
Introduction
Lung cancer is the leading cause of cancer death around the world. Late diagnosis and low response to therapeutic drugs are viewed as the causes of poor patient outcomes [1]. The five-year survival rate has remained at about 15% for the past three decades despite the development and use of several targeting drugs for lung cancer therapy [2]. Therefore, mechanistic studies to uncover the possible pathway(s) involved in drug resistance are essential for improving the outcomes and life quality in patients with this disease.
Cisplatin-based chemotherapy is still considered the first-line therapeutic strategy for lung cancer [3, 4]. Cisplatin induces cancer cell death via induction of double strand DNA breaks caused by the generation of reactive oxygen species (ROS) [5, 6]. Gene expression of antioxidants that eliminate ROS is promoted by an NF-E2-related factor 2 (Nrf2), which binds to antioxidant response elements (ARE) of the promoters of these genes [7-11]. Activation of Nrf2/ARE signaling has been demonstrated through mutations of Kelch-like ECH-associated protein 1 (Keap1) or Nrf2 to protect Nrf2 from degradation by Keap1 [12-17]. The Keap1/Nrf2 mutations in NSCLC patients were ranged from 3.2% to 60% and this variation may be due to the number of study subjects and histologic subtypes. In fact, the Keap1/Nrf2 mutation was frequently reported to be uncommon in NSCLC patients including Taiwanese ( < 3%); (Supplementary Table 1). High Nrf2 expression has been shown to promote resistance to different anticancer drugs in human cancers [18-22]. However, the underlying mechanism of an increase in Nrf2 expression is not fully understood although some mechanisms have been reported [23, 24]. For example, high Nrf2 expression driven by NF-κB signaling confers chemoresistance in human myeloid leukemia [23]. Mutant k-ras confers chemoresistance by upregulating Nrf2 transcription through a TPA response element [24].
The cross-talk between Nrf2 and p53 plays a crucial role in cellular homeostasis. Positive or negative co-regulation at the post-translational level has been reported between Nrf2 and p53 [25]. For example, binding of the p53 downstream p21 to Nrf2 inhibits Nrf2 degradation [26], whereas binding of the Nrf2 downstream NAD(P)H dehydrogenase quinone 1 (NQO1) to p53 prevents p53 degradation [27]. However, Nrf2 promotes MDM2 expression for degradation of p53 protein [28]. P53 inhibits Nrf2 downstream expression of genes, such as NQO1 and heme oxygenase 1 (HO-1) by directly interacting with ARE-containing promoters [29]. Therefore, we expected that the reciprocal regulation between Nrf2 and p53 may modulate cancer cell apoptosis induced by chemotherapeutic agents.
Our preliminary data from lung cancer patients showed that Nrf2 mRNA expression levels were higher in p53-mutant tumors than in p53-wild-type tumors. A software analysis revealed that putative binding sites for Sp1 and p53 could exist on the Nrf2 promoter (-1036/+1). We therefore hypothesized that mutant p53 might directly upregulate Nrf2 transcription to confer resistance of chemotherapeutic agents and consequently result in poor outcome in NSCLC patients.
Results
Nrf2 expression is suppressed by wild-type p53, but not by mutant p53 at the transcription level
Eight lung cancer cell lines were enrolled to test the hypothesis that Nrf2 expression is de-regulated at the transcription level by p53 status. The Nrf2/ARE downstream genes HO-1 and NQO1 expression were relatively lower in p53-wild-type cells than in p53-mutant cells (Figure 1A upper panel). Concomitantly, Nrf2 protein and its mRNA expression levels were significantly lower in p53-wild-type cells than in p53-mutant cells (Figure 1A). These results suggest that Nrf2 expression might be suppressed by wild-type p53, but not by mutant p53 at transcriptional level.
A software analyses predicted two p53 and four Sp1 putative binding sites located on the -1036/-1 Nrf2 promoter (http://www.genome.jp/tools/motif/). Three Nrf2 promoters (-1036/+1, -740/+1, and -229/+1) were constructed by PCR and deletion mutations for luciferase reporter assay (Figure 1B upper panel). These three Nrf2 promoters were respectively transfected into H1299, H1975, and CL3 cells. The reporter activity was significantly higher for the -1036/+1 Nrf2 promoter than for the other two Nrf2 promoters (-740/+1 and -229/+1) in these cells. These results revealed the possibility that p53 and Sp1 putative binding sites located near the transcriptional start site might play an important role in Nrf2 transcription in these three cell types.
The possibility that wild-type p53 could suppress Nrf2 promoter activity was explored using p53-null H1299 cells by transfection with wild-type p53 or different mutant p53 expression vectors including H179Y, L194R, S240R, R249S, A276D, and E286Q. Western blotting showed that p53 expression was detected in H1299 cells transfected with wild-type or mutant p53 expression vectors, but was not observed in H1299 cells with an empty vector transfection (VC) (Figure 1C upper panel). Nrf2 expression was almost completely suppressed by wild-type p53 expression vector, but was unchanged by mutant p53 expression vector transfections in H1299 cells. The luciferase reporter assay indicated a marked decrease in Nrf2 promoter activity following wild-type p53 expression vector transfection, but showed unchanged or relatively increased Nrf2 promoter activity following transfection with mutant p53 expression vectors when compared with the activity in VC cells (Figure 1C lower panel). These results suggest that Nrf2 expression in lung cancer cells is suppressed by wild-type p53, but not by mutant p53 at the transcriptional level.
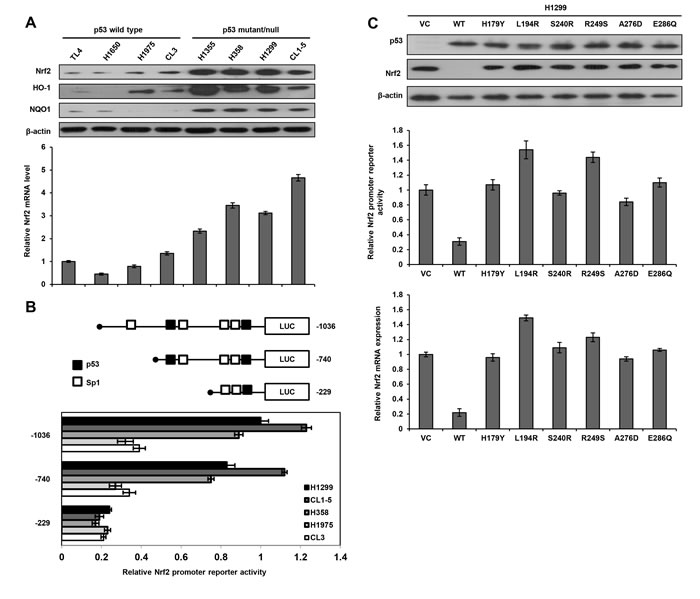
Figure 1: Nrf2 transcription is suppressed by wild-type p53. A. Western blotting analysis for Nrf2, HO-1, and NQO1 expression in various lung cancer cell lines, β-actin was used as a loading control. Real-time PCR analysis was performed to determine Nrf2 mRNA expression; GAPDH was served as an internal control. Nrf2 mRNA expression in TL4 cells (mRNA level = 1) was used as a reference to evaluate Nrf2 mRNA levels in other lung cancer cells. B. Diagram summarizing the positions of the p53 and Sp1 putative binding sites on Nrf2 promoter constructs (–1036~+1) predicted by a software analysis (http://www.atcc.org). An luciferase reporter assay was performed to evaluate the reporter activity of these three promoter fragments, including –1036/+1, –740/+1, and –229/+1. H1299, CL1-5, H358, H1975, and CL3 cells were separately transfected with these three promoters (5μg) for the luciferase reporter assay; β-gal was served as an internal control. The reporter activity of the Nrf2 (–1036/+1) promoter in H1299 cells served as a reference (activity = 1). C. Number of p53 wild-type/mutants plasmid and Nrf2 promoters plasmid (-740/+1) were co-transfected into H1299 cells, the cells lysates were separated by SDS-PAGE for the evaluation p53 expression by a specific antibody using western blotting. Luciferase reporter assay was performed to evaluate the reporter activity of Nrf2 promoter. Real-time PCR analysis was performed to determine Nrf2 mRNA expression.
Nrf2 transcription is down-regulated by wild-type p53, but not by mutant p53 via decreased Sp1 binding to the Nrf2 promoter
We next examined the possibility that wild-type p53 could interact with Sp1 to suppress Nrf2 transcription. The p53-null H1299 cells were transfected with two doses of wild-type p53 or mutant p53 expression vectors (H179Y and L194R). The expression of p53 protein in H1299 cells with wild-type or mutant p53 expression vector transfection was revealed by western blotting, but Nrf2 expression was almost completely suppressed by a high dose of wild-type p53 transfection, but was nearly unchanged by mutant p53 expression vector transfections in H1299 cells (Figure 2A upper panel). The luciferase reporter assay and real-time PCR analysis revealed a dose-dependent decrease in Nrf2 promoter activity and its mRNA levels in response to wild-type p53 transfection in H1299 cells. However, Nrf2 promoter activity and its mRNA level were nearly unchanged in H1299 cells transfected with H179Y or L194R expression vector, whereas Nrf2 promoter activity was elevated by high transfection doses of L194R expression vector (Figure 2A middle panel). ChIP assay showed that wild-type p53 was indeed bound, but the binding of mutant p53 was not revealed on the p53 binding site of Nrf2 promoter. The binding of Sp1 to the Nrf2 promoter in H1299 cells with wild-type p53 transfection almost completely eliminated when compared with VC and H1299 cells transfected with mutant p53 expression vectors (Figure 2A lower panel). On the other hand, Nrf2 protein, Nrf2 promoter activity, and its mRNA expression were elevated by p53 knockdown in p53-wild-type H1975 and CL3 cells. The binding of p53 and Sp1 to the Nrf2 promoter was decreased and increased by p53 silencing in both cell types (Figure 2B). A ChIP analysis further indicated that the binding of p53 to the Nrf2 promoter was decreased by p53 silencing, but the binding of Sp1 to the Nrf2 promoter was increased in p53-knockdown H1975 and CL3 cells in a dose-dependent manner (Figure 2B lower panel). These results suggest that p53 may interfere Sp1 binding to the Nrf2 promoter to reduce its promoter activity.
We then constructed Nrf2 promoters mutated at the p53 or Sp1 binding site by site-directed mutagenesis to verify whether wild-type p53 could interact with Sp1 to suppress Sp1 binding to the Nrf2 promoter. The Nrf2 promoter activity was unchanged by transfection with an Nrf2 promoter mutated at the p53 binding site in H1299 VC cells and H1299 cells with mutant p53 expression vector transfections, but the promoter activity was significantly elevated in H1299 cells transfected with the Nrf2 wild-type promoter. However, the Nrf2 promoter activity was elevated by Nrf2 wild-type promoter transfection in mutant p53-tranfected H1299 cells, but this promoter activity was completely eliminated by transfection with an Nrf2 promoter mutated at the Sp1 binding site in H1299 cells subjected to different treatments (Figure 2C). On the other hand, the Nrf2 promoter activity was markedly increased by transfection with an Nrf2 promoter mutated at the p53 binding site, but the increase in the Nrf2 promoter activity in p53-knockdown H1975 and CL3 cells was nearly completely reversed by transfection with an Nrf2 promoter mutated in the Sp1 binding site (Figure 2D). These results clearly indicate that Nrf2 transcription in lung cancer cells is predominately down-regulated by wild-type p53 via decreased Sp1 binding to the Nrf2 promoter, but is not affected by mutant p53.
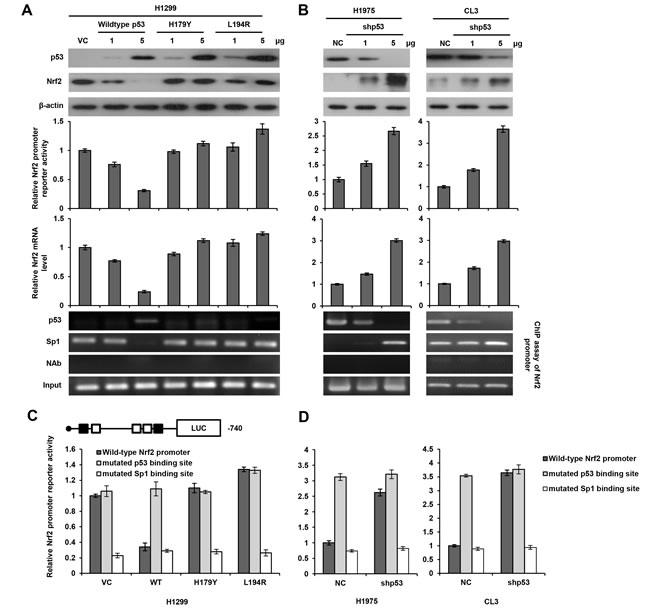
Figure 2: Nrf2 transcription is down-regulated by wild-type p53 via decreased Sp1 binding to the Nrf2 promoter. A. The Nrf2 (–740/+1) promoter activity in H1299 cells, which were transfected with p53 wild-type/mutants plasmid. The cells lysates were separated by SDS-PAGE for the evaluation p53 and Nrf2 expression by specific antibodies using western blotting. An luciferase reporter assay was performed to evaluate the reporter activity of Nrf2 promoter. A ChIP assay was performed to evaluate the binding ability of p53 and Sp1 to the putative binding site of the Nrf2 promoter region. The products were amplified by PCR and the as gel electrophoresis results are presented. B. The reporter activity of the Nrf2 (–740/+1) promoter in H1975 and CL3 cells, which were transfected with shp53 plasmid. The cells lysates were separated by SDS-PAGE for the evaluation p53 and Nrf2 expression by specific antibodies using western blotting. Luciferase reporter assay was performed to evaluate the reporter activity of Nrf2 promoter. A ChIP assay was performed to evaluate the binding ability of p53 and Sp1 to the putative binding site of the Nrf2 promoter region. The products were amplified by PCR and the gel electrophoresis results are presented. C. The reporter activity of the wild-type-, p53 binding site mutated-, and Sp1 binding site mutated-Nrf2 (–740/+1) promoter in H1299, which were transfected with p53 wild-type/mutants plasmid. An luciferase reporter assay was performed to evaluate the reporter activity of Nrf2 promoter. D. The reporter activity of the wild-type-, p53 binding site mutated-, and Sp1 binding site mutated-Nrf2 (–740/+1) promoter in H1975 and CL3 cells, which were transfected with shp53 plasmid. An luciferase reporter assay was performed to evaluate the reporter activity of Nrf2 promoter.
Nrf2-mediated Bcl-2, Bcl-xL, and HO-1 expression is dependent on p53 status and may confer cisplatin resistance
We investigated whether Nrf2 expression could determine cisplatin sensitivity in lung cancer cells that had different p53 status. Two types of p53-null cells (H1299 and H358) and three types of p53-wild-type cells (H1975, CL3, and TL4) were collected to evaluate the inhibitory concentration yielding 50% cell viability (IC50) for cisplatin using the MTT assay. Western blotting showed that Nrf2 expression was decreased by Nrf2 knockdown in H1299 and H358 cells, but was dose-dependently increased by Nrf2 overexpression in H1975, CL3, and TL4 cells (Figure 3A upper panel). Intriguingly, the IC50 value for cisplatin was concomitantly decreased by Nrf2-knockdown in H1299 and H358 cells, but was increased in Nrf2-overexpressing H1975, CL3, and TL4 cells (Figure 3A lower panel). However, the IC50 value for cisplatin was nearly unchanged by different mutant p53 expression vector transfections in H1299 cells when compared with VC cells; however, the IC50 value for cisplatin was almost completely rescued by Nrf2 knockdown in H1299 cells transfected with different mutant p53 expression vectors (Supplementary Figure 1). These results clearly indicated that Nrf2 expression is dependent on p53 status and may confer cisplatin resistance in lung cancer cells.
Nrf2 upregulates Bcl-2 and Bcl-xL transcription and in turn confers drug resistance [22, 30]. HO-1 is a downstream gene of the Nrf2/ARE signaling that promotes tumor drug resistance [31-33]. The possibility that wild-type p53-mediated Nrf2 reduction could promote cisplatin sensitivity was explored by transfecting wild-type p53 into H1299 cells. Western blotting indicated that Nrf2 expression was nearly completely eliminated by wild-type p53 transfection in H1299 cells when compared with VC cells (Figure 3B upper panel). As expected, the decrease in Nrf2 expression in wild-type p53-transfected H1299 cells was gradually increased by ectopic expression of Nrf2. As expected, p53-downstream p21 expression was increased by wild-type p53 overexpression in H1299 cells, but was decreased by wild-type p53 knockdown in H1975 cells. The expression of Bcl-2, Bcl-xL, and HO-1 was concomitantly elevated by ectopic expression of Nrf2 in wild-type p53-transfected H1299 cells. The IC50 value for cisplatin in H1299 cells was markedly decreased by wild-type p53 transfection, but the IC50 value was gradually increased by co-transfection with Nrf2 expression vector in H1299 cells (19.6 μM vs. 8.6 μM vs. 13.2 μM vs. 18.4 μM; Figure 3B left lower panel). The increase in Nrf2, Bcl-2, Bcl-xL, and HO-1 expressions were observed in p53-knockdown H1975 cells, but the increases of these three molecules were reversed by Nrf2 silencing in p53-knockdown H1975 cells (Figure 3B, right upper panel). The IC50 value of p53-knockdown H1975 cells was increased to 16.2 μM when compared with H1975 cells with non-specific shRNA transfection (NC) (5.6 μM). However, the increase in the IC50 value in p53-knockdown H1975 cells was suppressed by Nrf-2 silencing in a dose-dependent manner (Figure 3B, right lower panel). Annexin-V/PI assay further indicated that the percentages of cell apoptosis in both cell types were elevated and reduced by p53 manipulation (Supplementary Figure 2). These results suggest that Nrf2-mediated Bcl-2, Bcl-xL and HO-1 is dependent on p53 status and may confer cisplatin resistance via apoptotic machinery.
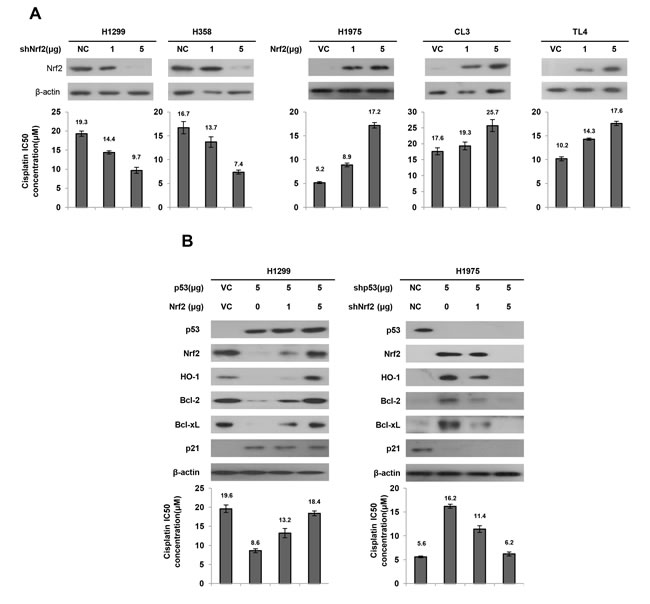
Figure 3: Nrf2 expression is responsible for cisplatin sensitivity in lung cancer cells. A. shNrf2 plasmids were transfected into high Nrf2-expressing (H1299 and H358) cell lines compared with both cell types transfected with a non-specific shRNA (NC), Nrf2 expression plasmids were transfected into low Nrf2-expressing (H1975 and CL3) cell lines compared with both cell types transfected with an empty vector (VC). After 24h, the indicated cells were incubated with or without cisplatin (0, 2, 4, 8, 16, 32 μM) for 48 h for MTT assay. The cell lysates were separated by SDS-PAGE for the evaluation Nrf2 expression by specific antibodies using western blotting. The MTT assay was used to determine the 50% inhibition concentration (IC50) of cisplatin. B. H1299 cells were transfected with p53 and/or Nrf2 plasmid. H1975 cells were transfected with shp53 and/or shNrf2 plasmid. After 24h, the indicated cells were incubated with or without cisplatin for 48 h for MTT assay. The cell lysates were separated by SDS-PAGE for the evaluation p53, Nrf2, HO-1, Bcl-2 and Bcl-xL expression by specific antibodies using western blotting. The MTT assay was used to determine the IC50 of cisplatin.
Bcl-2 and Bcl-xL are more involved than HO-1 in Nrf2-mediated cisplatin resistance
We next examined which genes regulated by Nrf2 were more involved in cisplatin resistance. High Nrf2 expressing H1299 cells were selected for transfection with shHO-1, shBcl-2, or shBcl-xL, and low Nrf2 expressing H1975 cells were transfected with the Nrf2 expression vector and then co-transfected with shHO-1, shBcl-2, or shBcl-xL. The MTT assay results showed that the IC50 value of H1299 and Nrf2-overexpressing H1975 cells was markedly decreased by shHO-1, shBcl-2, or shBcl-xL, when compared with H1299 NC cells and Nrf2-overexpressing H1975 cells (Figure 4). Interestingly, the IC50 value was lower for both cell types transfected with shBcl-2 or shBcl-xL than for both cell types transfected with shHO-1 (Figure 4). These results clearly indicated that Bcl-2 and Bcl-xL are more involved than HO-1 in Nrf2-mediated cisplatin resistance.
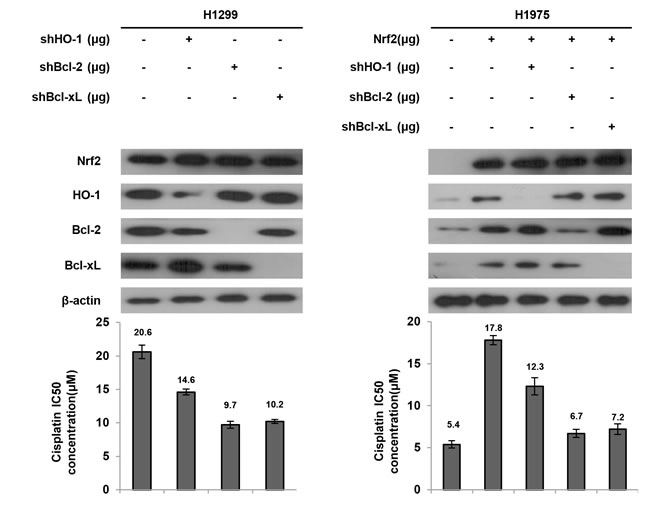
Figure 4: Bcl-2 and Bcl-xL are more involved than HO-1 on Nrf2-mediated cisplatin resistance. H1299 cells were transfected with shHO-1, shBcl-2 or shBcl-xL plasmid. H1975 cells were transfected with Nrf2, shHO-1, shBcl-2 or shBcl-xL plasmid. After 24h, the indicated cells were incubated with or without cisplatin for 48 h for MTT assay. The cell lysates were separated by SDS-PAGE for the evaluation Nrf2, HO-1, Bcl-2 and Bcl-xL expression by specific antibodies using western blotting. The MTT assay was used to determine the IC50 of cisplatin.
Nrf2 mRNA levels are associated with p53 status and related to tumor responses to cisplatin-based chemotherapy in NSCLC patients
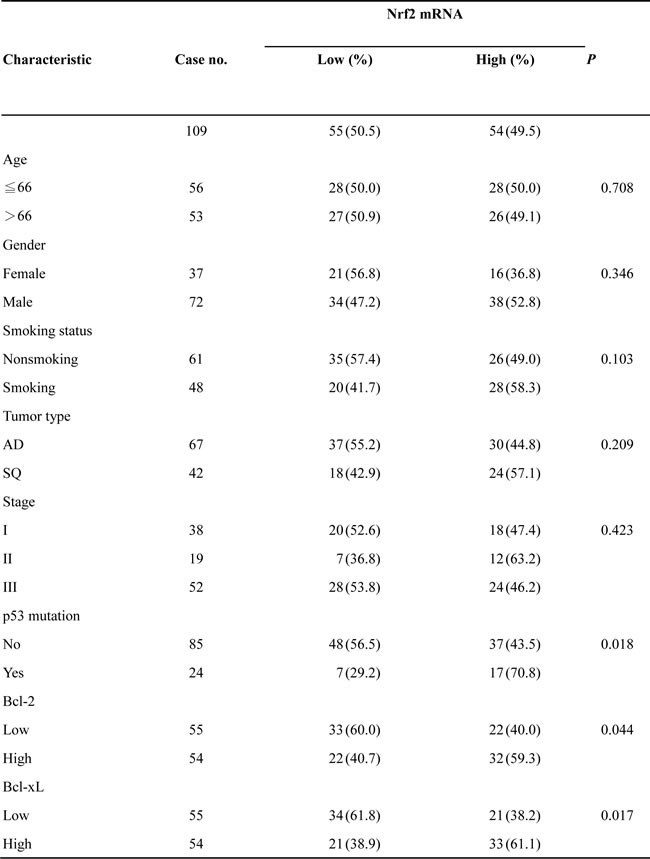
We examined whether Nrf2 mRNA expression levels could be associated with p53 status in NSCLC patients. In total, 114 tumors from surgically resected NSCLC patients, who were determined not to have keap1 and Nrf2 mutation (n = 109, Supplementary Table 1), were evaluated for Nrf2 mRNA expression levels using real-time PCR. The median value of Nrf2 mRNA expression levels in lung tumors was used as a cutoff point to divide patients into high-Nrf2 and low-Nrf2 subgroups and the categories were further confirmed by a box plot analysis (Supplementary Figure 3). The p53 mutation data were obtained from our previous reports [34, 35]. Nrf2 mRNA expression was not associated with clinico-pathological parameters, including age, gender, smoking status, tumor histology, and stage. Interestingly, high-Nrf2 mRNA tumors were more commonly observed in p53-mutant patients than in p53-wild-type patients (70.8% vs. 43.5%, p = 0.018; Table 1). High Bcl-2 and high Bcl-xL mRNA expression were more prevalently occurred in high-Nrf2 mRNA tumors than in low-Nrf2 mRNA tumors (59.3% vs. 40%, p = 0.044 for Bcl-2; 61.1% vs. 38.2%, p = 0.017; Table 1).
Table 1: Relationships between Nrf2 mRNA expression and clinical-pathological parameters NSCLC patients
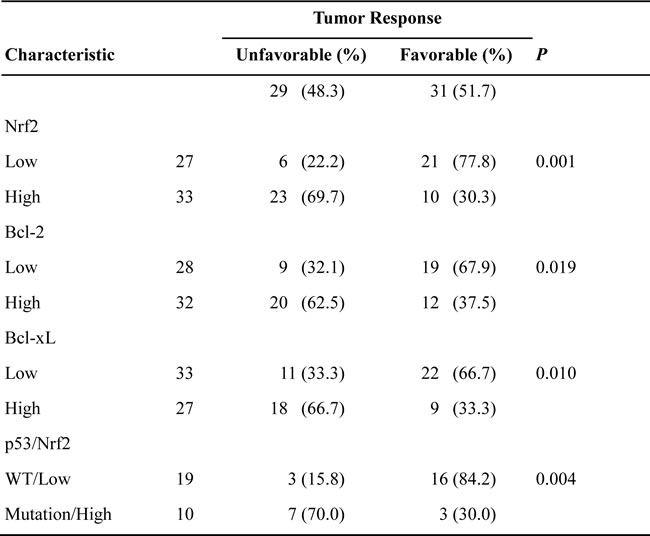
We next examined the possibility that Nrf2 mRNA expression levels could be associated with the tumor response to cisplatin-based chemotherapy. In total, 60 of the 109 patients were available for this retrospective study, and data indicated that an unfavorable response to cisplatin-based chemotherapy was more likely in patients with high-Nrf2 mRNA tumors than with low-Nrf2 mRNA tumors (71.9% vs. 21.4%, p = 0.001; Table 2). Similar findings in an unfavorable response to cisplatin-based chemotherapy were more commonly observed in high-Bcl-2 or high-Bcl-xL mRNA tumors than their counterparts (62.5% vs. 32.1% for Bcl-2; 66.7% vs. 33.3%, p = 0.01; Table 2). We further showed that an unfavorable response to cisplatin-based chemotherapy was more common in p53-mutant patients who harbored high-Nrf2 mRNA tumors than low-Nrf2 tumors (70% vs. 15.8%, p = 0.004; Table 2). These results suggested that high-Nrf2 mRNA patients whose tumors harbored p53 mutations may more frequently show an unfavorable response to cisplatin-based chemotherapy when compared with patients whose tumors harbored wild-type p53. Bcl-2 and Bcl-xL expression may be partially contributive to Nrf2-mediated unfavorable response to cisplatin-based chemotherapy in NSCLC patients.
Table 2: Association of Nrf2, Bcl-2 and Bcl-xL mRNA in lung tumors with tumor response to cisplatin-based chemotherapy in NSCLC patients with tumor recurrence and/or metastasis after surgical resection.
Nrf2 mRNA expression levels are associated with overall survival (OS) and relapse free survival (RFS) in NSCLC patients
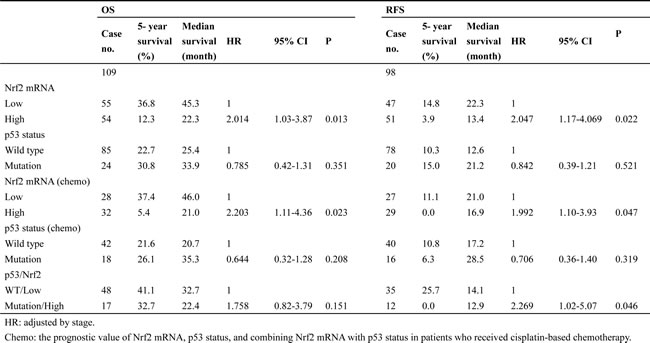
We next examined the possibility that Nrf2 mRNA expression levels could be associated with OS and RFS in NSCLC patients. Cox regression analysis using all studied population (n = 109) indicated that high-Nrf2 mRNA patients exhibited worse OS and RFS than low-Nrf2 mRNA patients (hazard ratio, HR, 2.014, 95% CI, 1.03-3.87, p = 0.013 for OS; HR, 2.047, 95% CI, 1.17-4.069, p = 0.022 for RFS; Table 3). The five-year survival rate and median survival month for OS and RFS were lower and shorter in high-Nrf2 mRNA patients than in low-Nrf2 mRNA patients (OS: 12.3% vs. 36.8% for five-year survival rate, 22.3 months vs. 45.3 months; RFS: 3.9% vs. 14.8% for five-year survival rate, 13.4 months vs. 22.3 months).
A prognostic significance of Nrf2 mRNA expression levels on OS and RFS was still observed in 60 patients who have received cisplatin-based chemotherapy (HR, 2.203, 95% CI, 1.11-4.36, p = 0.023 for OS; HR, 1.992, 95% CI, 1.10-3.93, p = 0.047; Table 3). However, a prognostic significance of p53 status on OS and RFS was not revealed in all studied cases or in the 60 patients who had received cisplatin-based chemotherapy (Table 2). We also found worse RFS in p53-mutant patients who harbored high-Nrf2 tumors rather than low-Nrf2 mRNA tumors (HR, 2.269, 95% CI, 1.02-5.07, p = 0.046; Table 3), but a prognostic value for OS was not observed in the high-Nrf2 patients. The chemotherapeutic regimens for these sixty patients are listed in Supplementary Table 2. These patients received cisplatin alone (8.3%) and/or combined with other chemotherapeutic agents including gemcitabine (73.3%), vp16 (8.3%), taxol (8.3%), and mitomycin C (1.7%). These results suggest that high Nrf2 mRNA expression levels may be useful for prediction of poorer OS and RFS in NSCLC patients. A prognostic significance of Nrf2 mRNA levels on OS and RFS was also observed in patients who had received cisplatin-based chemotherapy.
Table 3: Cox regression analysis for the influence of Nrf2 mRNA, p53 status, and combining Nrf2 mRNA with p53 status on OS and RFS in NSCLC patients.
Discussion
The present study provides evidence that suppression of Nrf2 transcription by wild-type p53, occurring via decreased Sp1 binding to the Nrf2 promoter, may confer cisplatin sensitivity, a favorable chemo-response, thereby leading to favorable outcomes in lung cancer patients. Moreover, a decrease in Nrf2 mRNA expression by wild-type p53 corresponded with its protein expression. These findings suggest that Nrf2 expression is predominately regulated by wild-type p53 at the transcription level. Conversely, Nrf2 mRNA and its protein expression levels in H1299 cells were markedly elevated by different mutant p53 expression vector transfections when compared with VC cells (Figure 1C). A previous report has indicated that Nrf2 expression is driven by the NF-κB signaling pathway in acute myeloid leukemia [23]. Mutation of p53 gene prolongs NF-κB activation and promotes chronic inflammation and inflammation-associated colorectal cancer [36]. Therefore, mutant p53 not only confers drug resistance via upregulation of Nrf2 expression but it also may activate the NF-κB signaling pathway for additional enhancement of Nrf2 expression (Figure 1C).
The prevalence of “low” or “high” Nrf2 expression was not significantly revealed in p53-wild-type patients compared with p53-mutant patients. This conflicting may be due to wild-type p53 dysfunction by several mechanisms. For example, Nrf2 may promote MDM2 expression to increase p53 degradation [28]. An early report indicated that MDM2 mRNA expression may be used to predict p53 transcriptional function and patients’ prognosis in NSCLC [37]. We thus evaluated MDM2 mRNA expression in p53-wild-type patients by real-time PCR in this study population and data showed that high Nrf2 mRNA expression was more commonly occurred in low-MDM2 mRNA tumors than in high-MDM2 tumors (Supplementary Table 3). Therefore, wild-type p53 dysfunction by different mechanisms may cause p53 wild-type patients with high Nrf2 mRNA tumors, and consequently resulting in the prevalence of “low” or “high” Nrf2 expression to be not significantly revealed in p53-wild-type patients..
ROS levels are tightly controlled by the Nrf2/Keap1 pathway [38]. However, an increased in Nrf2 transcription, that were triggered by oncogenes such as mutations in K-rasG12D and BrafV619V and overexpression of MycERT2, promotes ROS detoxification and tumorigenesis [38]. K-ras increases Nrf2 gene transcription through a TPA response element located on the Nrf2 promoter [24]. In a mouse model of mutant K-rasG12D-induced lung cancer, suppressing the Nrf2 pathway with the chemical inhibitor Brusatol enhanced the antitumor efficacy of cisplatin [24]. These results strongly suggest that oncogenic K-ras promotes tumor malignancy as well as conferring cisplatin resistance in lung cancer through upregulation of Nrf2 transcription. However, K-ras mutations were not detected in this study population (n = 114, data not shown). In the present study, mutant p53 upregulated Nrf2 transcription by increased Sp1 binding to the Nrf2 promoter, thereby conferring cisplatin resistance. Consistent findings were also observed in wild-type p53 cells subjected to p53 silencing and in p53-null cells transfected with different mutant p53 expression vectors. We therefore suggest that mutant p53 may confer cisplatin resistance in lung cancer cells via upregulating Nrf2 transcription.
In summary, we provide evidence that upregulation of Nrf2 transcription by mutant p53 may confer cisplatin resistance, an unfavorable response to cisplatin-based chemotherapy, and poor outcomes in NSCLC patients. Therefore, we suggest that Bcl-2 antagonists might be helpful in improving cisplatin sensitivity and outcomes in p53-mutant NSCLC patients who harbor high-Nrf2 mRNA tumors.
Materials and Methods
Study subjects
Lung tumor specimens were collected from 114 primary lung cancer at the Department of Thoracic Surgery, Taichung Veterans General Hospital (Taichung, Taiwan), between 1998 and 2004. Patients were asked to submit written informed consent and were approved by the Institutional Review Board (Institutional Review Board, Chung Shan Medical University Hospital, CSMUH No: CS11177). The tumor type and stage of each collected specimen were histologically determined according to the World Health Organization classification system. Cancer relapse data were obtained by chart review and confirmed by thoracic surgeons. Clinical parameters and OS and RFS data were collected from chart reviews and the Taiwan Cancer Registry, Ministry of Health and Welfare, Executive Yuan, ROC.
Cell lines and culture conditions
H1299, H1355, H1650, H1975 and H358 cells were obtained from ATCC (Manassas, VA, USA) and were cultured as previously described (http://www.atcc.org). CL1-5 and CL3 cells were kindly provided by Dr. Pan-Chyr Yang (Department of Internal Medicine, National Taiwan University Hospital, Taiwan). TL4 cells were kindly provided by Dr. Ya-Wen Cheng (Graduate Institute of Cancer Biology and Drug Discovery, Taipei Medical University, Taipei, Taiwan). H1299 cells were grown in Dulbecco’s modified Eagle’s medium (Hyclone, Waltham, MA, USA) supplemented with 10% fetal bovine serum (Hyclone, Waltham, MA, USA). CL1-5, CL3, H1355, H1650, H1975, H358 and TL4 cells were grown in RPMI-1640 medium (Hyclone, Waltham, MA, USA) with 10% fetal bovine serum. The type of p53 mutations in eight cell lines has been listed in Supplementary Table 4. All cell lines were grown at 37°C in a 5% carbon dioxide atmosphere. These cells were cultured according to the suppliers’ instructions and were stored used at passages 5 to 20. Once resuscitated, cell lines were routinely authenticated (once every 6 months; cells were last tested in December 2012) through cell morphology monitoring, growth curve analysis, species verification by isoenzymology and karyotyping, identity verification using short tandem repeat profiling analysis, and contamination checks.
Plasmid constructs and transfection assays
Nrf2 cDNA was cloned into pcDNA3.1 Zeo(+) (Invitrogen, Carlsbad, CA, USA) by PCR amplification with newly created XhoI and BamHI sites attached on the Nrf2 5’ends of forward and reverse primers, using H1299 cDNA as template. The Nrf2 promoter reporter plasmid was constructed by inserting 1036, 740, and 229 bps KpnI/HidIII fragments into a KpnI/HidIII-treated pGL3 vector (Promega, Madison, WI, USA). The primer sequences are listed in Supplementary Table 5. The wild-type and mutant p53 constructs were kindly provided by Dr. Jiunn-Liang Ko (Institute of Medicine, Chung Shan Medical University, Taichung, Taiwan) [43]. The shRNA was purchased from National RNAi Core Facility, Academia Sinica, Taipei, Taiwan. Different concentrations of plasmids were transiently transfected into lung cancer cells (1 × 106 cells) using the Turbofect reagent (Fermentas, Waltham, MA, USA). After 48 h, cells were harvested and whole-cell extracts were used for subsequent experiments.
Real-time PCR analysis of mRNA expression levels
DNase I-treated total RNA (10 ng) was subjected to real-time PCR analysis with the Reverse Transcription Kit (Applied Biosystems, Foster City, CA), mRNA Assays (Applied Biosystems, Foster City, CA, USA), using a Real-Time Thermocycler 7500 (Applied Biosystems, Foster City, CA, USA). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as the mRNA reference housekeeping gene. The primers used for real-time PCR analysis of mRNA expression are presented in Supplementary Table 5.
Luciferase reporter assay
The luciferase reporter assay was conducted by transfecting appropriate numbers of cells with sufficient reporter plasmids, as determined from earlier studies [39].
ChIP assay
ChIP analysis was performed as described previously [39]. The primer sequences are presented in Supplementary Table 5.
3-(4,5-cimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) cytotoxicity assay
The cell lines were cultured in 96-well flat-bottomed microtiter plates supplemented with RPMI 1640 and DMEM containing 10% heat-inactivated fetal bovine serum, 100 units/mL penicillin, and 100 units/mL streptomycin in a humidified atmosphere containing 95% air and 5% CO2 at 37°C in a humidified incubator. Before cisplatin treatment(0, 2, 4, 8, 16, 32 μM), the cells cultured in the exponential growth phase were pretreated with shRNAs, p53 and Nrf2 overexpression plasmid for 24 h. After 48 h incubation, the in vitro cytotoxic effects of these treatments were determined by MTT assay (at 570 nm).
Statistical analysis
Statistical analysis was performed using the SPSS statistical software program Version 15.0 (SPSS Inc., USA). Survival plots were generated using the Kaplan-Meier method, and differences between patient groups were determined by a log-rank test. A multivariate Cox regression analysis was performed for overall survival and relapse free survival. The analysis was stratified for all known prognostic variables (age, sex, smoking status, tumor type, and stage) and for mRNA expression.
Acknowledgments
This work was supported by grants from the Ministry of Science and Technology (MOST103-2320-B-038-036-MY2, TMU101-AE1-B10) Taiwan.
conflicts of interest
The authors declare no conflict of interest.
References
1. Jin J, Sklar GE, Min Sen Oh V and Chuen Li S. Factors affecting therapeutic compliance: A review from the patient’s perspective. Therapeutics and clinical risk management. 2008; 4:269-286.
2. Molina JR, Yang P, Cassivi SD, Schild SE and Adjei AA. Non-small cell lung cancer: epidemiology, risk factors, treatment, and survivorship. Mayo Clinic proceedings. 2008; 83:584-594.
3. Tiseo M, Ardizzoni A and Boni L. First-Line Chemotherapy Treatment of Advanced Non-Small-Cell Lung Cancer: Does Cisplatin versus Carboplatin Make a Difference? Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2014; 9:e82.
4. Santana-Davila R, Szabo A, Arce-Lara C, Williams CD, Kelley MJ and Whittle J. Cisplatin versus carboplatin-based regimens for the treatment of patients with metastatic lung cancer. an analysis of Veterans Health Administration data. Journal of thoracic oncology. 2014; 9:702-709.
5. Itoh T, Terazawa R, Kojima K, Nakane K, Deguchi T, Ando M, Tsukamasa Y, Ito M and Nozawa Y. Cisplatin induces production of reactive oxygen species via NADPH oxidase activation in human prostate cancer cells. Free radical research. 2011; 45:1033-1039.
6. Marullo R, Werner E, Degtyareva N, Moore B, Altavilla G, Ramalingam SS and Doetsch PW. Cisplatin induces a mitochondrial-ROS response that contributes to cytotoxicity depending on mitochondrial redox status and bioenergetic functions. PloS one. 2013; 8:e81162.
7. Villeneuve NF, Sun Z, Chen W and Zhang DD. Nrf2 and p21 regulate the fine balance between life and death by controlling ROS levels. Cell cycle. 2009; 8:3255-3256.
8. Nguyen T, Nioi P and Pickett CB. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. The Journal of biological chemistry. 2009; 284:13291-13295.
9. Gorrini C, Harris IS and Mak TW. Modulation of oxidative stress as an anticancer strategy. Nature reviews Drug discovery. 2013; 12:931-947.
10. Kansanen E, Kuosmanen SM, Leinonen H and Levonen AL. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox biology. 2013; 1:45-49.
11. Zucker SN, Fink EE, Bagati A, Mannava S, Bianchi-Smiraglia A, Bogner PN, Wawrzyniak JA, Foley C, Leonova KI, Grimm MJ, Moparthy K, Ionov Y, Wang J, Liu S, Sexton S, Kandel ES, et al. Nrf2 amplifies oxidative stress via induction of Klf9. Molecular cell. 2014; 53:916-928.
12. Singh A, Misra V, Thimmulappa RK, Lee H, Ames S, Hoque MO, Herman JG, Baylin SB, Sidransky D, Gabrielson E, Brock MV and Biswal S. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS medicine. 2006; 3:e420.
13. Padmanabhan B, Tong KI, Ohta T, Nakamura Y, Scharlock M, Ohtsuji M, Kang MI, Kobayashi A, Yokoyama S and Yamamoto M. Structural basis for defects of Keap1 activity provoked by its point mutations in lung cancer. Molecular cell. 2006; 21:689-700.
14. Yoo NJ, Kim HR, Kim YR, An CH and Lee SH. Somatic mutations of the KEAP1 gene in common solid cancers. Histopathology. 2012; 60:943-952.
15. Miura S, Shibazaki M, Kasai S, Yasuhira S, Watanabe A, Inoue T, Kageshita Y, Tsunoda K, Takahashi K, Akasaka T, Masuda T and Maesawa C. A somatic mutation of the KEAP1 gene in malignant melanoma is involved in aberrant NRF2 activation and an increase in intrinsic drug resistance. The Journal of investigative dermatology. 2014; 134:553-556.
16. Kim YR, Oh JE, Kim MS, Kang MR, Park SW, Han JY, Eom HS, Yoo NJ and Lee SH. Oncogenic NRF2 mutations in squamous cell carcinomas of oesophagus and skin. The Journal of pathology. 2010; 220:446-451.
17. Shibata T, Kokubu A, Saito S, Narisawa-Saito M, Sasaki H, Aoyagi K, Yoshimatsu Y, Tachimori Y, Kushima R, Kiyono T and Yamamoto M. NRF2 mutation confers malignant potential and resistance to chemoradiation therapy in advanced esophageal squamous cancer. Neoplasia. 2011; 13:864-873.
18. Wang XJ, Sun Z, Villeneuve NF, Zhang S, Zhao F, Li Y, Chen W, Yi X, Zheng W, Wondrak GT, Wong PK and Zhang DD. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis. 2008; 29:1235-1243.
19. Akhdar H, Loyer P, Rauch C, Corlu A, Guillouzo A and Morel F. Involvement of Nrf2 activation in resistance to 5-fluorouracil in human colon cancer HT-29 cells. European journal of cancer. 2009; 45:2219-2227.
20. Homma S, Ishii Y, Morishima Y, Yamadori T, Matsuno Y, Haraguchi N, Kikuchi N, Satoh H, Sakamoto T, Hizawa N, Itoh K and Yamamoto M. Nrf2 enhances cell proliferation and resistance to anticancer drugs in human lung cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009; 15:3423-3432.
21. Ji L, Li H, Gao P, Shang G, Zhang DD, Zhang N and Jiang T. Nrf2 pathway regulates multidrug-resistance-associated protein 1 in small cell lung cancer. PloS one. 2013; 8:e63404.
22. Niture SK and Jaiswal AK. Nrf2-induced antiapoptotic Bcl-xL protein enhances cell survival and drug resistance. Free radical biology & medicine. 2013; 57:119-131.
23. Rushworth SA, Zaitseva L, Murray MY, Shah NM, Bowles KM and MacEwan DJ. The high Nrf2 expression in human acute myeloid leukemia is driven by NF-kappaB and underlies its chemo-resistance. Blood. 2012; 120:5188-5198.
24. Tao S, Wang S, Moghaddam SJ, Ooi A, Chapman E, Wong PK and Zhang DD. Oncogenic KRAS confers chemoresistance by upregulating NRF2. Cancer research. 2014; 74:7430-41.
25. Rotblat B, Melino G and Knight RA. NRF2 and p53: Januses in cancer? Oncotarget. 2012; 3:1272-1283.
26. Chen W, Sun Z, Wang XJ, Jiang T, Huang Z, Fang D and Zhang DD. Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Molecular cell. 2009; 34:663-673.
27. Asher G, Lotem J, Kama R, Sachs L and Shaul Y. NQO1 stabilizes p53 through a distinct pathway. Proceedings of the National Academy of Sciences of the United States of America. 2002; 99:3099-3104.
28. You A, Nam CW, Wakabayashi N, Yamamoto M, Kensler TW and Kwak MK. Transcription factor Nrf2 maintains the basal expression of Mdm2: An implication of the regulation of p53 signaling by Nrf2. Archives of biochemistry and biophysics. 2011; 507:356-364.
29. Faraonio R, Vergara P, Di Marzo D, Pierantoni MG, Napolitano M, Russo T and Cimino F. p53 suppresses the Nrf2-dependent transcription of antioxidant response genes. The Journal of biological chemistry. 2006; 281:39776-39784.
30. Niture SK and Jaiswal AK. Nrf2 protein up-regulates antiapoptotic protein Bcl-2 and prevents cellular apoptosis. The Journal of biological chemistry. 2012; 287:9873-9886.
31. Srisook K, Kim C and Cha YN. Molecular mechanisms involved in enhancing HO-1 expression: de-repression by heme and activation by Nrf2, the “one-two” punch. Antioxidants & redox signaling. 2005; 7:1674-1687.
32. Tan Q, Wang H, Hu Y, Hu M, Li X, Aodeng Q, Ma Y, Wei C and Song L. Src/STAT3-dependent HO-1 induction mediates chemoresistance of breast cancer cells to doxorubicin by promoting autophagy. Cancer science. 2015; 106:1023-32.
33. Berberat PO, Dambrauskas Z, Gulbinas A, Giese T, Giese N, Kunzli B, Autschbach F, Meuer S, Buchler MW and Friess H. Inhibition of heme oxygenase-1 increases responsiveness of pancreatic cancer cells to anticancer treatment. Clinical cancer research : an official journal of the American Association for Cancer Research. 2005; 11:3790-3798.
34. Wu JY, Wang J, Lai JC, Cheng YW, Yeh KT, Wu TC, Chen CY and Lee H. Association of O6-methylguanine-DNA methyltransferase (MGMT) promoter methylation with p53 mutation occurrence in non-small cell lung cancer with different histology, gender, and smoking status. Annals of surgical oncology. 2008; 15:3272-3277.
35. Chiou YH, Wong RH, Chao MR, Chen CY, Liou SH and Lee H. Nickel accumulation in lung tissues is associated with increased risk of p53 mutation in lung cancer patients. Environmental and molecular mutagenesis. 2014; 55:624-632.
36. Cooks T, Pateras IS, Tarcic O, Solomon H, Schetter AJ, Wilder S, Lozano G, Pikarsky E, Forshew T, Rosenfeld N, Harpaz N, Itzkowitz S, Harris CC, Rotter V, Gorgoulis VG and Oren M. Mutant p53 prolongs NF-kappaB activation and promotes chronic inflammation and inflammation-associated colorectal cancer. Cancer cell. 2013; 23:634-646.
37. Ko JL, Cheng YW, Chang SL, Su JM, Chen CY and Lee H. MDM2 mRNA expression is a favorable prognostic factor in non-small-cell lung cancer. International journal of cancer Journal international du cancer. 2000; 89:265-270.
38. DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, Mangal D, Yu KH, Yeo CJ, Calhoun ES, Scrimieri F, Winter JM, Hruban RH, Iacobuzio-Donahue C, Kern SE, Blair IA, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011; 475:106-109.
39. Wu DW, Liu WS, Wang J, Chen CY, Cheng YW and Lee H. Reduced p21(WAF1/CIP1) via alteration of p53-DDX3 pathway is associated with poor relapse-free survival in early-stage human papillomavirus-associated lung cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011; 17:1895-1905.