
Introduction
Wnt signaling is essential for stem cell regulation in development and tissue homeostasis [1]. Wnt signaling plays key roles in cell proliferation, cell fate determination, cell migration, and cell polarity during embryonic development and stem cell regulation [2]. Wnt ligands bind to Frizzled receptors and low-density lipoprotein receptor-related protein 5/6 co-receptors, which stabilizes β-catenin protein by inhibiting the protein destruction complex composed of adenomatous polyposis coli, Axin, casein kinase 1, and glycogen synthase kinase 3. Subsequently, stabilized β-catenin protein is translocated into the nucleus and replaces T cell factor (TCF)–associated corepressors with coactivators, which results in the transcriptional activation of the β-catenin target genes [2]. However, deregulated Wnt signaling leads to intestinal tumorigenesis. Frequent genetic mutations in Wnt signaling components have been closely associated with human colorectal cancer (CRC) [3]. In mouse models, genetic mutations that lead to the hyperactivation of Wnt signaling induce mammary tumors and intestinal adenomas [6], recapitulating the pivotal roles of Wnt signaling in intestinal tumor initiation. Such hyperactivation of Wnt/β-catenin signaling is driven by genetic mutations in core components of Wnt signaling. For example, more than 70 % of CRC cells display somatic mutation in Wnt signaling components including APC, β-catenin/CTNNB1, or AXIN1 [4, 5]. Additionally, it was shown that transcriptional silencing of Wnt signaling antagonists induces Wnt signaling hyperactivation in CRC cells [7-13]. Moreover, it was suggested that high expression of Wnt3A is associated with intestinal tumorigenesis [14]. Among 19 Wnt ligands in mammals, Wnt1, Wnt3A, Wnt8, and Wnt10 transduce Wnt signaling via β-catenin, called as canonical Wnt ligands [15]. Wnt5A and Wnt11 activate small GTPases (RhoA and Rac1), Ca2+ signaling, protein kinase C, or planar cell polarity, called as non-canonical Wnt signal transduction [16]. Due to diverse impacts of 19 Wnt ligands to cellular functions in combination with 10 Frizzled receptors, the effects of Wnt ligands on tumorigenesis still remains ambiguous.
Wnt2, a member of the WNT gene family, directs cell specification during development [17]. Wnt2 plays a critical role in development. In mouse models, genetic ablation of WNT2 induces vascular defects [18]. In Drosophila, Wnt2 is required for development of male reproductive tract [19]. Intriguingly, Wnt2 upregulation was observed in various human cancers [20-23]. It was shown that Wnt2 plays tumorigenic roles in several cancers including non-small-cell lung cancer, pancreatic cancer, ovary cancer, esophageal cancer, and gastro-intestinal cancer [24-29]. Also, it was suggested that upregulation of Wnt2 is likely to be an early event during intestinal tumorigenesis [21]. In cancer cells, Wnt2 expression is associated with anchorage-independent cell survival, metastasis, and tumor invasion [24, 30]. Moreover, the expression of Wnt2 is implicated in activating/stabilizing β-catenin, similar to other canonical (β-catenin-mediated) Wnt ligands [27, 31-35]. And, the blockade of Wnt2 destabilizes β-catenin protein in CRC cells [36]. It was shown that Wnt2 is enriched in circulating pancreatic cancer cells [24]. Despite significant implication of Wnt2 in malignant cancer, it remains unclear how Wnt2 contributes to tumorigenesis.
Herein, we identified Wnt2 as a key ligand that complements Wnt/β-catenin signaling activity in CRC. To understand the pathologic impacts of Wnt ligands to intestinal tumorigenesis, we analyzed the expression of 19 Wnt ligands in CRC cells, and found that Wnt2 is significantly upregulated in CRC and hyperactivates β-catenin. Depletion of endogenous Wnt2 inhibits CRC cell proliferation, accompanied with the decreased Wnt/β-catenin signaling activity. We also found that Polycomb Repressive Complex 2 (PRC2) epigenetically controls expression of WNT2. Our results suggest that Wnt2 enhances Wnt/β-catenin signaling in an autocrine manner, which contributes to intestinal tumorigenesis.
Results
Expression of Wnt2 in CRC
We initially sought to investigate the impacts of each Wnt ligand to intestinal tumorigenesis. To do this, we assessed expression of Wnt ligands in CRC cells. We performed in silico expression analysis of Wnt ligands in CRC cells, using publicly available database (www.oncomine.org). We found that, among 19 Wnt ligands, WNT2 and WNT5A are highly upregulated in colon mucinous adenocarcinoma, rectal adenocarcinoma, colon adenocarcinoma, and cecum adenocarcinoma (Figure 1A). Additionally, analysis of 26 independent datasets showed that expression of WNT2 and WNT5A is highly elevated in CRC, while underexpressed or not expressed in normal intestine (Figure 1B). These results were further validated by immunostaining of CRC tissue microarray for Wnt2 expression. We observed that unlike normal colorectum tissues, 60% (21/35) of human CRC tissues expressed high level of Wnt2 (Figure 1C). Consistently, immunoblot (IB) analyses confirmed that Wnt2 is highly expressed in CRC cell lines (Figure 1D). These results suggest that Wnt2 expression is significantly upregulated in CRC.
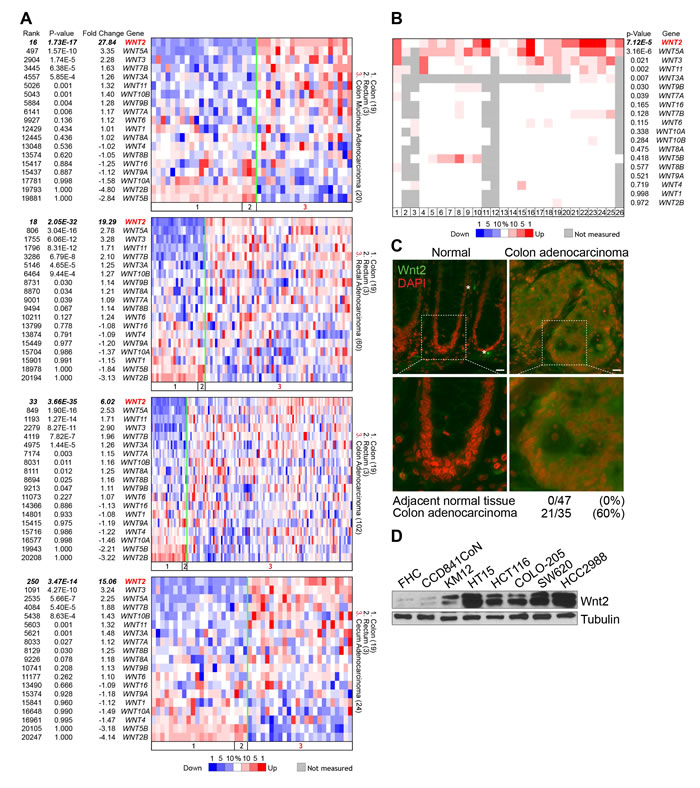
Figure 1: Expression of Wnt2 in CRC. A. Oncomine analysis of WNT ligands in CRC. P value < 0.0001; fold change > 2. TCGA CRC datasets. B. Oncomine analysis of WNT2 expression in CRC datasets. P value < 0.0001; fold change > 2. Dataset information: 1. Rectal Adenocarcinoma vs. Normal / Gaedcke Colorectal, Genes Chromosomes Cancer, 2010; 2. Colorectal Adenoma Epithelia vs. Normal / Gaspar Colon, Am J Pathol, 2008; 3. Colorectal Carcinoma vs. Normal / Graudens Colon, Genome Biol, 2006; 4. Colorectal Carcinoma vs. Normal / Hong Colorectal, Clin Exp Metastasis, 2010; 5. Cecum Adenocarcinoma vs. Normal / Kaiser Colon, Genome Biol, 2007; 6. Colon Adenocarcinoma vs. Normal / Kaiser Colon, Genome Biol, 2007; 7. Colon Mucinous Adenocarcinoma vs. Normal / Kaiser Colon, Genome Biol, 2007; 8. Rectal Adenocarcinoma vs. Normal / Kaiser Colon, Genome Biol, 2007; 9. Rectal Mucinous Adenocarcinoma vs. Normal / Kaiser Colon, Genome Biol, 2007; 10. Rectosigmoid Adenocarcinoma vs. Normal / Kaiser Colon, Genome Biol, 2007; 11. Colon Adenocarcinoma vs. Normal / Ki Colon, Int J Cancer, 2007; 12. Colon Adenocarcinoma vs. Normal / Notterman Colon, Cancer Res, 2001; 13. Colon Adenoma vs. Normal / Sabates-Bellver Colon, Mol Cancer Res, 2007; 14. Rectal Adenoma vs. Normal / Sabates-Bellver Colon, Mol Cancer Res, 2007; 15. Colorectal Adenocarcinoma vs. Normal / Skrzypczak Colorectal, PLoS One, 2010; 16. Colorectal Carcinoma vs. Normal / Skrzypczak Colorectal, PLoS One, 2010; 17. Colon Adenoma Epithelia vs. Normal / Skrzypczak Colorectal 2, PLoS One, 2010; 18. Colon Adenoma vs. Normal / Skrzypczak Colorectal 2, PLoS One, 2010; 19. Colon Carcinoma Epithelia vs. Normal / Skrzypczak Colorectal 2, PLoS One, 2010; 20. Colon Carcinoma vs. Normal / Skrzypczak Colorectal 2, PLoS One, 2010; 21. Cecum Adenocarcinoma vs. Normal / TCGA Colorectal, Nature, 2012; 22. Colon Adenocarcinoma vs. Normal / TCGA Colorectal, Nature, 2012; 23. Colon Mucinous Adenocarcinoma vs. Normal / TCGA Colorectal, Nature, 2012; 24. Rectal Adenocarcinoma vs. Normal / TCGA Colorectal, Nature, 2012; 25. Rectal Mucinous Adenocarcinoma vs. Normal / TCGA Colorectal, Nature, 2012; 26. Colon Carcinoma vs. Normal / Zou Colon, Oncogene, 2002. C. Wnt2 expression in human CRC tissues. Human CRC tissue microarray was analyzed for Wnt2 expression by immunofluorescent staining. 4’,6-diamidino-2-phenylindole, dihydrochloride (DAPI): nuclear counterstaining. D. Expression of Wnt2 protein in non-CRC (FHC and CCD841CoN) and CRC cell lines. IB analysis.
Wnt2 enhances Wnt/β-catenin signaling activity in CRC
While Wnt5A transduces non-canonical (not mediated by β-catenin) Wnt signaling [37], Wnt2 activates canonical Wnt signaling and upregulates β-catenin target genes [34, 35, 38, 39]. This led us to test whether Wnt2 is required for full activation of Wnt/β-catenin signaling activity in CRC cells. Thus, we examined the correlation between WNT2 and Wnt signaling activation by assessing the co-expression of WNT2 and AXIN2, a well-established β-catenin target gene [40, 41]. Analyses of Oncomine datasets showed that WNT2 expression is strongly co-related with expression of AXIN2, compared with other Wnt ligands (Figure 2A and Supplementary Figure 1), implying that Wnt2 might be associated with Wnt signaling in CRC cells.
Next, to examine the effect of Wnt2 on Wnt/β-catenin signaling in CRC, we employed the loss-of-function approaches by transducing lentiviruses encoding short-hairpin RNA (shRNA) against green fluorescence (GFP; shGFP) or Wnt2 (shWnt2) (Figure 2B), as previously performed [42, 43]. We found that Wnt2 knockdown (KD) markedly downregulates β-catenin reporter activity (TOPFLASH luciferase) in HCT116 cells, which was rescued by ectopic expression of Wnt2 (Figure 2C). iCRT14, an inhibitor for β-catenin:TCFs binding, served as a positive control. Additionally, other CRC cell lines (HCC2998, HT15, and SW620) similarly displayed the downregulation of β-catenin reporter activity by endogenous Wnt2 KD (Figure 2D). To better confirm the effects of Wnt2 depletion on Wnt signaling, we employed the clustered regularly interspaced short palindromic repeat (CRISPR) gene targeting system using lentivirus encoding Cas9 and guide RNA (gRNA). IB assays showed that two different gRNAs successfully target WNT2 alleles (Figure 2E), which was also validated by genomic DNA sequencing (data not shown). Similar to the effects of Wnt2 KD, WNT2 knockout (KO) cells displayed significantly reduced expression of β-catenin target genes (AXIN2, CD44, and CD133) (Figure 2F) and β-catenin reporter activity (Figure 2G), compared to WNT2 wild-type (WT) cells. We also analyzed the effects of WNT2 KO on activity of each Wnt signaling component. We found that WNT2 KO cells exhibited decreased phosphorylation of LRP6, compared to WNT2 WT cells (Figure 2H). Next, we tested whether Wnt2 activates β-catenin in non-CRC cells. Indeed, ectopic expression of Wnt2 upregulates β-catenin protein in 293T and CCD841CoN intestinal epithelial cells (IECs) (Figure 2I). These results suggest that Wnt2 is required for maintenance of Wnt signaling activity in CRC cells.
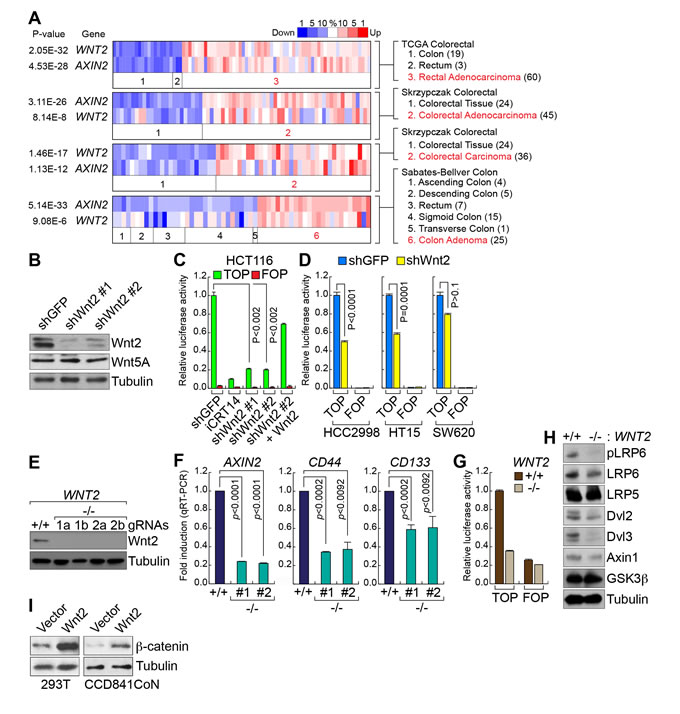
Figure 2: Wnt2 is required for β-catenin target gene activation in CRC cells. A. Co-expression of WNT2 with AXIN2 in CRC. Co-expression analysis of WNT2 with AXIN2, using Oncomine database. P value < 0.0001; fold change > 2. B. Depletion of endogenous Wnt2 by shRNAs. Lentiviruses encoding two different shRNAs (#1 and #2) were stably transduced into HCT116 cells. IB assays. Of note, Wnt5A expression was not affected by Wnt2 depletion. C. and D. Downregulation of β-catenin transcriptional activity by Wnt2 KD in CRC cells. HCT116 cells (shGFP or shWnt2) were transiently transfected with pMegaTOPFLASH (TOP) or pMegaFOPFLASH (FOP) β-catenin luciferase reporter plasmids, with pRenilla plasmids (internal control). 48 hours after transfection, cells were analyzed for luciferase activity C.. Other CRC cell lines D.. iCRT14 served as a positive control for inhibition of β-catenin activity. Wnt2-expressing plasmids were co-transfected for rescue assays. E. KO of WNT2 alleles by CRISPR gene targeting system. HCT116 cells were transduced with lentiviruses encoding Cas9 and gRNAs (1 and 2 indicate two different gRNAs). IB analysis. F. Downregulation of β-catenin target genes by WNT2 KO. HCT116 (WNT2 WT vs. WNT2 KO) cells were analyzed by quantitative reverse transcriptase PCR (qRT-PCR) of AXIN2, CD44, and CD133. G. Suppression of β-catenin transcriptional activity by WNT2 KO. β-catenin reporter assays of HCT116 (WNT2 WT vs. WNT2 KO). H. Dephosphorylation of LRP6 by WNT2 KO. IB analysis of HCT116 (WNT2 WT vs. WNT2 KO). I. Upregulation of β-catenin protein by Wnt2. 293T and CCD841CoN cells were transfected with plasmids encoding Wnt2. 24 hours after transfection, cells were analyzed for IB of β-catenin.
Wnt2 is required for CRC cell proliferation
To address the pathologic role of Wnt2 in intestinal tumorigenesis, we tested the impacts of WNT2 expression to IEC proliferation. We stably transfected Wnt2 expression plasmids into CCD841CoN and assessed cell proliferation. We found that Wnt2-expressing CCD841CoN displayed the increased cell proliferation compared to empty vector transfected control cells (Figure 3A and 3B). Next, we examined the effects of WNT2 KO on CRC cell proliferation. In contrast to the results from Wnt2 overexpression in IECs, WNT2 KO inhibited cell proliferation of HCT116 cells (Figure 3C). Cell cycle analysis showed that WNT2 KO HCT116 cells displayed the increase in S phase and the decrease in G0/G1 phase, compared to WT cells, as also shown in iCRT14-treated cells (Figure 3D). Similarly, Wnt2 KD also inhibits cell proliferation of CRC cell lines (HCT116, KM12, and HT15). However, HCC2998 and SW620 CRC cell lines did not show the cell growth inhibition by Wnt2 KD (Figure 3E and 3F). Given the high expression of WNT2 in CRC cells (Figure 1), we also tested whether Wnt2 induces cell proliferation in an autocrine manner. We collected conditioned medium (CM) from KM12 cells transfected with empty vector (control), shWnt2, or Wnt3A, and treated HCT116 with each CM. Then, we analyzed cell proliferation of HCT116 cells. Similar to the results from Wnt2 knockdown in CRC cells (see Figure 3A and 3B), cells treated with CM collected from Wnt2-depleted KM12 cells showed the decrease in cell proliferation (Figure 3G). However, CM from Wnt3A-overexpresing cells did not affect cell proliferation (Figure 3G). Next, we blocked the secreted Wnt2 ligands using anti-Wnt2 antibody (Wnt2 nAb). As shown in WNT2 KO results, neutralizing Wnt2 by treatment with Wnt2 nAb downregulates AXIN2 expression (Figure 3H) and inhibits HCT116 cell proliferation (Figure 3I). We also co-treated HCT116 cells with Wnt2 nAb and Wnt3A, a canonical Wnt ligand. We observed that Wnt2 nAb-induced cell growth inhibition was reverted by Wnt3A treatment (Figure 3J), indicating that canonical Wnt signaling mediates Wnt2-induced CRC cell proliferation. These results suggest that secreted Wnt2 is required for CRC cell proliferation.
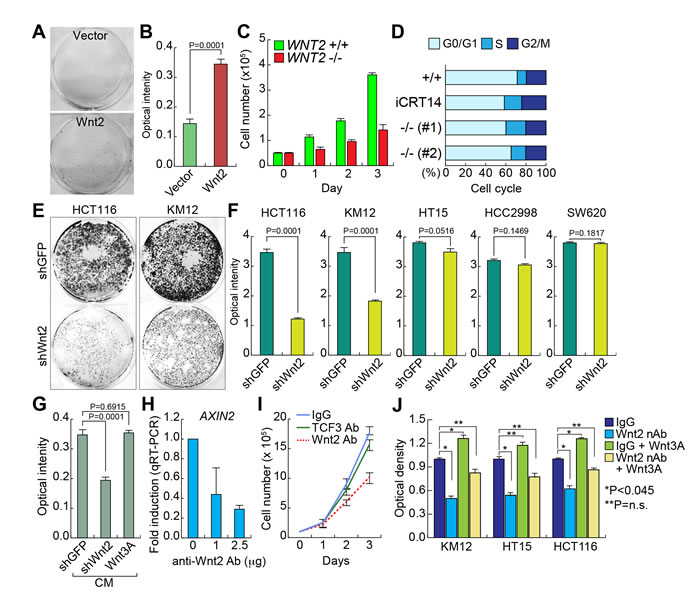
Figure 3: Wnt2 is required for CRC cell proliferation. A. and B. Increased cell proliferation by Wnt2. CCD841CoN IECs were stably transfected with Wnt2-expressing plasmid. Then, the equal number of cells were plated and cultured for 20 days. Crystal violet staining A.; quantification of optical intensity of crystal violet stained cells B.. C. Decreased cell proliferation by WNT2 KO. The equal number of HCT116 (WNT2 WT vs. WNT2 KO) cells were plated and counted in a different time course. D. Cell cycle analysis of WNT2 KO cells. HCT116 cells were analyzed using flow cytometry. E. and F. Depletion of Wnt2 reduced CRC cell proliferation. CRC cells were stably transduced with lentivirus encoding shRNAs (shGFP: control; shWnt2). Then, the equal number of cells were plated and grown for 14 days for crystal violet staining E. and quantification F.. G. Decreased cell proliferation by Wnt2 KD CM. KM12 cells were transfected with plasmids (empty vector: control; shWnt2; Wnt3A). After transfection, KM12 cells were incubated with serum free DMEM for collection of CM. Then, each CM was treated to HCT116 cells. Cell proliferation (crystal violet staining quantification) was monitored after were monitored for 20 days of CM treatment. H. Downregulation of AXIN2 by neutralizing Wnt2 protein. HCT116 cells were treated with anti-Wnt2 antibody. 24 hours after treatment, cells were analyzed for qRT-PCR of AXIN2. HPRT served as an internal control. I. Inhibition of cell proliferation by neutralization of Wnt2. The equal number of HCT116 cells were plated and treated with antibodies (IgG and TCF3 antibodies: negative controls; Wnt2 antibody). Cell proliferation was analyzed by cell counting. J. Rescue of Wnt nAb-induced cell growth inhibition by Wnt3A. CRC cells were treated with Wnt2 nAb and/or Wnt3A (100 ng/ml). Cell proliferation was analyzed by crystal violet staining and quantification using plate reader.
PRC2-mediated transcriptional repression of WNT2
Next, to understand how WNT2 expression is upregulated in CRC, we analyzed WNT2 promoter using VISTA genome browser (http://pipeline.lbl.gov/cgi-bin/gateway2). Interestingly, we found that WNT2 proximal promoter is dominantly occupied by PRC2 (EZH2 and SUZ12) and H3K27me3 histone modification, a marker for gene repression mediated by PRC2 [44] (Figure 4A). These findings led us to test whether PRC2 represses WNT2 transcription. Previously, it was shown that PRC2 components (EZH2, SUZ12, and EED) are highly expressed in CRC [23, 45, 46]. While PRC2 components are expressed in both 293T and HCT116 cells, the expression of EZH2, EED, and SUZ12 is slightly higher in HCT116 cells than 293T cells (Figure 4B). However, chromatin immunoprecipitation assay (ChIP) assays of WNT2 promoter showed that EZH2 did not occupy to WNT2 promoter in CRC cells (HCT116, KM12, and HT15), while it occupies WNT2 promoter in 293T and IECs (CCD841CoN) (Figure 4C). These results suggest that the loss of EZH2 binding to WNT2 promoter might be involved in WNT2 de-repression in CRC cells. To test whether EZH2 is associated with transcriptional repression of WNT2, we ectopically expressed WT or F681I, catalytically inactive mutant, EZH2 in CRC cells (HCT116, KM12, and HT15). qRT-PCR results showed that EZH2 overexpression downregulates WNT2 expression, whereas EZH2 F681I mutant does not (Figure 4D). Of note, the expression of Wnt3, another highly expressed Wnt ligand, was not affected by ectopic expression of EZH2 (data not shown). To complement gain-of-function approach, we also asked whether blockade of EZH2 transcriptionally activates WNT2 in non-CRC cells. We treated 293T cells with GSK343, a specific inhibitor of EZH2 [47]. We found that GSK343 treatment induces the significant de-repression (upregulation) of WNT2 expression in 293T cells (Figure 4E). These results suggest that WNT2 expression is transcriptionally repressed by PRC2 in non-CRC cells and de-repressed by the loss of PRC2’s promoter occupancy in CRC cells.
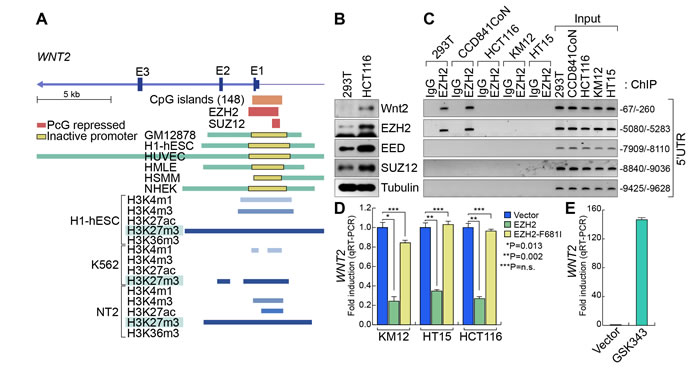
Figure 4: De-repression of WNT2 in CRC. A. WNT2 promoter analysis. VISTA genome browser. ChIP-Seq analysis of EZH2, SUZ12, and histone modification markers in various cell lines. Of note, ChIP-Seq data show that enrichment of PRC2 components and H3K27me3 modification on Wnt2 proximal promoter. B. Expression of PRC2 components. PRC2 components are highly expressed in HCT116. 293T and HCT116 cells were harvested and subjected to IB analysis for PRC2 components. Tubulin was used as an internal control. C. EZH2’s occupancy of WNT2 promoter. Non-CRC (293T and CCD841CoN) and CRC (HCT116, KM12, and HT15) cells were analyzed for ChIP assays. Five different regions of 5’UTR were analyzed (-67/-260; -5080/-5283; -7909/-8110; -8840/-9036; -9425/-9628). Of note, EZH2 is associated with only the proximal promoter regions (-67/-260; -5080/-5283) in non-CRC cells. D. Downregulation of WNT2 by ectopic expression of EZH2 in CRC. CRC cells were transfected with EZH2 (WT and F681 mutant). 48 hours after transfection, cells were analyzed for qRT-PCR. E. De-repression of WNT2 by EZH2 inhibition. 293T cells were treated with GSK343 for 48 hours and analyzed for qRT-PCR.
Discussion
In this study, we found that Wnt2 is highly upregulated in CRC and complements Wnt/β-catenin signaling for CRC cell proliferation (Figure 5). Despite homogeneous genetic mutations in APC, CTNNB1, or AXIN2, CRC cells show heterogeneous nuclear localization of β-catenin [48, 49], which is called ‘β-catenin paradox’. Accumulating evidences have supported this model: (a) Rather than having an absolutely higher level of β-catenin protein, fold-induction of β-catenin is critical for β-catenin target gene activation [50]. (b) Mutant APC protein partially functions in protein destruction complex to target β-catenin protein [14]. (c) Blockade of Wnt ligands induces growth inhibition and apoptosis in cells harboring APC mutation [14, 51], so does secreted frizzled-related proteins (SFRPs) (secreted Wnt antagonists)[48, 52]. (d) Genetic mutation in E3 ligases (RNF43 and ZNRF3) targeting Wnt receptors contributes to CRC [53, 54]. (e) Moreover, Tankyrase inhibitor (blocking Axin1 degradation for subsequent β-catenin inhibition) suppresses CRC cell proliferation [55]. (f) It was shown that Ras/MAPK signaling is required for Wnt/β-catenin signaling activation [56, 57]. Therefore, we hypothesized that additional stimuli at the upstream of protein destruction complex (APC, GSK3, Axin, and CK1) complement Wnt/β-catenin signaling activity in CRC. Among 19 Wnt ligands, we found that Wnt2 is most significantly upregulated in CRC, which led us to investigate the potential tumorigenic roles of Wnt2 in CRC.
We initially questioned whether hyperactivated β-catenin by genetic mutations is further activated by additional Wnt signaling stimuli. To address this, we examined whether GSK3 inhibition stabilizes (activates) β-catenin protein. We inhibited GSK3, a component of protein destruction complex targeting β-catenin for ubiquitin-mediated protein degradation, using LiCl. Interestingly, LiCl treatment not only upregulates β-catenin protein but also activates β-catenin reporter activity in CRC cells despite genetic mutations in CTNNB1/β-catenin or APC of CRC cells (Supplementary Figure 2). Interestingly, HCT116 and KM12 cells show the significant decrease in cell proliferation by depletion of Wnt2 (Figure 3E and 3F), which correlates with LiCl-induced stabilization of β-catenin protein (Supplementary Figure 2). Of note is that SW620 and HCC2998 CRC cells did not exhibit cell growth inhibition by Wnt2 depletion (Figure 3F), which is also consistent with the results that LiCl does not stabilize beta-catenin protein and not activate beta-catenin reporter activity (Supplementary Figure 2). Considering the high expression of Wnt2 in all CRC cells (Figure 1D), it is plausible that other Wnt ligands such as Wnt3 might compensate the loss of Wnt2 in SW620 or HCC2998 cells. Thus, comprehensive analysis of highly expressed Wnt ligands (Wnt2, Wnt3, Wnt5A, and Wnt7b) is necessary. Nonetheless, these results imply that additional stimuli can further activate Wnt signaling in CRC cells. In regard to this, our finding strongly suggests that Wnt2 is a crucial factor for maintaining Wnt signaling activation in CRC, in addition to genetic activation of Wnt signaling. It is noteworthy that APC mutations are mainly observed in CRC cells regardless of mutations or aberrant expression of AXIN2, CTNNB1 (Supplementary Figure 3), which indicates the insufficiency of APC mutation per se in fully competent Wnt signaling hyperactivation during intestinal tumorigenesis. In line with this, Voloshanenko et al. recently showed that APC mutation is not sufficient to completely activate Wnt signaling in CRC cells. They proposed that Wnt3A is required for complete activation of Wnt signaling [14]. However, our in silico analysis indicates that WNT2, WNT5A, and WNT3, but not WNT3A are significantly upregulated in CRC (Figure 1A and 1B). Moreover, depletion of endogenous Wnt2 downregulates Wnt signaling activity and inhibits CRC cell proliferation (Figures 2 and 3). Thus, Wnt2 is more likely to be associated with the maintenance of Wnt signaling activity in CRC. To better address this, it is necessary to directly compare the impacts of Wnt2 and Wnt3A to Wnt signaling activation and CRC cell proliferation. Additionally, K-Ras mutation was shown to be required for activation of β-catenin in CRC [57]. However, CRC cell lines with mutations in either K-Ras or BRAF (Supplementary Figure 4) also exhibit Wnt2 dependency on cell proliferation and β-catenin activation, which suggests that the impact of Wnt2 to β-catenin stabilization might be independent of Ras/MAPK signaling.
We found that EZH2 regulates transcriptional repression of WNT2. EZH2, a core component of PRC2, silences target gene transcription by histone modification (H3K27me3). EZH2 and SUZ12 specifically occupy the proximal promoter of WNT2 and induce H3K27me3, which leads to transcriptional inactivation of WNT2 in non-CRC cells. However, CRC cells exhibit the loss of EZH2 association with WNT2 promoter, which leads to de-repression of WNT2 expression. Due to the expression of PRC2 components (EZH2, EED, and SUZ12) in both non-CRC and CRC cells (Figure 4B), it is plausible that EZH2’s activity or target gene selection might be specifically modulated by additional factors. For example, posttranslational modification of EZH2 was shown to modulate EZH2-mediated histone modification [58, 59], which might facilitate release of EZH2 from WNT2 promoter and induces de-repression of WNT2 transcription. Another possible explanation of regulatory mechanism of EZH2 is the sequestration of EZH2 from PRC2 to β-catenin transcriptional complex. Previously, we found that PAF (PCNA-associated factor)/KIAA0101 is specifically expressed in CRC cells but not in normal IECs. PAF binds to β-catenin and sequesters EZH2 from PRC2 to β-catenin transcriptional complex, resulting in transcriptional activation of β-catenin target genes in CRC [42]. Thus, it is highly likely that PRC2-mediated repression of WNT2 is diminished by the loss of EZH2 in PRC2, which subsequently leads to de-repression of WNT2 in CRC. This should be addressed in future studies. Although the expression of PRC2 components is elevated in CRC, it is noteworthy that WNT2 expression is mutually exclusive with the expression of EZH2, EED, and SUZ12 (Supplementary Figure 5). These in silico results support our model that PRC2 mediates repression and de-repression of WNT2 in normal cells and CRC cells, respectively. Importantly, the multiple Kaplan-Meier survival analyses suggest that high expression of EZH2 is associated with relapse-free survival [46], indicating that high EZH2 expression correlates with improved survival of CRC patients. These pathologic results also support our finding that EZH2 negatively regulates the expression of WNT2, another key factor for CRC cell proliferation.
Taken together, our findings suggest that Wnt2 plays a critical role in complementing Wnt signaling in CRC. Furthermore, our study proposes that rather than blockage of core-components of Wnt signaling, molecular intervention of Wnt2 might be beneficial to translation into CRC therapy, by minimizing normal tissue damages.
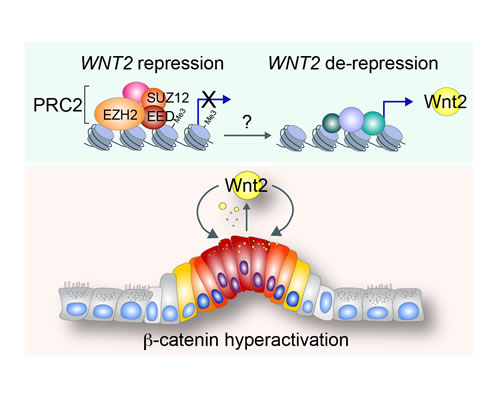
Figure 5: Illustration of working model. In normal intestine, WNT2 expression is repressed by PRC2-induced H3K27me3 histone modification of WNT2 proximal promoter. By unknown mechanisms, PRC2’s repressive function on WNT2 expression is inhibited and WNT2 expression is de-repressed. Subsequently, Wnt2 activates canonical Wnt signaling during intestinal tumorigenesis, which leads to maintenance of hyperactivation of Wnt/β-catenin signaling for CRC cell proliferation.
Materials and Methods
Oncomine database analysis
cDNA microarray datasets of colon adenocarcinoma and normal tissue samples were analyzed using Oncomine database (www.oncomine.org). P < 0.0001; fold change >2; 10% top ranked.
Immunohistochemistry
Human CRC tissue microarray was purchased from Biomax (Co1002), and immunostained with anti-Wnt2 antibody (SantaCruz), as previously performed [60]. For immunostaining of tumor samples from xenograft transplantation, samples were collected and fixed with 10% formalin. After processing for paraffin embedding, sectioned samples were immunostained followed by standard protocols.
Mammalian cell culture and constructs
All cell lines were purchased from American Type Culture Collection (ATCC) and maintained in Dulbecco’s modified Eagle medium (containing 10% fetal bovine serum and 1% penicillin-streptomycin). For gene depletion, shRNA lentiviruses (shGFP or shWnt2) (Sigma; MISSION shRNA) were stably transduced into target cells using puromycin selection (1 to 2 μg/ml). Wnt2 expressing plasmid was purchased from Addgene. GSK343 was purchased from Sigma.
Immunoblotting
Proteins were obtained as previously described [42], and the following antibodies were used for Immunoblotting: β-catenin (Cell Signaling), active β-catenin (Millipore), Tubulin (Sigma), FLAG (M2; Sigma), Wnt2 (SantaCruz), Wnt3a (Cell Signaling), Wnt5a (Cell Signaling), pLRP6 (Cell signaling), LRP6 (Cell signaling), LRP5 (Cell signaling), Dvl2 (Cell signaling), Dvl3 (Cell signaling), Axin1 (Cell signaling) and GSK3β (BD Bioscience), EZH2 (Cell signaling), EED (Millipore), and SUZ12 (Abcam).
Reporter assays
The reporter plasmids, pMegaTOPFLASH and pMegaFOPFLASH, were transiently transfected with pSV40-Renilla and analyzed using Dual luciferase assay system (Promega), as previously performed [42].
WNT2 somatic gene targeting
The KO cells were generated by CRISPR using a lentiviral vector [61]. The lentiviral plasmid contains two expression cassettes, hSpCas9 and the chimeric gRNA. gRNAs were designed based on the protospacer adjacent motif (PAM) on the target site, using standard cloning methodology generating lentiCRISPR. The lentiCRISPR plasmids were transfected into HEK293T cells along with packaging plasmids pCMV-ΔR8.2 dvpr and pCMV-VSVG for lentiviral packaging. CRC cell lines were then transduced with the lentivirus and selected in puromycin for 72 hours. The KO was confirmed by immunoblotting and genomic DNA sequencing. gRNA sequences: #1: 5’- CCATG AAGAG TTGAC CTCGG-3’; #2: 5’- ACCAT GAAGA GTTGA CCTCG-3’.
Gene expression analysis
RNAs were extracted by TRIzol (Invitrogen) and converted into cDNAs using SuperScript II (Invitrogen) with random hexamer. For gene expression analysis, qRT-PCR was performed. qRT-PCR results were quantified by comparative 2-ΔΔCt methods. For internal controls, HPRT was used. qRT-PCR primer sequence information: Wnt2 forward/reverse: 5’CCC ACA GCA CAT GAC TTC AC3’/5’CTG TAT CAG GGA CCG AGA GG3’; Axin2 forward/reverse: 5’ CTC CTT GGA GGC AAG AGC3’/5’ GGC CAC GCA GCA CCG CTG3’; HPRT forward/reverse: 5’GCT ATA AAT TCT TTG CTG ACC TGC TG3’/5’AAT TAC TTT TAT GTC CCC TGT TGA CTG G3’
Cell proliferation analysis
For FACS analysis, cells were fixed with 70% ethanol and stained with propidium iodide, then subjected to cell cycle analysis using FACSCalibur (Becton Dickinson FACSCalibur), as previously performed [62]. For cell proliferation analysis, fourteen days after seeding the cells, we fixed the cells with 10% formalin and stained them with crystal violet for 30 min. For quantitative analysis, cells stained with crystal violet were subjected to lysis with 1% sodium dodecyl sulfate (SDS), and absorbance was measured at 590 nm using a 96-well microplate reader (BioTek microplate reader).
ChIP assay
ChIP assays were performed as previously performed [42]. The antibody against EZH2 was purchased from cell signaling. ChIP primer sequence information:
-9425/-9628 forward/reverse: 5’ctctt gaatc tgggc aggtc3’/5’gaaag attga ctggg ctgga3’; -8840/-9036 forward/reverse: 5’cgtta atccc accac tttgg3’/5’tacaa cctct gcctc cttgg3’; -7909/-8110 forward/reverse: 5’ttttg ccttt cacca aaacc3’/5’tagtg agtgg ccagg agacc3’; -5080/-5283 forward/reverse: 5’ttcag gtttt tggcg tctct3’/5’atgcg cctgt gtgta tgtgt3’; -67/-260 Forward/reverse: 5’tgctt tggca gatac tgctg3’/5’ctgaa gctgg gatga agagc3’
Statistical analysis
The Student’s t-test was used for comparisons of two groups (n ≥ 3). P values less than 0.05 were considered significant. Error bars indicate standard deviation.
Acknowledgments
We thank Joan Shang, and Esther Lien for helpful comments on the manuscript. This work was supported by the Cancer Prevention and Research Institute of Texas (RP140563), the National Institutes of Health (NCI R01 CA193297-01), the Department of Defense Peer Reviewed Cancer Research Program (CA140572), the Duncan Family Institute Research Program, the University Cancer Foundation (IRG-08-061-01), the Center for Stem Cell and Developmental Biology (The University of Texas MD Anderson Cancer Center), an Institutional Research Grant (MD Anderson Cancer Center), a New Faculty Award (MD Anderson Cancer Center Support Grant), a Metastasis Research Center Grant (MD Anderson Cancer Center), and SPORE in ovarian cancer (P50 CA83639). The Flow Cytometry and Cellular Imaging Facility is supported by the NIH through the MD Anderson Cancer Center Support Grant (CA016672).
Conflicts of Interest
Authors declared no conflict of interest.
References
1. Clevers H, Loh KM and Nusse R. Stem cell signaling. An integral program for tissue renewal and regeneration: Wnt signaling and stem cell control. Science. 2014; 346:1248012.
2. Clevers H and Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012; 149:1192-1205.
3. Clements WM, Lowy AM and Groden J. Adenomatous polyposis coli/beta-catenin interaction and downstream targets: altered gene expression in gastrointestinal tumors. Clinical colorectal cancer. 2003; 3:113-120.
4. Polakis P. Wnt signaling in cancer. Cold Spring Harbor perspectives in biology. 2012; 4.
5. Polakis P. The many ways of Wnt in cancer. Current opinion in genetics & development. 2007; 17:45-51.
6. Su LK, Kinzler KW, Vogelstein B, Preisinger AC, Moser AR, Luongo C, Gould KA and Dove WF. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science. 1992; 256:668-670.
7. Caldwell GM, Jones C, Gensberg K, Jan S, Hardy RG, Byrd P, Chughtai S, Wallis Y, Matthews GM and Morton DG. The Wnt antagonist sFRP1 in colorectal tumorigenesis. Cancer Res. 2004; 64:883-888.
8. Oberwalder M, Zitt M, Wontner C, Fiegl H, Goebel G, Zitt M, Kohle O, Muhlmann G, Ofner D, Margreiter R and Muller HM. SFRP2 methylation in fecal DNA—a marker for colorectal polyps. International journal of colorectal disease. 2008; 23:15-19.
9. Mounier CM, Wendum D, Greenspan E, Flejou JF, Rosenberg DW and Lambeau G. Distinct expression pattern of the full set of secreted phospholipases A2 in human colorectal adenocarcinomas: sPLA2-III as a biomarker candidate. Br J Cancer. 2008; 98:587-595.
10. Qi J, Zhu YQ, Luo J, Tao WH and Zhang JM. [Hypermethylation and regulation of expression of secreted frizzled-related protein genes in colorectal tumor]. Zhonghua zhong liu za zhi [Chinese journal of oncology]. 2007; 29:842-845.
11. Huang Y, Stewart TM, Wu Y, Baylin SB, Marton LJ, Perkins B, Jones RJ, Woster PM and Casero RA, Jr. Novel oligoamine analogues inhibit lysine-specific demethylase 1 and induce reexpression of epigenetically silenced genes. Clin Cancer Res. 2009; 15:7217-7228.
12. Lavergne E, Hendaoui I, Coulouarn C, Ribault C, Leseur J, Eliat PA, Mebarki S, Corlu A, Clement B and Musso O. Blocking Wnt signaling by SFRP-like molecules inhibits in vivo cell proliferation and tumor growth in cells carrying active beta-catenin. Oncogene. 2011; 30:423-433.
13. Suzuki H, Watkins DN, Jair KW, Schuebel KE, Markowitz SD, Chen WD, Pretlow TP, Yang B, Akiyama Y, Van Engeland M, Toyota M, Tokino T, Hinoda Y, Imai K, Herman JG and Baylin SB. Epigenetic inactivation of SFRP genes allows constitutive WNT signaling in colorectal cancer. Nature genetics. 2004; 36:417-422.
14. Voloshanenko O, Erdmann G, Dubash TD, Augustin I, Metzig M, Moffa G, Hundsrucker C, Kerr G, Sandmann T, Anchang B, Demir K, Boehm C, Leible S, Ball CR, Glimm H, Spang R, et al. Wnt secretion is required to maintain high levels of Wnt activity in colon cancer cells. Nature communications. 2013; 4:2610.
15. MacDonald BT, Tamai K and He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009; 17:9-26.
16. Semenov MV, Habas R, Macdonald BT and He X. SnapShot: Noncanonical Wnt Signaling Pathways. Cell. 2007; 131:1378.
17. Peng T, Tian Y, Boogerd CJ, Lu MM, Kadzik RS, Stewart KM, Evans SM and Morrisey EE. Coordination of heart and lung co-development by a multipotent cardiopulmonary progenitor. Nature. 2013; 500:589-592.
18. Monkley SJ, Delaney SJ, Pennisi DJ, Christiansen JH and Wainwright BJ. Targeted disruption of the Wnt2 gene results in placentation defects. Development. 1996; 122:3343-3353.
19. Kozopas KM, Samos CH and Nusse R. DWnt-2, a Drosophila Wnt gene required for the development of the male reproductive tract, specifies a sexually dimorphic cell fate. Genes & development. 1998; 12:1155-1165.
20. Vider BZ, Zimber A, Chastre E, Prevot S, Gespach C, Estlein D, Wolloch Y, Tronick SR, Gazit A and Yaniv A. Evidence for the involvement of the Wnt 2 gene in human colorectal cancer. Oncogene. 1996; 12:153-158.
21. Park JK, Song JH, He TC, Nam SW, Lee JY and Park WS. Overexpression of Wnt-2 in colorectal cancers. Neoplasma. 2009; 56:119-123.
22. Holcombe RF, Marsh JL, Waterman ML, Lin F, Milovanovic T and Truong T. Expression of Wnt ligands and Frizzled receptors in colonic mucosa and in colon carcinoma. Molecular pathology : MP. 2002; 55:220-226.
23. Benard A, Goossens-Beumer IJ, van Hoesel AQ, Horati H, Putter H, Zeestraten EC, van de Velde CJ and Kuppen PJ. Prognostic value of polycomb proteins EZH2, BMI1 and SUZ12 and histone modification H3K27me3 in colorectal cancer. PloS one. 2014; 9:e108265.
24. Yu M, Ting DT, Stott SL, Wittner BS, Ozsolak F, Paul S, Ciciliano JC, Smas ME, Winokur D, Gilman AJ, Ulman MJ, Xega K, Contino G, Alagesan B, Brannigan BW, Milos PM, et al. RNA sequencing of pancreatic circulating tumour cells implicates WNT signalling in metastasis. Nature. 2012; 487:510-513.
25. Bravo DT, Yang YL, Kuchenbecker K, Hung MS, Xu Z, Jablons DM and You L. Frizzled-8 receptor is activated by the Wnt-2 ligand in non-small cell lung cancer. BMC cancer. 2013; 13:316.
26. Katoh M. WNT2 and human gastrointestinal cancer (review). International journal of molecular medicine. 2003; 12:811-816.
27. Katoh M. Frequent up-regulation of WNT2 in primary gastric cancer and colorectal cancer. International journal of oncology. 2001; 19:1003-1007.
28. Ricken A, Lochhead P, Kontogiannea M and Farookhi R. Wnt signaling in the ovary: identification and compartmentalized expression of wnt-2, wnt-2b, and frizzled-4 mRNAs. Endocrinology. 2002; 143:2741-2749.
29. Fu L, Zhang C, Zhang LY, Dong SS, Lu LH, Chen J, Dai Y, Li Y, Kong KL, Kwong DL and Guan XY. Wnt2 secreted by tumour fibroblasts promotes tumour progression in oesophageal cancer by activation of the Wnt/beta-catenin signalling pathway. Gut. 2011; 60:1635-1643.
30. Le Floch N, Rivat C, De Wever O, Bruyneel E, Mareel M, Dale T and Gespach C. The proinvasive activity of Wnt-2 is mediated through a noncanonical Wnt pathway coupled to GSK-3beta and c-Jun/AP-1 signaling. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2005; 19:144-146.
31. Blasband A, Schryver B and Papkoff J. The biochemical properties and transforming potential of human Wnt-2 are similar to Wnt-1. Oncogene. 1992; 7:153-161.
32. Hollmann CA, Kittrell FS, Medina D and Butel JS. Wnt-1 and int-2 mammary oncogene effects on the beta-catenin pathway in immortalized mouse mammary epithelial cells are not sufficient for tumorigenesis. Oncogene. 2001; 20:7645-7657.
33. Roelink H, Wagenaar E and Nusse R. Amplification and proviral activation of several Wnt genes during progression and clonal variation of mouse mammary tumors. Oncogene. 1992; 7:487-492.
34. Shimizu H, Julius MA, Giarre M, Zheng Z, Brown AM and Kitajewski J. Transformation by Wnt family proteins correlates with regulation of beta-catenin. Cell growth & differentiation : the molecular biology journal of the American Association for Cancer Research. 1997; 8:1349-1358.
35. Karasawa T, Yokokura H, Kitajewski J and Lombroso PJ. Frizzled-9 is activated by Wnt-2 and functions in Wnt/beta -catenin signaling. The Journal of biological chemistry. 2002; 277:37479-37486.
36. Shi Y, He B, Kuchenbecker KM, You L, Xu Z, Mikami I, Yagui-Beltran A, Clement G, Lin YC, Okamoto J, Bravo DT and Jablons DM. Inhibition of Wnt-2 and galectin-3 synergistically destabilizes beta-catenin and induces apoptosis in human colorectal cancer cells. International journal of cancer Journal international du cancer. 2007; 121:1175-1181.
37. Nishita M, Enomoto M, Yamagata K and Minami Y. Cell/tissue-tropic functions of Wnt5a signaling in normal and cancer cells. Trends in cell biology. 2010; 20:346-354.
38. Goss AM, Tian Y, Tsukiyama T, Cohen ED, Zhou D, Lu MM, Yamaguchi TP and Morrisey EE. Wnt2/2b and beta-catenin signaling are necessary and sufficient to specify lung progenitors in the foregut. Developmental cell. 2009; 17:290-298.
39. Cho SH and Cepko CL. Wnt2b/beta-catenin-mediated canonical Wnt signaling determines the peripheral fates of the chick eye. Development. 2006; 133:3167-3177.
40. Lustig B, Jerchow B, Sachs M, Weiler S, Pietsch T, Karsten U, van de Wetering M, Clevers H, Schlag PM, Birchmeier W and Behrens J. Negative feedback loop of Wnt signaling through upregulation of conductin/axin2 in colorectal and liver tumors. Molecular and cellular biology. 2002; 22:1184-1193.
41. Jho EH, Zhang T, Domon C, Joo CK, Freund JN and Costantini F. Wnt/beta-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Molecular and cellular biology. 2002; 22:1172-1183.
42. Jung HY, Jun S, Lee M, Kim HC, Wang X, Ji H, McCrea PD and Park JI. PAF and EZH2 induce Wnt/beta-catenin signaling hyperactivation. Mol Cell. 2013; 52:193-205.
43. Jun S, Lee S, Kim HC, Ng C, Schneider AM, Ji H, Ying H, Wang H, DePinho RA and Park JI. PAF-mediated MAPK signaling hyperactivation via LAMTOR3 induces pancreatic tumorigenesis. Cell Rep. 2013; 5:314-322.
44. Di Croce L and Helin K. Transcriptional regulation by Polycomb group proteins. Nature structural & molecular biology. 2013; 20:1147-1155.
45. Fussbroich B, Wagener N, Macher-Goeppinger S, Benner A, Falth M, Sultmann H, Holzer A, Hoppe-Seyler K and Hoppe-Seyler F. EZH2 depletion blocks the proliferation of colon cancer cells. PloS one. 2011; 6:e21651.
46. Fluge O, Gravdal K, Carlsen E, Vonen B, Kjellevold K, Refsum S, Lilleng R, Eide TJ, Halvorsen TB, Tveit KM, Otte AP, Akslen LA, Dahl O and Norwegian Gastrointestinal Cancer G. Expression of EZH2 and Ki-67 in colorectal cancer and associations with treatment response and prognosis. British journal of cancer. 2009; 101:1282-1289.
47. Verma SK, Tian X, LaFrance LV, Duquenne C, Suarez DP, Newlander KA, Romeril SP, Burgess JL, Grant SW, Brackley JA, Graves AP, Scherzer DA, Shu A, Thompson C, Ott HM, Aller GS, et al. Identification of Potent, Selective, Cell-Active Inhibitors of the Histone Lysine Methyltransferase EZH2. ACS medicinal chemistry letters. 2012; 3:1091-1096.
48. Brabletz T, Jung A, Hermann K, Gunther K, Hohenberger W and Kirchner T. Nuclear overexpression of the oncoprotein beta-catenin in colorectal cancer is localized predominantly at the invasion front. Pathology, research and practice. 1998; 194:701-704.
49. Vermeulen L, De Sousa EMF, van der Heijden M, Cameron K, de Jong JH, Borovski T, Tuynman JB, Todaro M, Merz C, Rodermond H, Sprick MR, Kemper K, Richel DJ, Stassi G and Medema JP. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nature cell biology. 2010; 12:468-476.
50. Goentoro L and Kirschner MW. Evidence that fold-change, and not absolute level, of beta-catenin dictates Wnt signaling. Molecular cell. 2009; 36:872-884.
51. He B, Reguart N, You L, Mazieres J, Xu Z, Lee AY, Mikami I, McCormick F and Jablons DM. Blockade of Wnt-1 signaling induces apoptosis in human colorectal cancer cells containing downstream mutations. Oncogene. 2005; 24:3054-3058.
52. McGimpsey JG, Webb CH and McCrea PH. Comparison of serum erythromycin concentration following administration of 100 mg erythromycin ethyl succinate intramuscularly and 500 mg erythromycin stearate orally. International journal of oral surgery. 1981; 10:198-202.
53. Koo BK, Spit M, Jordens I, Low TY, Stange DE, van de Wetering M, van Es JH, Mohammed S, Heck AJ, Maurice MM and Clevers H. Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature. 2012; 488:665-669.
54. Hao HX, Xie Y, Zhang Y, Charlat O, Oster E, Avello M, Lei H, Mickanin C, Liu D, Ruffner H, Mao X, Ma Q, Zamponi R, Bouwmeester T, Finan PM, Kirschner MW, et al. ZNRF3 promotes Wnt receptor turnover in an R-spondin-sensitive manner. Nature. 2012; 485:195-200.
55. Huang SM, Mishina YM, Liu S, Cheung A, Stegmeier F, Michaud GA, Charlat O, Wiellette E, Zhang Y, Wiessner S, Hild M, Shi X, Wilson CJ, Mickanin C, Myer V, Fazal A, et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature. 2009; 461:614-620.
56. Horst D, Chen J, Morikawa T, Ogino S, Kirchner T and Shivdasani RA. Differential WNT activity in colorectal cancer confers limited tumorigenic potential and is regulated by MAPK signaling. Cancer research. 2012; 72:1547-1556.
57. Phelps RA, Chidester S, Dehghanizadeh S, Phelps J, Sandoval IT, Rai K, Broadbent T, Sarkar S, Burt RW and Jones DA. A two-step model for colon adenoma initiation and progression caused by APC loss. Cell. 2009; 137:623-634.
58. Cha TL, Zhou BP, Xia W, Wu Y, Yang CC, Chen CT, Ping B, Otte AP and Hung MC. Akt-mediated phosphorylation of EZH2 suppresses methylation of lysine 27 in histone H3. Science. 2005; 310:306-310.
59. Wei Y, Chen YH, Li LY, Lang J, Yeh SP, Shi B, Yang CC, Yang JY, Lin CY, Lai CC and Hung MC. CDK1-dependent phosphorylation of EZH2 suppresses methylation of H3K27 and promotes osteogenic differentiation of human mesenchymal stem cells. Nature cell biology. 2011; 13:87-94.
60. Jun S, Lee S, Kim HC, Ng C, Schneider AM, Ji H, Ying H, Wang H, Depinho RA and Park JI. PAF-Mediated MAPK Signaling Hyperactivation via LAMTOR3 Induces Pancreatic Tumorigenesis. Cell Rep. 2013.
61. Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG and Zhang F. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014; 343:84-87.
62. Jung HY, Wang X, Jun S and Park JI. Dyrk2-associated EDD-DDB1-VprBP E3 ligase inhibits telomerase by TERT degradation. J Biol Chem. 2013; 288:7252-7262.