
Introduction
Epigenetic modifications are a common hallmark of all human cancers [1] and can include alterations in methylation patterns where methyl groups are added or removed from CpG sites in DNA. In hematopoietic malignancies, the promoter CpG islands of tumor-suppressor genes are often hypermethylated and thus epigenetically silenced [2]. Myelodysplastic syndrome (MDS) is a group of clonal stem cell disorders characterized by ineffective hematopoiesis, a potential for progression to acute myeloid leukemia (AML) and which is connected to deregulation of innate immune and inflammatory signaling [3]. Abnormal cellular differentiation in MDS can frequently be linked to aberrant genome-wide and loci-specific DNA hypermethylation as well as mutations in genes that regulate epigenetic programs [4].
Several epigenetic drugs have been developed for cancer treatment, including the cytosine analogs 5-azacytidine (5-aza) and 5-aza-2’-deoxycytidine (decitabine). 5-aza has been shown to induce a clinical response in around 50% of patients with MDS and treatment is associated with prolonged survival [5]. For the last 10 years, 5-aza has been the first-line treatment for patients with high-risk MDS and acute myeloid leukemia (AML) with dysplastic features and 20-29% marrow blasts [6]. 5-aza and decitabine lack methylation sites thus effectively acting as DNA methyltransferase inhibitors (DNMTs). 5-aza particularly inhibits DNMT1, which is completely depleted after 5-aza exposure due to the formation of an irreversible covalent bond, leading to loss of DNA methylation marks [7, 8]. In this way, methylation-dependent silencing in cancer cell lines is reversed in a dose-dependent manner, where only low doses that do not disturb replication show hypomethylating effects [9].
Besides demethylation, administration of 5-aza may also delay the progression of MDS by influencing the immunological control of the malignant clone [10]. Natural killer (NK) cells express a vast array of activating receptors that sense cellular stress, including NKG2D, DNAM-1 and natural cytotoxicity receptors (NCRs) [11]. Stressed and transformed cells upregulate MHC class I chain related proteins A and B (MICA/B) and UL16-binding proteins (ULBPs) (NKG2DL) that interact with NKG2D, one of the major NK-cell activating receptors involved in tumor cell recognition [12]. Absence of transcription of NKG2DL, due to high levels of DNA methylation, can be detected in tumor cell lines but expression can be restored by treatment with demethylating agents [13, 14].
In addition to synergistic signaling through activating receptors, NK cell cytotoxicity is tightly controlled by the expression of MHC class I binding receptors including CD94/NKG2A and killer cell immunoglobulin-like receptors (KIRs). Variegated expression of these receptors in the NK cell repertoire confers tolerance to normal cells but also contributes to determine the functional potential of the cells in a process termed education [15, 16]. Interactions between inhibitory KIRs and/or CD94/NKG2A and their cognate HLA class I ligands at steady state influence the ability of the cell to respond to environmental changes, including the recognition of cells with low expression of HLA class I [17, 18]. KIRs are acquired late during NK cell differentiation and are also commonly expressed on terminally differentiated CD56+ T cells [19]. In both T and NK cells, KIR expression is regulated at the transcriptional level by epigenetic changes in a complex and synchronized fashion determined by the activities from multiple promoters [20]. KIR genes are consistently demethylated in cells expressing KIRs and methylated in cells without KIR expression [21, 22]. The transcriptional regulation of KIR genes leads to a stochastic expression of KIR proteins at the cell surface, where some NK cells do not express a single KIR while others express combinations of multiple KIRs [23, 24]. In vitro, NK cell lines and primary ex vivo expanded NK cells upregulate KIRs on their cell surface during decitabine [21, 22] and 5-aza stimulation [25].
In spite of existing in vitro data on the effects of hypomethylating agents on the NK cell compartment, little is known regarding the possible effects of 5-aza on NK cells in vivo. Given the high turn-over of NK cells in peripheral blood [26] we hypothesized that incorporation of 5-aza during NK cell homeostasis might lead to altered expression of KIR, thereby affecting their functionality through education and possibly impact their ability to mediate anti-tumor immunity. Moreover, we reasoned that changes in KIR expression might serve as a sensitive tool to monitor uptake of 5-aza into circulating cells in vivo, the evidence for which is sparse [27]. Our data show that expression of multiple KIRs was rapidly induced on NK cells during in vitro culture with physiologically relevant low doses of 5-aza. This effect was tightly linked to IL-2 driven cellular proliferation and therefore most prominent in less differentiated cells with high proliferative capacity. Longitudinal assessment of NK cells in MDS patients undergoing systemic 5-aza treatment revealed increased frequencies of KIR expression in Ki-67+ NK cells, indicative of 5-aza uptake during cell division in vivo. Interestingly, cycling NK cells exposed to 5-aza in vitro had higher degranulation and IFN-γ production in response to K562 target cells suggesting enhanced function post-5-aza exposure. Our data reveal an imprint of 5-aza on NK cells in vivo and support the notion that the therapeutic effects of 5-aza may be partially mediated via epigenetic remodeling of the immune system.
Results
5-aza increases KIR expression on proliferating NK cells in vitro
To assess the effect of the hypomethylating drug 5-aza on the NK cell KIR repertoire of healthy individuals, NK cells were isolated from PBMC and cultured in vitro with IL-2 in the presence or absence of 5-aza. 5-aza was added consecutively to the culture at dose-levels in the range of those observed in plasma of patients receiving systemic treatment [28]. After six days the frequency of cells expressing KIRs was analyzed using a flow-cytometry panel that enabled identification of cells expressing single KIRs or combination thereof (Figure 1A). Addition of 5-aza significantly increased the frequency of total KIR-expressing NK cells, of NK cells co-expressing 2, 3 or 4 KIRs and of each of the analyzed inhibitory KIRs (Figure 1B-1D). In the three donors with group B KIR haplotype, a similar increase in the expression of KIR2DS1 was noted (as illustrated by one donor in Figure 1A).
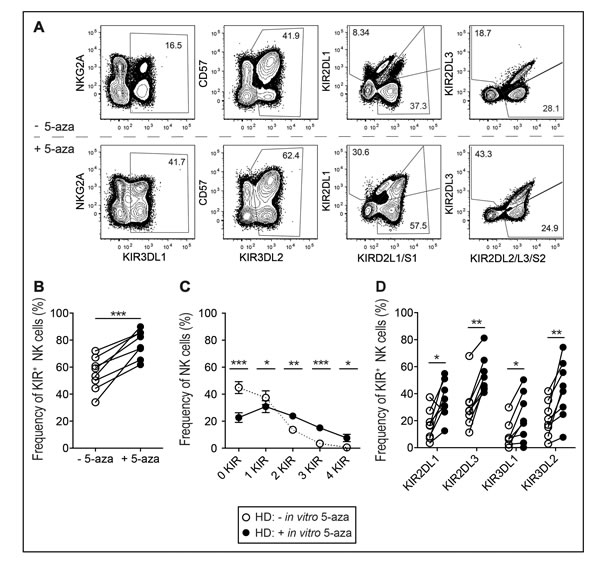
Figure 1: KIR repertoires in the NK cell population after in vitro 5-aza addition. NK cells were isolated from healthy donor PBMC and cultured in 500U/ml of IL-2 for six days with or without the addition of 5-aza for the first four consecutive days. A. Gating scheme to identify subsets of CD56+ NK cells expressing single KIR and combinations thereof. Doublet cells were excluded based on an FSC-area versus FSC-height gate. Gates were set on live CD3− cells as determined by staining with a dead cell marker (DCM) and anti-CD3. Shown in B. frequency of KIR+CD56+ NK cells, C. the number of expressed inhibitory KIRs and in D. each investigated KIR. HD n = 8.
As the hypomethylating effects of 5-aza require incorporation into DNA during cell division [29], we stratified the analysis based on the number of cell divisions (Figure 2A) induced by IL-2. The effect of 5-aza on KIR expression was most evident in cycling cells, where nearly 100% of the cells expressed at least one KIR following three or more cell divisions (Figure 2B). This was in sharp contrast to cultures without 5-aza where we observed a gradual decline in KIR expression, presumingly due to the preferential proliferation of less differentiated KIR- NK cells [30]. Notably, late generation NK cells also co-expressed multiple KIRs, which was rarely seen in non-dividing cells (Figure 2C-2E). To assess if 5-aza preferentially induced specific combinations of KIRs, we resolved the KIR repertoire of NK cells in generation 3+. Again, the frequency of NK cells expressing three or more KIRs was higher with addition of 5-aza, although no specific pattern in the KIR repertoire was noticed (Figure 2F).
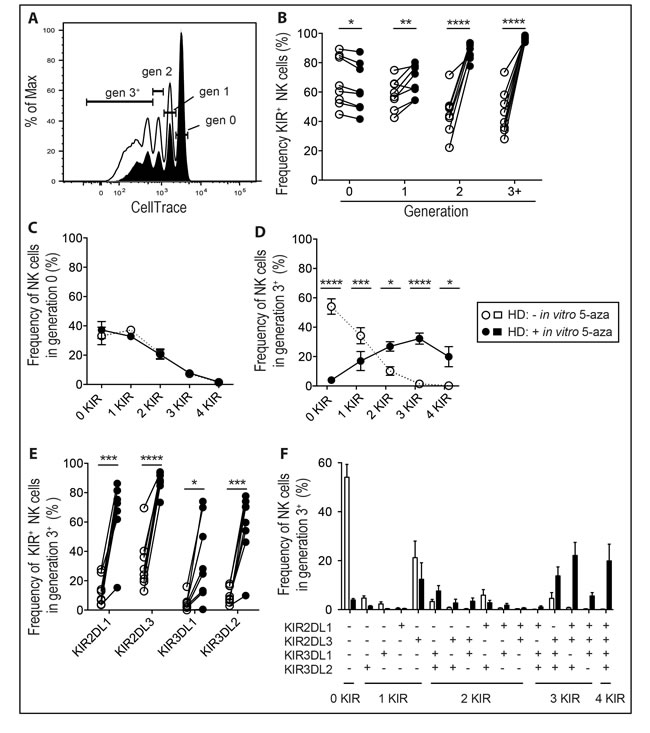
Figure 2: 5-aza induces expression of multiple KIRs on proliferating NK cells. NK cells were isolated from healthy donor PBMC and cultured in 500U/ml of IL-2 for six days with or without the addition of 5-aza for the first four consecutive days. Shown in A. CellTrace dilution of NK cells from one representative donor, open histogram represents cultures without 5-aza and black histogram with 5-aza. In B. the frequency of KIR+CD56+ NK cells in each generation, in C. the number of expressed KIRs in non-dividing NK cells or D. in those that had undergone three or more cell divisions (generation 3+). In E. the frequency of each investigated KIR in NK cells in generation 3+ and in F. resolution of their KIR repertoires. HD n = 8.
5-aza-induced KIR expression is most evident in NKG2A+CD57- NK cells
To further explore the effect of 5-aza on NK cells, we stratified the analysis based on the stage of NK cell differentiation as determined by the expression of NKG2A and CD57. In agreement with previous results [30] less differentiated NKG2A+CD57- NK cells proliferated the most in response to IL-2 (Figure 3A). Although 5-aza induced a significant increase in the expression of multiple KIRs in cycling cells of both subsets, the difference was most evident on NKG2A+CD57- NK cells that had lower initial KIR expression and higher proliferation rates (Figure 3B).
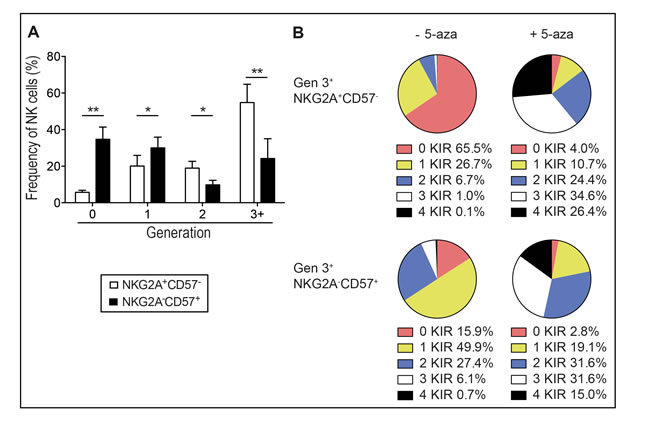
Figure 3: 5-aza induced KIR expression on proliferating immature and mature NK cells. NK cells were isolated from healthy donor PBMC and cultured in 500U/ml of IL-2 for six days with or without the addition of 5-aza for the first four consecutive days. In A. the frequency of immature NKG2A+CD57- and mature NKG2A-CD57+ NK cells within each generation. In B. the mean frequencies of generation 3+ NKG2A+CD57- and NKG2A-CD57+ NK cell subsets expressing 0-4 KIRs. HD n = 8.
KIR repertoires of proliferating NK cells of MDS patients after in vivo 5-aza treatment
Next, we set out to test whether systemic 5-aza treatment had an influence on the NK cell repertoire in a cohort of high-risk MDS patients. To this end we first compared the baseline frequencies of NK cell subsets in healthy donors and patients. The overall frequency of NK cells and CD56bright NK cells were similar in healthy controls and patients pre-treatment (Supplementary Figure 1A-1B). The distribution of NKG2A+CD57- and NKG2A-CD57+ subsets varied greatly among patients with a tendency for higher frequencies of more immature subsets (Supplementary Figure 1C-1D) in line with previous findings [31]. Notably, however, the frequency of KIR2DL3+ and KIR3DL1+ NK cells, as well as NK cells expressing multiple KIRs, was significantly lower in MDS patients than in healthy donors (Figure 4A-4B). To follow dynamic changes during therapy, patients were samples at day one and day five in standard treatment regimes. On bulk NK cells the KIR repertoire was similar regardless of 5-aza, even when patients had undergone a total of 4 cycles of treatment (data not shown). However, as the in vitro data clearly showed that cell division was needed for 5-aza-induced KIR expression, we focused our analysis on the pool of cells expressing Ki-67, a surrogate marker for cells having undergone recent in vivo activation/proliferation [32]. Before treatment, patients displayed increased levels of Ki-67+ NK cells that returned to baseline on day five post-treatment (Figure 4C). In line with the in vitro proliferation data, more Ki-67+ cells were found within the less differentiated NKG2A+CD57- NK cell subset than in the terminally differentiated NKG2A-CD57+ NK cell in healthy donors as well as patients (Supplementary Figure 1E). Notably, there was an increased frequency of Ki-67+ NK cells expressing KIR2DL3 after five days of 5-aza administration (Figure 4D). Furthermore, the frequency of cells expressing multiple KIRs was also increased (Figure 4E). When the Ki-67+ NK cells were stratified into NKG2A+CD57- and NKG2A-CD57+ subsets, the effect of 5-aza was greater in the NKG2A+CD57- subset (Figure 4F) concurrent with the in vitro data (Figure 3B). Thus, these data demonstrate that 5-aza treatment increases the total pool of KIR expressing NK cells, thereby partly restoring a mature NK repertoire in MDS patients.
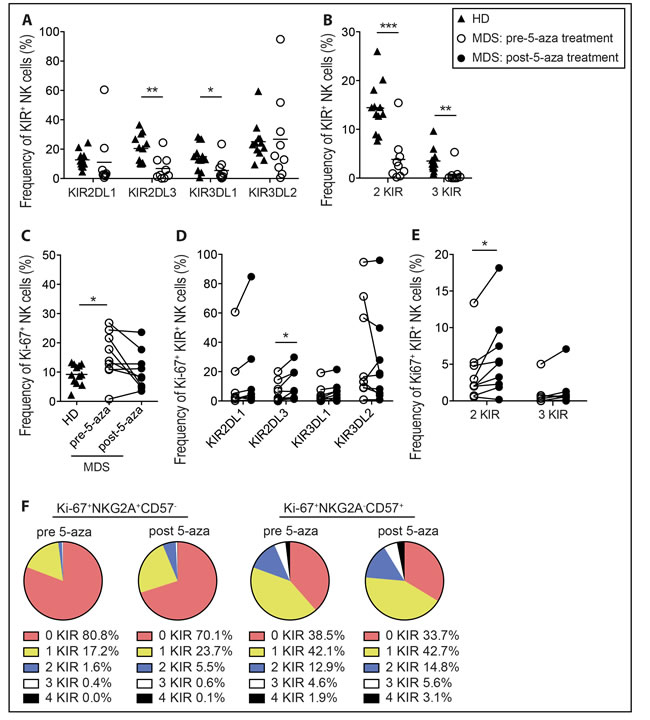
Figure 4: NK cell KIR repertoires of proliferating NK cells in high-risk MDS patients after 5-aza treatment. PBMC were collected from healthy donors (HD), and MDS patients on day one pre-5-aza treatment and on day five post-5-aza treatment of cycle one. NK cells were analyzed directly ex vivo without culture. Shown for HD and pre-treatment patients in A. CD3-CD56+KIR2DL1+, -KIR2DL3+, -KIR3DL1+ and -KIR3DL2+ and in B. NK cells expressing multiple KIRs. Further, in C. the frequency of Ki-67+ NK cells in all groups, and in D. and E. the frequency of KIR2DL1+, -KIR2DL3+, -KIR3DL1+ and -KIR3DL2+ NK cells and cells expressing multiple KIRs within the Ki-67+ NK cell population in MDS patients, respectively. In F. the mean frequencies of Ki-67+NKG2A+CD57- and Ki-67+NKG2A-CD57+ NK cell subsets expressing 0-4 KIRs in MDS patients. HD n = 12, MDS n = 9.
5-aza does not accumulate in NK cells to give KIR expression upon induced proliferation
As 5-aza has a short half-life in vivo [28], cellular uptake and storage could play an important role in its mechanism of action. To address whether NK cells had been loaded with 5-aza in vivo but not yet undergone cell division and demethylation of KIR promoters, we monitored KIR repertoires in pre- and post 5-aza treatment patient samples after IL-2-induced proliferation in vitro. Proliferation rates of NK cells in patients were comparable to healthy donors and in most donors; systemic treatment with 5-aza lowered the ability of NK cells to proliferate in response to IL-2 (Supplementary Figure 2A). There was no effect on KIR expression regardless of the number of cell divisions during in vitro culture (Supplementary Figure 2B-2E), suggesting that NK cells had not accumulated intracellular stores of 5-aza.
Moreover, in vitro 5-aza addition to NK cells from MDS patients sampled pre-treatment induced KIR expression, although not to the extent observed in NK cells of healthy donors. In MDS patient samples, around 20% of NK cells that had divided three or more times in the presence of IL-2 and 5-aza still did not express any KIRs (Supplementary Figure 3A).
5-aza confers increased target cell responses of proliferating NK cells
Short-term exposure to demethylating agents has been reported to affect NK cell functionality [25, 33]. Here we monitored functional responses in discrete NK cell subsets in healthy donors following exposure to 5-aza in vitro and stratified the results based on cellular proliferation. Following the six-day IL-2 driven proliferation with or without 5-aza, we removed IL-2 for two days before subjecting the NK cells to a K562 tumor target cell assay. This rest period allowed for the induced effects of 5-aza to act independently of IL-2 induced hyper-responsiveness. As expected, KIR+ NK cells were more frequently CD107a+ and produced IFN-γ as compared to KIR- cells following K562 stimulation (data not shown). 5-aza-treatment of NK cells led to significantly higher IFN-γ responses (Figure 5A and 5C), which was linked to proliferation (Figure 5D). Generation 3+ NK cells also had higher fractions of CD107a+ cells as compared to the same generation from untreated cultures (Figure 5E). Because of the variable frequencies of NK cells reaching generation 3, increased degranulation after 5-aza treatment was only visible at the bulk level in some donors (Figure 5B), correlating with killing of target cells as measured in a FACS-based killing assay (Supplementary Figure 4A-4B). Both KIR- and KIR+ NK cells displayed enhanced responses to target cell stimulation following 5-aza treatment, although the KIR+ subset showed the largest increase in IFN-γ production (Figure 5F-5G). These results revealed that 5-aza had profound and replication-dependent effects on NK cell functionality, which were only partly linked to acquisition of inhibitory KIRs.
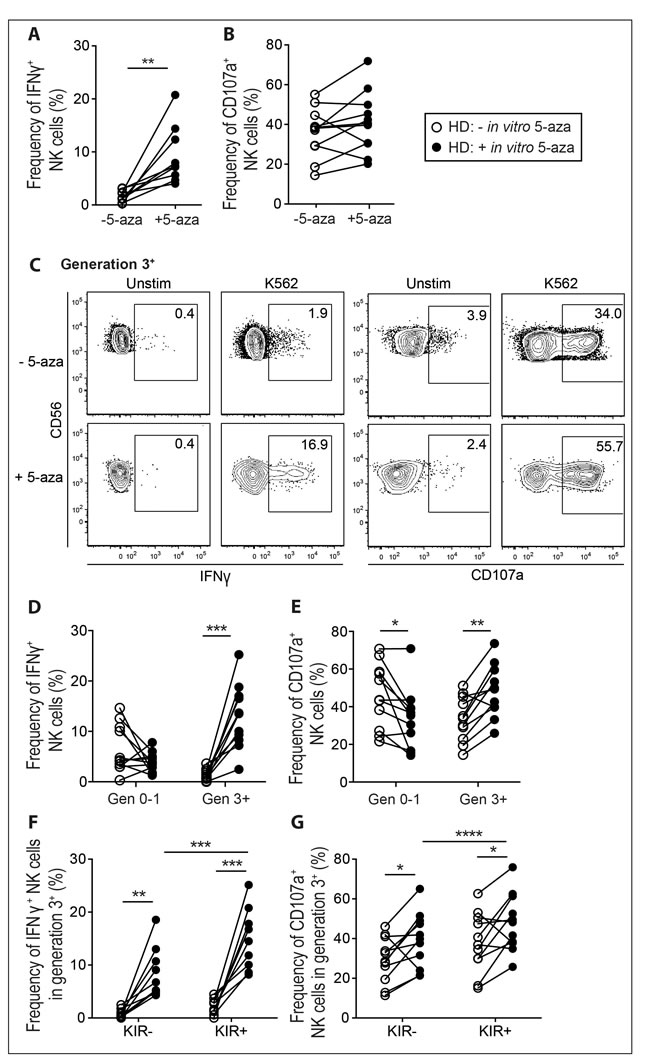
Figure 5: 5-aza enhances function of proliferating NK cells towards tumor target cells. NK cells were isolated from healthy donor PBMC and cultured in 500U/ml of IL-2 for six days with or without the addition of 5-aza for the first four consecutive days. Subsequently cells were cultured in IL-2 free medium for 48 h followed by a 4 h K562 degranulation assay. In A. and B. the frequency of responding cells in terms of IFN-γ production and CD107a mobilization within the total CD3-CD56+ NK cell population. In C. functional responses in generation 3+ NK cells from one representative donor, and quantified for generation 0-1 and 3+ for all donors in D. and E. In F. and G. stratification of generation 3+ NK cells into KIR-/+ subsets. HD n = 11.
Discussion
MDS patients present with disturbed hematopoiesis in the myeloid lineage and defects in the number and subtypes of peripheral cells of multiple lymphoid lineages [31, 34-36]. MDS NK cells have impaired responses against tumor targets and immature subsets are overrepresented [31, 34, 36]. Here we show that 5-aza treatment increases expression of multiple KIRs in proliferating NK cells both in vitro and in MDS patients undergoing 5-aza treatment, and that 5-aza exposed proliferating NK cells display a higher functionality and may thus contribute to a beneficial anti-leukemic effect in 5-aza treated patients.
Previous reports demonstrated a profound effect on KIR expression in NK cells in vitro by the 5-aza related drug decitabine [21, 22]. However, decitabine has a largely different mode of action compared to 5-aza, involving incorporation only into DNA in contrast to the dual integration into both RNA and DNA of 5-aza [37]. Extending the results of Gao et al. [25] our high-resolution analysis of the NK cell KIR repertoire showed that low-dose 5-aza treatment of NK cells induced a significant expression of multiple inhibitory KIRs but only in proliferating cells. Thus, 5-aza was able to affect KIR promoter methylation despite the relatively lower degree of incorporation into DNA compared to decitabine. This is consistent with the need for only minor substitution of 5-aza for cytosine in DNA to inactivate more than 95% of DNMT1 [38]. We observed no preferential induction of specific KIRs or combinations of KIRs, suggesting a non-discriminate demethylation of the CpG islands adjacent to the KIR gene promotors, in line with the global demethylation by 5-aza observed in primary MDS CD34+ bone marrow cells [39].
Despite detailed knowledge on the effect of 5-aza on various immune cell subsets and cancer cell lines in vitro, little is known regarding 5-aza effects on the immune cell compartment in vivo. MDS patients had lower fractions of KIR-expressing NK cells pre-treatment and also tended to have lower frequencies of the most mature NKG2A-CD57+ NK cell subset. These data are consistent with those previously reported by Hejazi et al. [31], albeit they noted higher levels of KIR2DL3 in MDS patients. Interestingly, although we observed lower levels of all KIRs before therapy, the proliferating Ki-67+ fraction of NK cells displayed a selective increase in KIR2DL3, also manifested as higher frequencies of NK cells expressing multiple inhibitory KIRs. It was previously shown that the haplotype C1-specific KIR2DL2/3 is acquired earlier and with higher frequency than the C2-specific KIR2DL1 during NK cell differentiation [40-42] potentially explaining why the effect was most pronounced in this NK cell subset. Together these data indicate uptake of the drug into circulating cells and a demethylating effect of the drug in vivo. Both in vitro and in vivo when proliferating NK cells were stratified into subsets based on NKG2A and CD57 expression the effect of 5-aza was greater in the NKG2A+CD57- subset, suggesting an overall skewing of the NK cell pool towards a more mature profile [30] under 5-aza influence. Notably, the in vivo effect of 5-aza was noted in the absence of IL-2 under steady-state. It is possible that the induction of KIR will be more pronounced if 5-aza is administrated together with immunomodulatory therapies or during the early reconstitution following stem cell transplantation.
When a standard dose of 5-aza is administered subcutaneously to patients, the highest levels measurable in plasma are similar, or slightly higher, to those that can induce demethylation in vitro [28, 43]. However, 5-aza is rapidly cleared from circulation with a half time in plasma of less than one hour [28] partly due to the action of the enzyme cytidine deaminase, which catalyzes the deamination of 5-aza, thereby destabilizing the drug and reducing the in vivo half-life [44]. Administration of 5-aza to MDS patients decreases the number of highly methylated loci but, surprisingly, the maximal effects on methylation levels appear one week after the end of drug administration [45] indicating that shifts in the NK cell KIR repertoire may be more noticeable at later time-points.
NK cells can contribute to the killing of MDS blast cells and disease progression may be associated with evasion of NK-cell mediated immune surveillance [46, 47]. Previous studies have demonstrated a strong association between reduced numbers and function of both peripheral and bone-marrow NK cells in MDS patients and disease severity [31, 34, 36, 47]. Here proliferating NK cells exposed to 5-aza in vitro exhibited considerably higher responses against K562 cells, suggesting that NK cells with an active uptake of 5-aza might mount improved responses against tumor targets. It is important to point out that this beneficial effect of 5-aza on NK cell cytotoxicity may only be relevant in conditions of intensified proliferation since resting NK cells are negatively affected even at low doses of 5-aza.
NK-cell education regulates cytotoxic responses against cellular targets and determines the functional potential of NK cells [15, 16, 30]. Surprisingly, the potentiating effect of 5-aza was evident in all cycling NK cells, irrespective of KIR status. This could possibly be a result of epigenetic remodeling of e.g. the IFN-γ locus, in similarity to what is observed during NK cell differentiation [48]. However, we observed no effect of 5-aza on the expression of granulysin, perforin or granzyme B or the activating receptors NKG2D or DNAM-1 (data not shown), indicating a lack of general upregulation of markers involved in NK cell activation/effector pathways. It is also possible that NK cells that had acquired KIR expression during cytokine-induced proliferation lost expression during the two days of IL-2 ‘rest’ and thus contributed to the response of the KIR- population. Indeed we noticed a drop in KIR expressing cells between day 6 and 8 in generation 3+ NK cells.
Other studies have shown that short-term treatment of resting NK cells with high doses of 5-aza lowers NK cell reactivity [25, 33]. Such replication-independent negative influence of 5-aza on NK cells that had not undergone cell division was also noted in our study despite the lower doses of 5-aza used. However, Schmiedel found that although initially impaired, IFN-γ production was recovered and even increased as compared to controls when NK cells were treated for 4 days with 5-aza under IL-2 stimulation [33] thereby supporting our results that proliferating cells with active incorporation of 5-aza acquire a heightened functionality. A recent study also demonstrated that decitabine treatment can depolarize Th2 cells to effectively secrete IFN-γ, signal via T-bet, and achieve demethylation of critical Th1 specific promoters [49]. The net effect of 5-aza on NK cell anti-tumor responses was recently investigated in B cell precursor acute lymphoblastic leukemia (BCP-ALL) bearing humanized mice. It was shown that 5-aza treatment reduced the tumor burden, which was attributed to improved NK cell functionality rather than direct effects of the drug on the tumor [50].
The prospect of engraftment of autologous or allogeneic NK cells for the treatment of hematopoietic malignancies has been highlighted in recent years [51]. The effects of 5-aza on the NK cell population may provide an avenue of experimental modification of KIR expression in this context. By selecting NK cells from donors with a given KIR genotype, and culturing these in the presence of 5-aza, it may be possible to increase KIR expression and thereby obtain cultures with high density of cells with enhanced function. Although 5-aza appears to induce expression of all KIRs, co-culture in the presence or absence of specific ligands may allow skewing of the KIR repertoires towards desired specificities [52]. Such efforts will require careful titration of the 5-aza doses, since the drug affects proliferative responses even at lower concentrations. 5-aza modulation of NK cells to yield specific subsets has been suggested for therapeutic applications for placental disorders associated with altered NK cell biology [53] and for pre-treatment of patients suffering from ALL [50].
In conclusion, these findings suggest that 5-aza remodeling of components of the immune system may act in concert with demethylating/cytotoxic effects on the malignant clones in MDS patients and warrants further investigations in NK cells of patients undergoing 5-aza treatment.
Materials and Methods
Patients and healthy controls
The study was approved by the regional ethics committee [Stockholm, Sweden, approval number 2006/229-31/3] and was conducted in accordance with the Declaration of Helsinki. Patients (n=11) were recruited from the Hematology unit at the Karolinska University Hospital in Huddinge, Sweden. All patients gave their informed consent. Median age of the cohort was 74 years (range 45-83 years). According to the dosing schedule of the Nordic MDS guidelines, patients were administered 100 mg/m2 of 5-azacytidine (5-aza) subcutaneously in cycles consisting of one dose of 5-aza for five consecutive days followed by three weeks of rest. Blood samples were collected at day one (pre-treatment) and day five (post-treatment) of cycle one, and in some cases of cycle four. Healthy adult donor buffy coats were obtained from the blood bank at Karolinska University Hospital, Huddinge, Sweden. The frequencies of KIR A and B haplotypes were similar in healthy donors and patients (data not shown) as estimated based on expression patterns obtained with a 16-color KIR phenotyping panel, which resolves KIR2DL1, KIR2DS1, KIR2DL2/S2 expression [54, 55]. No KIR genotyping was performed.
Cell processing
Peripheral blood mononuclear cells (PBMC) from healthy controls and patients were isolated by density gradient centrifugation (Ficoll-Hypaque; GE Healthcare) and NK cell phenotyping was performed directly ex vivo without culturing. The remaining PBMC were rested o.n. in culture medium (RPMI 1640 supplemented with 10% heat-inactivated human AB+ serum and 2 mM L-glutamine), followed by next-day negative isolation of NK cells (magnetic-activated cell sorting; Miltenyi Biotech). Isolated NK cells were labeled with 0.5 µM CellTrace Violet (Life Technologies), re-suspended in culture medium supplemented with 500 U/ml IL-2 (Peprotech) at a concentration of 1.5x106 cells/ml and cultured at 37°C and 5% CO2 for six days. For in vitro 5-aza stimulation of healthy donor cultures or MDS patient samples, NK cells were treated with 1µM 5-azacytidine (Sigma) for the first 4 consecutive days of the culture. K562 cells were culture in RPMI 1640 supplemented with 10% FBS and 5 mM L-glutamine at 37°C and 5% CO2.
Flow cytometry
For flow cytometry the following fluorochrome-conjugated specific antibodies were used: from Beckman Coulter; anti-CD3 PC5 (UHCT1), anti-CD56 ECD (NHK-1), anti-CD158a,h (KIR2DL1, KIR2DS1) PC7 (EB6B), anti-CD158b1/b2,j PC5.5 (GL183), custom made anti-NKG2A APC-AF750 (Z199). From BD Biosciences; anti-CD19 V500 (HIB19), anti-IgM eF650 (R6-60.2), anti-CD107a PE (H4A3), anti-Ki-67 PE (B56). From BioLegend; anti-IFN-γ BV785 (4S.B3), anti-KIR3DL1 AF700 (Dx9) and anti-CD56 BV421 (HCD56). From eBiosciences; purified anti-CD57 (TB01). From R&D systems; anti-KIR2DL1 APC (143211), anti-KIR2DL3 FITC (180701). From MabTech; anti-KIR3DL2 biotin (Dx31) with streptavidin QD605 (Invitrogen). From Life Technologies; CellTrace Violet™ Cell Proliferation Kit and LIVE/DEAD® Fixable Aqua Dead Cell Stain Kit. From MBL: FITC conjugated caspase-3 substrate (FITC-VAD-FMK). Data were acquired in FACSDiva software on a BD LSR Fortessa equipped with a 488-nm laser, a 633-nm laser, a 405-nm laser, and a 562-nm laser. Acquired data were analyzed in FlowJo software (TreeStar, USA). Gating was performed on live CD19-CD3-CD56+ single cells within the lymphocyte gate or on CD56- K562 cells within a larger forward and side scatter gate for killing assays.
Functional assays
NK cell function was evaluated after the six-day IL-2-driven proliferation -/+ in vitro 5-aza (as described above) followed by a period of rest (48 h) in IL-2-free culture medium. NK cells were then mixed at a 1:1 ratio with K562 target cells and incubated in a V-bottom 96-well plate for 4 h at 37C and 5% CO2. For measurements of NK cell degranulation and intracellular cytokine production, Monensin (Golgi Stop, 1:1500, BD Biosciences), Brefeldin A (GolgiPlug, 1:1000, BD Biosciences) and anti-CD107a PE were added at the start of the 4 h co-incubation. At harvest, the cells were surface stained, followed by fixation and permeabilization (Fix/Perm kit, eBioscience) and subsequent intracellular staining for IFN-γ. For FACS-based evaluation of NK cell-mediated target cell killing, FITC conjugated caspase-3 substrate (FITC-VAD-FMK)(C3) was added at the start of the 4 h co-incubation. At harvest, cells were stained with LIVE/DEAD® Fixable Aqua Dead Cell Stain (DCM) and CD56 BV421. The percentage of lysed target cells was determined as CD56-DCM+C3+ K562 cells. For all functional assays, values from unstimulated cultures were subtracted from stimulated cultures and subsequent statistical analysis was performed.
Statistical analyses
Statistical differences were evaluated by Students t-test (non-paired or paired). All analyses were performed with GraphPad Software. Statistical significance was assumed when p < 0.05; **** indicates p < 0.0001, *** indicates p < 0.001, ** indicates p < 0.01, and * indicates p < 0.05.
Acknowledgments
We thank all the MDS patients for their participation in the study and Gunilla Walldin for kind assistance with sample collection.
conflicts of interest
The authors declare no conflict of interest.
Grant support
This work was supported by grants from the Swedish Research Council, the Swedish Children’s Cancer Society, the Swedish Cancer Society, the Karolinska Institutet, Oslo University Hospital, the Norwegian Research Council, the KG Jebsen Foundation, the South-Eastern Norway Regional Health Authority and the Norwegian Cancer Foundation. The authors declare no conflict of interest.
References
1. Esteller M. Epigenetic gene silencing in cancer: the DNA hypermethylome. Human Molecular Genetics. 2007; 16 Spec No 1: R50-9.
2. McDevitt MA. Clinical applications of epigenetic markers and epigenetic profiling in myeloid malignancies. Seminars in Oncology. 2012; 39: 109-122.
3. Ganan-Gomez I, Wei Y, Starczynowski DT, Colla S, Yang H, Cabrero-Calvo M, Bohannan ZS, Verma A, Steidl U, Garcia-Manero G. Deregulation of innate immune and inflammatory signaling in myelodysplastic syndromes. Leukemia. 2015.
4. Kulasekararaj AG, Mohamedali AM, Mufti GJ. Recent advances in understanding the molecular pathogenesis of myelodysplastic syndromes. British Journal of Haematology. 2013; 162: 587-605.
5. Fenaux P, Mufti GJ, Hellstrom-Lindberg E, Santini V, Finelli C, Giagounidis A, Schoch R, Gattermann N, Sanz G, List A, Gore SD, Seymour JF, Bennett JM, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. The Lancet Oncology. 2009; 10: 223-232.
6. Malcovati L, Hellstrom-Lindberg E, Bowen D, Ades L, Cermak J, Del Canizo C, Della Porta MG, Fenaux P, Gattermann N, Germing U, Jansen JH, Mittelman M, Mufti G, et al. Diagnosis and treatment of primary myelodysplastic syndromes in adults: recommendations from the European LeukemiaNet. Blood. 2013; 122: 2943-2964.
7. Santi DV, Norment A, Garrett CE. Covalent bond formation between a DNA-cytosine methyltransferase and DNA containing 5-azacytosine. Proceedings of the National Academy of Sciences of the United States of America. 1984; 81: 6993-6997.
8. Ghoshal K, Datta J, Majumder S, Bai S, Kutay H, Motiwala T, Jacob ST. 5-Aza-deoxycytidine induces selective degradation of DNA methyltransferase 1 by a proteasomal pathway that requires the KEN box, bromo-adjacent homology domain, and nuclear localization signal. Molecular and Cellular Biology. 2005; 25: 4727-4741.
9. Qin T, Jelinek J, Si J, Shu J, Issa JP. Mechanisms of resistance to 5-aza-2’-deoxycytidine in human cancer cell lines. Blood. 2009; 113: 659-667.
10. Goodyear O, Agathanggelou A, Novitzky-Basso I, Siddique S, McSkeane T, Ryan G, Vyas P, Cavenagh J, Stankovic T, Moss P, Craddock C. Induction of a CD8+ T-cell response to the MAGE cancer testis antigen by combined treatment with azacitidine and sodium valproate in patients with acute myeloid leukemia and myelodysplasia. Blood. 2010; 116: 1908-1918.
11. Long EO, Kim HS, Liu D, Peterson ME, Rajagopalan S. Controlling natural killer cell responses: integration of signals for activation and inhibition. Annual Review of Immunology. 2013; 31: 227-258.
12. Diefenbach A, Jensen ER, Jamieson AM, Raulet DH. Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature. 2001; 413: 165-171.
13. Rohner A, Langenkamp U, Siegler U, Kalberer CP, Wodnar-Filipowicz A. Differentiation-promoting drugs up-regulate NKG2D ligand expression and enhance the susceptibility of acute myeloid leukemia cells to natural killer cell-mediated lysis. Leukemia Research. 2007; 31: 1393-1402.
14. Baragano Raneros A, Martin-Palanco V, Fernandez AF, Rodriguez RM, Fraga MF, Lopez-Larrea C, Suarez-Alvarez B. Methylation of NKG2D ligands contributes to immune system evasion in acute myeloid leukemia. Genes and Immunity. 2015; 16: 71-82.
15. Anfossi N, Andre P, Guia S, Falk CS, Roetynck S, Stewart CA, Breso V, Frassati C, Reviron D, Middleton D, Romagne F, Ugolini S, Vivier E. Human NK cell education by inhibitory receptors for MHC class I. Immunity. 2006; 25: 331-342.
16. Fauriat C, Ivarsson MA, Ljunggren HG, Malmberg KJ, Michaelsson J. Education of human natural killer cells by activating killer cell immunoglobulin-like receptors. Blood. 2010; 115: 1166-1174.
17. Joncker NT, Fernandez NC, Treiner E, Vivier E, Raulet DH. NK cell responsiveness is tuned commensurate with the number of inhibitory receptors for self-MHC class I: the rheostat model. Journal of Immunology. 2009; 182: 4572-4580.
18. Yawata M, Yawata N, Draghi M, Partheniou F, Little AM, Parham P. MHC class I-specific inhibitory receptors and their ligands structure diverse human NK-cell repertoires toward a balance of missing self-response. Blood. 2008; 112: 2369-2380.
19. Chan WK, Rujkijyanont P, Neale G, Yang J, Bari R, Das Gupta N, Holladay M, Rooney B, Leung W. Multiplex and genome-wide analyses reveal distinctive properties of KIR+ and CD56+ T cells in human blood. Journal of Immunology. 2013; 191: 1625-1636.
20. Cichocki F, Miller JS, Anderson SK. Killer immunoglobulin-like receptor transcriptional regulation: a fascinating dance of multiple promoters. Journal of Innate Immunity. 2011; 3: 242-248.
21. Santourlidis S, Trompeter HI, Weinhold S, Eisermann B, Meyer KL, Wernet P, Uhrberg M. Crucial role of DNA methylation in determination of clonally distributed killer cell Ig-like receptor expression patterns in NK cells. Journal of Immunology. 2002; 169: 4253-4261.
22. Chan HW, Kurago ZB, Stewart CA, Wilson MJ, Martin MP, Mace BE, Carrington M, Trowsdale J, Lutz CT. DNA methylation maintains allele-specific KIR gene expression in human natural killer cells. The Journal of Experimental Medicine. 2003; 197: 245-255.
23. Valiante NM, Uhrberg M, Shilling HG, Lienert-Weidenbach K, Arnett KL, D’Andrea A, Phillips JH, Lanier LL, Parham P. Functionally and structurally distinct NK cell receptor repertoires in the peripheral blood of two human donors. Immunity. 1997; 7: 739-751.
24. Cooley S, Xiao F, Pitt M, Gleason M, McCullar V, Bergemann TL, McQueen KL, Guethlein LA, Parham P, Miller JS. A subpopulation of human peripheral blood NK cells that lacks inhibitory receptors for self-MHC is developmentally immature. Blood. 2007; 110: 578-586.
25. Gao XN, Lin J, Wang LL, Yu L. Demethylating treatment suppresses natural killer cell cytolytic activity. Molecular Immunology. 2009; 46: 2064-2070.
26. Lutz CT, Karapetyan A, Al-Attar A, Shelton BJ, Holt KJ, Tucker JH, Presnell SR. Human NK cells proliferate and die in vivo more rapidly than T cells in healthy young and elderly adults. Journal of Immunology. 2011; 186: 4590-4598.
27. Derissen EJ, Hillebrand MJ, Rosing H, Otten HM, Laille E, Schellens JH, Beijnen JH. Quantitative determination of azacitidine triphosphate in peripheral blood mononuclear cells using liquid chromatography coupled with high-resolution mass spectrometry. Journal of Pharmaceutical and Biomedical analysis. 2014; 90: 7-14.
28. Marcucci G, Silverman L, Eller M, Lintz L, Beach CL. Bioavailability of azacitidine subcutaneous versus intravenous in patients with the myelodysplastic syndromes. Journal of Clinical Pharmacology. 2005; 45: 597-602.
29. Stresemann C, Lyko F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. International Journal of Cancer. 2008; 123: 8-13.
30. Bjorkstrom NK, Riese P, Heuts F, Andersson S, Fauriat C, Ivarsson MA, Bjorklund AT, Flodstrom-Tullberg M, Michaelsson J, Rottenberg ME, Guzman CA, Ljunggren HG, Malmberg KJ. Expression patterns of NKG2A, KIR, and CD57 define a process of CD56dim NK-cell differentiation uncoupled from NK-cell education. Blood. 2010; 116: 3853-3864.
31. Hejazi M, Manser AR, Frobel J, Kundgen A, Zhao X, Schonberg K, Germing U, Haas R, Gattermann N, Uhrberg M. Impaired cytotoxicity associated with defective natural killer cell differentiation in myelodysplastic syndromes. Haematologica. 2015.
32. Scholzen T, Gerdes J. The Ki-67 protein: from the known and the unknown. Journal of Cellular Physiology. 2000; 182: 311-322.
33. Schmiedel BJ, Arelin V, Gruenebach F, Krusch M, Schmidt SM, Salih HR. Azacytidine impairs NK cell reactivity while decitabine augments NK cell responsiveness toward stimulation. International journal of Cancer. 2011; 128: 2911-2922.
34. Epling-Burnette PK, Bai F, Painter JS, Rollison DE, Salih HR, Krusch M, Zou J, Ku E, Zhong B, Boulware D, Moscinski L, Wei S, Djeu JY, et al. Reduced natural killer (NK) function associated with high-risk myelodysplastic syndrome (MDS) and reduced expression of activating NK receptors. Blood. 2007; 109: 4816-4824.
35. Zou JX, Rollison DE, Boulware D, Chen DT, Sloand EM, Pfannes LV, Goronzy JJ, Bai F, Painter JS, Wei S, Cosgrove D, List AF, Epling-Burnette PK. Altered naive and memory CD4+ T-cell homeostasis and immunosenescence characterize younger patients with myelodysplastic syndrome. Leukemia. 2009; 23: 1288-1296.
36. Kiladjian JJ, Bourgeois E, Lobe I, Braun T, Visentin G, Bourhis JH, Fenaux P, Chouaib S, Caignard A. Cytolytic function and survival of natural killer cells are severely altered in myelodysplastic syndromes. Leukemia. 2006; 20: 463-470.
37. Li LH, Olin EJ, Buskirk HH, Reineke LM. Cytotoxicity and mode of action of 5-azacytidine on L1210 leukemia. Cancer research. 1970; 30: 2760-2769.
38. Creusot F, Acs G, Christman JK. Inhibition of DNA methyltransferase and induction of Friend erythroleukemia cell differentiation by 5-azacytidine and 5-aza-2’-deoxycytidine. The Journal of Biological Chemistry. 1982; 257: 2041-2048.
39. Grovdal M, Karimi M, Tobiasson M, Reinius L, Jansson M, Ekwall K, Ungerstedt J, Kere J, Greco D, Hellstrom-Lindberg E. Azacitidine induces profound genome-wide hypomethylation in primary myelodysplastic bone marrow cultures but may also reduce histone acetylation. Leukemia. 2014; 28: 411-413.
40. Schonberg K, Sribar M, Enczmann J, Fischer JC, Uhrberg M. Analyses of HLA-C-specific KIR repertoires in donors with group A and B haplotypes suggest a ligand-instructed model of NK cell receptor acquisition. Blood. 2011; 117: 98-107.
41. Miller JS, McCullar V. Human natural killer cells with polyclonal lectin and immunoglobulinlike receptors develop from single hematopoietic stem cells with preferential expression of NKG2A and KIR2DL2/L3/S2. Blood. 2001; 98: 705-713.
42. Fischer JC, Ottinger H, Ferencik S, Sribar M, Punzel M, Beelen DW, Schwan MA, Grosse-Wilde H, Wernet P, Uhrberg M. Relevance of C1 and C2 epitopes for hemopoietic stem cell transplantation: role for sequential acquisition of HLA-C-specific inhibitory killer Ig-like receptor. Journal of Immunology. 2007; 178: 3918-3923.
43. Qin T, Castoro R, El Ahdab S, Jelinek J, Wang X, Si J, Shu J, He R, Zhang N, Chung W, Kantarjian HM, Issa JP. Mechanisms of resistance to decitabine in the myelodysplastic syndrome. PloS one. 2011; 6: e23372.
44. Mahfouz RZ, Jankowska A, Ebrahem Q, Gu X, Visconte V, Tabarroki A, Terse P, Covey J, Chan K, Ling Y, Engelke KJ, Sekeres MA, Tiu R, et al. Increased CDA expression/activity in males contributes to decreased cytidine analog half-life and likely contributes to worse outcomes with 5-azacytidine or decitabine therapy. Clinical Cancer Research. 2013; 19: 938-948.
45. Garcia-Manero G, Gore SD, Cogle C, Ward R, Shi T, Macbeth KJ, Laille E, Giordano H, Sakoian S, Jabbour E, Kantarjian H, Skikne B. Phase I study of oral azacitidine in myelodysplastic syndromes, chronic myelomonocytic leukemia, and acute myeloid leukemia. Journal of Clinical Oncology. 2011; 29: 2521-2527.
46. Chamuleau ME, Westers TM, van Dreunen L, Groenland J, Zevenbergen A, Eeltink CM, Ossenkoppele GJ, van de Loosdrecht AA. Immune mediated autologous cytotoxicity against hematopoietic precursor cells in patients with myelodysplastic syndrome. Haematologica. 2009; 94: 496-506.
47. Carlsten M, Baumann BC, Simonsson M, Jadersten M, Forsblom AM, Hammarstedt C, Bryceson YT, Ljunggren HG, Hellstrom-Lindberg E, Malmberg KJ. Reduced DNAM-1 expression on bone marrow NK cells associated with impaired killing of CD34+ blasts in myelodysplastic syndrome. Leukemia. 2010; 24: 1607-1616.
48. Luetke-Eversloh M, Cicek BB, Siracusa F, Thom JT, Hamann A, Frischbutter S, Baumgrass R, Chang HD, Thiel A, Dong J, Romagnani C. NK cells gain higher IFN-gamma competence during terminal differentiation. European Journal of Immunology. 2014; 44: 2074-2084.
49. Dubovsky JA, Powers JJ, Gao Y, Mariusso LF, Sotomayor EM, Pinilla-Ibarz JA. Epigenetic repolarization of T lymphocytes from chronic lymphocytic leukemia patients using 5-aza-2’-deoxycytidine. Leukemia Research. 2011; 35: 1193-1199.
50. Kubler A, Woiterski J, Witte KE, Buhring HJ, Hartwig UF, Ebinger M, Oevermann L, Mezger M, Herr W, Lang P, Handgretinger R, Munz C, Andre MC. Both mature KIR+ and immature KIR- NK cells control pediatric acute B-cell precursor leukemia in NOD.Cg-Prkdcscid IL2rgtmWjl/Sz mice. Blood. 2014; 124: 3914-3923.
51. Ljunggren HG, Malmberg KJ. Prospects for the use of NK cells in immunotherapy of human cancer. Nature Reviews Immunology. 2007; 7: 329-339.
52. Igarashi T, Wynberg J, Srinivasan R, Becknell B, McCoy JP,Jr, Takahashi Y, Suffredini DA, Linehan WM, Caligiuri MA, Childs RW. Enhanced cytotoxicity of allogeneic NK cells with killer immunoglobulin-like receptor ligand incompatibility against melanoma and renal cell carcinoma cells. Blood. 2004; 104: 170-177.
53. Cerdeira AS, Rajakumar A, Royle CM, Lo A, Husain Z, Thadhani RI, Sukhatme VP, Karumanchi SA, Kopcow HD. Conversion of peripheral blood NK cells to a decidual NK-like phenotype by a cocktail of defined factors. Journal of Immunology. 2013; 190: 3939-3948.
54. Beziat V, Liu LL, Malmberg JA, Ivarsson MA, Sohlberg E, Bjorklund AT, Retiere C, Sverremark-Ekstrom E, Traherne J, Ljungman P, Schaffer M, Price DA, Trowsdale J, et al. NK cell responses to cytomegalovirus infection lead to stable imprints in the human KIR repertoire and involve activating KIRs. Blood. 2013; 121: 2678-2688.
55. Beziat V, Traherne J, Malmberg JA, Ivarsson MA, Bjorkstrom NK, Retiere C, Ljunggren HG, Michaelsson J, Trowsdale J, Malmberg KJ. Tracing dynamic expansion of human NK-cell subsets by high-resolution analysis of KIR repertoires and cellular differentiation. European Journal of Immunology. 2014; 44: 2192-2196.