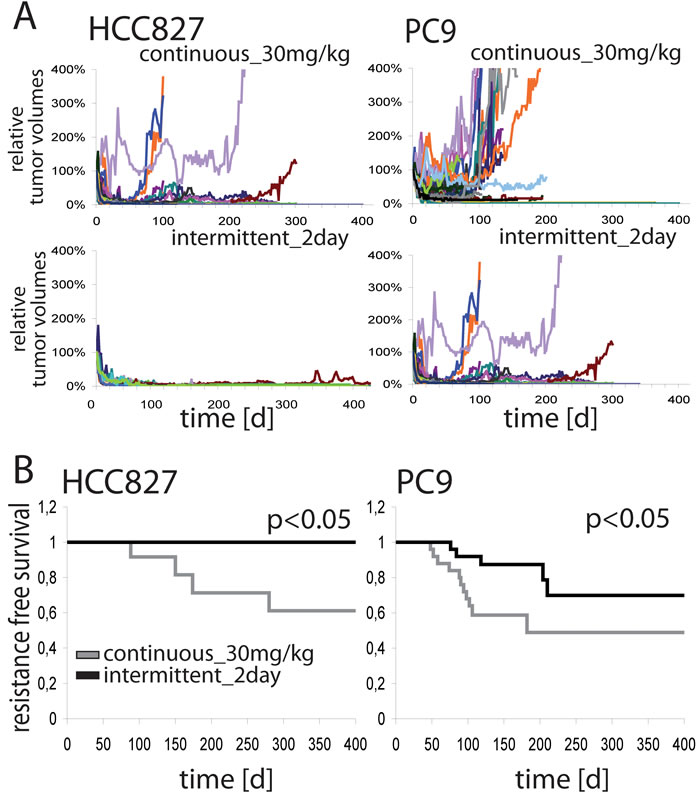
Figure 4: A. Tumor volumes of HCC827- (left panel) and PC9- (right panel) xenograft harboring mice treated long term with erlotinib either 30mg/kg daily (upper row) or 200mg/kg every 2nd day (lower row). Each colored line represents the relative volume of one single tumor. B. Kaplan Meier curves of resistance free survival of mice harboring HCC827- (left) and PC9- (right) xenografts treated long term with erlotinib. Shown are the treatment schedules with 30mg/kg erlotinib daily or 200mg/kg erlotinib every 2nd day. p indicates statistical significance by log-rank-test.
Discussion
Here we present a strategy for improving therapeutic efficacy in a xenograft model of EGFR-mutant lung cancer by intermittent high-dose scheduling of erlotinib. This schedule was tolerable, but enhanced both the magnitude and the duration of response. Despite the high dose, efficacy was due to on-target effects of erlotinib as the treatment effects were not observed in cells bearing the T790M resistance mutation.
We could show that plasma peak levels of erlotinib after a dose of 200mg/kg were only two fold higher than after 30mg/kg indicating saturation of absorption. By contrast, the AUC was about 6-fold higher suggesting that the duration of target suppression was increased several-fold. As a consequence we found for pulsatile high-dose treatment of erlotinib stronger and more durable target suppression by pEGFR-staining, and could show sufficient cell-cycle-arrest in-vivo by 18F-FLT-PET.
Consequently, intermittent high-dose treatment improved the progression-free survival in two EGFR-mutant xenografts in a long-term treatment study. While emergence of resistance was completely abolished in HCC827 xenografts, resistance occurred less frequently and at a later time in PC9 xenografts. The latter are known to become resistant to erlotinib over time due to emergence of the T790M resistance mutation; thus, the efficacy of reversible quinazoline EGFR inhibitors will always be limited in these tumors. In line with these findings we could detect the T790M mutation in resistant PC9 tumors, but not in HCC827 tumors. However, the observation that PFS could still be improved in PC9 tumors may arguably be due to the possibility that high doses of erlotinib are still capable of inhibiting T790M-mutant EGFR to some extent (note that the biochemical IC50 of erlotinib for EGFRT790M is still in the nanomolar range).
In a first study in humans treated with an intermittent high-dose schedule of erlotinib, patient suffered only moderate side-effects [30]. The limited efficacy in this study is likely due to the lack of patient selection based on EGFR mutations. Thus, patients with EGFR-mutant lung cancer may benefit from high-dose treatment with EGFR inhibitors at reasonable toxicity. In line with this notion, Riely and colleagues showed in a recent phase II study, that OS of patients can be prolonged by a high-dose pulse of erlotinib before chemotherapy, without increasing toxic side effects [31]. Patients on this trial were not selected based on EGFR mutations either. Thus, patients with EGFR-mutant lung cancer may derive further benefit from such intermittent high-dose schedule. However, in these studies no pharmacokinetic analyses in patients treated with high-dose erlotinib were performed. Further studies are required to determine peak and AUC levels during high dose erlotinib treatment and to evaluate whether these high dose erlotinib levels in mice are tolerable in humans. Finally, these high doses of erlotinib may penetrate the blood-brain barrier and induce tumor shrinkage in EGFR-mutant brain metastases, which are usually not tractable by standard dosing of erlotinib [32].
In summary, we provide evidence that intermittent high-dose treatment of EGFR-mutant tumors with erlotinib enables enhanced tumor shrinkage and prolonged PFS, while limiting toxicity in a mouse xenograft study. This effect is associated with high trough and peak levels, which induced more durable shutdown of EGFR-associated oncogenic signaling. While treatment is increasingly tailored to each patient based on tumor genotyping, the established 150mg once daily dose of erlotinib may simply not be the ideal schedule for optimal target shutdown and tumor control. Thus, protocols testing such intermittent high dose pulses of erlotinib specifically in patients with EGFR-mutant lung cancer warrant clinical exploration.
Materials and Methods
Cell-culture and reagents
The human NSCLC celllines PC9, HCC827 and H1975 were obtained from the American Type Culture Collection (ATCC), HCC827GR were kindly provided by the laboratory of Jeff Engelman. Cells were cultured in RPMI-1640 medium with 10% FCS and 1% Penicillin+Streptomycin.
Erlotinib was purchased from LC Labs, USA. For in-vitro studies erlotinib was prepared in stock solution of 10mM in DMSO (Sigma Aldrich, Germany) and stored at -20°C. For in-vivo studies erlotinib was dissolved in 6% Captisol® (CyDex Inc., USA) in concentrations of 10mg/ml (continuous schedules) or 30mg/ml (intermittent schedules) and stored in a rotating device at 4°C.
Immunoblotting
Immunoblotting was performed using the following antibodies: pEGFR (Y1068), EGFR, pAkt (S473), AKT, pErk 44/42 (Thr202/Tyr204), ERK 44/42, Caspase-3 (Cell Signaling Technologies, USA), ß-actin (clone C4) (MP Biomedicals LLC, USA), anti-rabbit-HRP- and anti-mouse-HRP-antibody (Millipore, Germany).
Annexin V-flow-cytometry
Flow cytometryofHCC827, PC9, H1975 and HCC827GR cells treated with erlotinib (0.1µM, 1µM or 10µM) or DMSO for either 20 minutes or 24 hours, was performed using the AnnexinV-FITC Apoptosis Detection Kit I (BD Pharmingen, Germany) according to the manufactures protocol and measured by Gallios Flow Cytometer (Beckmann Coulter), detecting at least 100,000 events per probe. Data was evaluated by setting appropriate gates in Kaluza analysis software (Beckman Coulter) and apoptosis was calculated as the difference between sample and DMSO control.
In vivo experiments
All animal procedures were approved by the local animal protection committee and the local authorities.
All experiments were performed in 10 to 15 week old male athymic NMRI-nude-mice (Janvier, Europe) as described recently [26, 33]. For creation of xenografts tumor cells (HCC827, PC9 and H1975) were implanted subcutaneously. Therapy was started when tumors reached a size of approx. 100mm³. Mice were treated by oral gavage of erlotinib in these schedules: 15mg/kg daily, 30mg/kg daily, 200mg/kg every 2nd day or 200mg/kg every 4th day or 100µl vehicle detergent daily.
Tumor size was monitored every 2nd day by calimetric measurement. Tumor volumes were calculated by the modified ellipsoid formula [V = 0.5 x (long diameter) x (short diameter)²]. All absolute tumor volumes were set relative to day 0.
Toxicity of erlotinib in mice
Human and murine EGFR were aligned using the ClustalW-alignment (http://www.ebi.ac.uk/Tools/msa/clustalw2) and performed an analysis of erlotinib binding to the ATP-binding pocket of the EGFR. To assess the tolerable therapy schedules of erlotinib we treated non-tumor harboring mice with these schedules of erlotinib: 15mg/kg, 30mg/kg, 50mg/kg or 100mg/kg daily, or with 200mg/kg every 2nd or 4th day, as well as with vehicle detergent alone daily. Weight was measured every 2nd day and toxic side effects (diarrhea, rash and death) in all treated mice were monitored.
Pharmacokinetics of erlotinib
For pharmacokinetic analyses mice were treated with a single dose of either 30mg/kg or 200mg/kg erlotinib. Blood samples were taken from the tail-vein and erlotinib plasma concentrations were quantified using liquid chromatography tandem mass spectrometry with a lower limit of quantification of 18ng/ml.
Mass spectrometry of erlotinib from tumor lysates
For determination of erlotinib concentration in the tumor, HCC827, PC9 and H1975 xenografts were used. Mice were treated with a single dose of either 30mg/kg or 200mg/kg erlotinib and sacrificed 6, 12, 24 or 48 hours after treatment or without treatment. Then tumors were resected and lysed for determination of erlotinib and OSI-420 concentrations within the tumor by mass spectrometry. Erlotinib and OSI-420 concentration in the lysates were set relative to the protein concentration of the lysate.
Immunohistochemistry
For pEGFR-immunohistochemistry tumors were resected 12 hours after treatment with either 30mg/kg or 200mg/kg erlotinib or without treatment. Tumors were fixed in 4% formaldehyde for 24 hours and transferred to PBS. Tissues were embedded in paraffin, were cut and stained with pEGFR Tyr 1068 primary antibodies (1:100 over night, pretreatment pH6 20min), Corresponding secondary antibody detection kits for reduced background on murine tissue were used (Histofine Simple Stain Mouse MAX PO and Histofinemousestain kit, medac) and stained on an automated stainer (LabVisionAutostainer 480S, Thermo Scientific).
18F-FLT-PET imaging
Synthesis of 18F-fluoro-L-thymidine (18F-FLT) and PET measurement protocols were performed as described elsewhere [26, 34, 35].
Mice harboring HCC827, PC9 and H1975 xenografts were measured one day before start of therapy (day -1), at the second day of therapy (d = 1) and at day 6 and 8 using a R4 microPET scanner (Concord Microsystems, Inc, Knoxville, TN). Quantitative analysis was done using the in-house software VINCI using a region of interest (ROI) analysis. All data were decay corrected.
Statistical analysis
For statistical analyses we used Sigma Plot 11.0 (Systat Software, USA). We used student’s t-test (unpaired, 2-sided), χ2-test and log-rank-test. p < 0.05 was considered statistical significant.
For further details we refer to Supplementary Methods.
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft through SFB832 (Z2 to R.T. Ullrich and B. Neumaier; TP6 to R.T. Ullrich; TP5 to L.C. Heukamp; and Z1 to L.C. Heukamp), by the DFG (Grant No.: UL379/1-1 to R.T. Ullrich) and by the Nachwuchsforschungsgruppen-NRW (Grant No.: 1411ng005 to R.T. Ullrich).
Conflicts of interest
RTU received consulting fees from Novartis.
References
1. Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y, Nishiwaki Y, Ohe Y, Yang JJ, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med, 2009; 361: 947-57.
2. Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science, 2004; 304: 1497-500.
3. Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. The New England journal of medicine, 2004; 350: 2129-39.
4. Sequist LV, Martins RG, Spigel D, Grunberg SM, Spira A, Janne PA, Joshi VA, McCollum D, Evans TL, Muzikansky A, Kuhlmann GL, Han M, Goldberg JS, et al. First-line gefitinib in patients with advanced non-small-cell lung cancer harboring somatic EGFR mutations. Journal of clinical oncology : official journal of the American Society of Clinical Oncology, 2008; 26: 2442-9.
5. Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG and Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS medicine, 2005; 2: e73.
6. Balak MN, Gong Y, Riely GJ, Somwar R, Li AR, Zakowski MF, Chiang A, Yang G, Ouerfelli O, Kris MG, Ladanyi M, Miller VA and Pao W. Novel D761Y and common secondary T790M mutations in epidermal growth factor receptor-mutant lung adenocarcinomas with acquired resistance to kinase inhibitors. Clinical cancer research : an official journal of the American Association for Cancer Research, 2006; 12: 6494-501.
7. Costa DB, Schumer ST, Tenen DG and Kobayashi S. Differential responses to erlotinib in epidermal growth factor receptor (EGFR)-mutated lung cancers with acquired resistance to gefitinib carrying the L747S or T790M secondary mutations. Journal of clinical oncology : official journal of the American Society of Clinical Oncology, 2008; 26: 1182-4; author reply 1184-6.
8. Bean J, Riely GJ, Balak M, Marks JL, Ladanyi M, Miller VA and Pao W. Acquired resistance to epidermal growth factor receptor kinase inhibitors associated with a novel T854A mutation in a patient with EGFR-mutant lung adenocarcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research, 2008; 14: 7519-25.
9. Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, Kosaka T, Holmes AJ, Rogers AM, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science, 2007; 316: 1039-43.
10. Bean J, Brennan C, Shih J-Y, Riely G, Viale A, Wang L, Chitale D, Motoi N, Szoke J, Broderick S, Balak M, Chang W-C, Yu C-J, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proceedings of the National Academy of Sciences of the United States of America, 2007; 104: 20932-7.
11. Sos ML, Koker M, Weir BA, Heynck S, Rabinovsky R, Zander T, Seeger JM, Weiss J, Fischer F, Frommolt P, Michel K, Peifer M, Mermel C, et al. PTEN loss contributes to erlotinib resistance in EGFR-mutant lung cancer by activation of Akt and EGFR. Cancer research, 2009; 69: 3256-61.
12. Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, McDermott U, Azizian N, Zou L, Fischbach MA, Wong KK, Brandstetter K, Wittner B, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell, 2010; 141: 69-80.
13. Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T, Abdel-Rahman M, Wang X, Levine AD, Rho JK, Choi YJ, Choi CM, Kim SW, et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nature genetics, 2012; 44: 852-60.
14. Takezawa K, Pirazzoli V, Arcila ME, Nebhan CA, Song X, de Stanchina E, Ohashi K, Janjigian YY, Spitzler PJ, Melnick MA, Riely GJ, Kris MG, Miller VA, et al. HER2 amplification: a potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFRT790M mutation. Cancer discovery, 2012; 2: 922-33.
15. Terai H, Soejima K, Yasuda H, Nakayama S, Hamamoto J, Arai D, Ishioka K, Ohgino K, Ikemura S, Sato T, Yoda S, Satomi R, Naoki K, et al. Activation of the FGF2-FGFR1 Autocrine Pathway: A Novel Mechanism of Acquired Resistance to Gefitinib in NSCLC. Molecular cancer research : MCR, 2013.
16. Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, Bergethon K, Shaw AT, Gettinger S, Cosper AK, Akhavanfard S, Heist RS, Temel J, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Science translational medicine, 2011; 3: 75ra26.
17. Hammerman PS, Janne PA and Johnson BE. Resistance to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Non-Small Cell Lung Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research, 2009; 15: 7502-7509.
18. Godin-Heymann N, Ulkus L, Brannigan BW, McDermott U, Lamb J, Maheswaran S, Settleman J and Haber DA. The T790M „gatekeeper“ mutation in EGFR mediates resistance to low concentrations of an irreversible EGFR inhibitor. Molecular cancer therapeutics, 2008; 7: 874-9.
19. Shah NP, Kasap C, Weier C, Balbas M, Nicoll JM, Bleickardt E, Nicaise C and Sawyers CL. Transient potent BCR-ABL inhibition is sufficient to commit chronic myeloid leukemia cells irreversibly to apoptosis. Cancer cell, 2008; 14: 485-93.
20. Amin DN, Sergina N, Ahuja D, McMahon M, Blair JA, Wang D, Hann B, Koch KM, Shokat KM and Moasser MM. Resiliency and vulnerability in the HER2-HER3 tumorigenic driver. Science translational medicine, 2010; 2: 16ra7.
21. Das Thakur M, Salangsang F, Landman AS, Sellers WR, Pryer NK, Levesque MP, Dummer R, McMahon M and Stuart DD. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature, 2013; 494: 251-5.
22. Chmielecki J, Foo J, Oxnard GR, Hutchinson K, Ohashi K, Somwar R, Wang L, Amato KR, Arcila M, Sos ML, Socci ND, Viale A, de Stanchina E, et al. Optimization of dosing for EGFR-mutant non-small cell lung cancer with evolutionary cancer modeling. Science translational medicine, 2011; 3: 90ra59.
23. Foo J, Chmielecki J, Pao W and Michor F. Effects of pharmacokinetic processes and varied dosing schedules on the dynamics of acquired resistance to erlotinib in EGFR-mutant lung cancer. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer, 2012; 7: 1583-93.
24. Gleeson MP, Hersey A, Montanari D and Overington J. Probing the links between in vitro potency, ADMET and physicochemical parameters. Nature reviews. Drug discovery, 2011; 10: 197-208.
25. Swinney DC and Anthony J. How were new medicines discovered? Nature reviews. Drug discovery, 2011; 10: 507-19.
26. Ullrich RT, Zander T, Neumaier B, Koker M, Shimamura T, Waerzeggers Y, Borgman CL, Tawadros S, Li H, Sos ML, Backes H, Shapiro GI, Wolf J, et al. Early detection of erlotinib treatment response in NSCLC by 3’-deoxy-3’-[F]-fluoro-L-thymidine ([F]FLT) positron emission tomography (PET). PloS one, 2008; 3: e3908.
27. Zander T, Scheffler M, Nogova L, Kobe C, Engel-Riedel W, Hellmich M, Papachristou I, Toepelt K, Draube A, Heukamp L, Buettner R, Ko YD, Ullrich RT, et al. Early prediction of nonprogression in advanced non-small-cell lung cancer treated with erlotinib by using [(18)F]fluorodeoxyglucose and [(18)F]fluorothymidine positron emission tomography. Journal of clinical oncology : official journal of the American Society of Clinical Oncology, 2011; 29: 1701-8.
28. Sherley JL and Kelly TJ. Regulation of human thymidine kinase during the cell cycle. The Journal of biological chemistry, 1988; 263: 8350-8.
29. Leyton J, Latigo JR, Perumal M, Dhaliwal H, He Q and Aboagye EO. Early detection of tumor response to chemotherapy by 3’-deoxy-3’-[18F]fluorothymidine positron emission tomography: the effect of cisplatin on a fibrosarcoma tumor model in vivo. Cancer research, 2005; 65: 4202-10.
30. Milton DT, Azzoli CG, Heelan RT, Venkatraman E, Gomez JE, Kris MG, Krug LM, Pao W, Rizvi NA, Dunne M and Miller VA. A phase I/II study of weekly high-dose erlotinib in previously treated patients with nonsmall cell lung cancer. Cancer, 2006; 107: 1034-41.
31. Riely GJ, Rizvi NA, Kris MG, Milton DT, Solit DB, Rosen N, Senturk E, Azzoli CG, Brahmer JR, Sirotnak FM, Seshan VE, Fogle M, Ginsberg M, et al. Randomized phase II study of pulse erlotinib before or after carboplatin and paclitaxel in current or former smokers with advanced non-small-cell lung cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology, 2009; 27: 264-70.
32. Grommes C, Oxnard GR, Kris MG, Miller VA, Pao W, Holodny AI, Clarke JL and Lassman AB. “Pulsatile” high-dose weekly erlotinib for CNS metastases from EGFR mutant non-small cell lung cancer. Neuro-oncology, 2011; 13: 1364-9.
33. Chatterjee S, Heukamp LC, Siobal M, Schottle J, Wieczorek C, Peifer M, Frasca D, Koker M, Konig K, Meder L, Rauh D, Buettner R, Wolf J, et al. Tumor VEGF:VEGFR2 autocrine feed-forward loop triggers angiogenesis in lung cancer. The Journal of clinical investigation, 2013; 123: 1732-40.
34. Kobus D, Giesen Y, Ullrich R, Backes H and Neumaier B. A fully automated two-step synthesis of an (18)F-labelled tyrosine kinase inhibitor for EGFR kinase activity imaging in tumors. Appl Radiat Isot, 2009; 67: 1977-84.
35. Shields AF, Grierson JR, Dohmen BM, Machulla HJ, Stayanoff JC, Lawhorn-Crews JM, Obradovich JE, Muzik O and Mangner TJ. Imaging proliferation in vivo with [F-18]FLT and positron emission tomography. Nature medicine, 1998; 4: 1334-6.