
INTRODUCTION
Multidrug resistance (MDR) formation and metastasis are the important issues for lung cancer therapeutic failure [1]. Tumors [2–5] or stromal cells [6–8] expressing interleukin 6 (IL-6) have been documented to promote tumor metastasis [1, 9–10]. Our previous study reveals that IL-6 contribute to lung cancer chemotherapeutic resistance [11]. The good correlation of IL-6 level and poor clinical outcome of lung cancer, breast cancer and pancreatic cancer patients indicates that IL-6 might be a pivotal molecule to overcome inflammation-correlated lung cancer metastasis [12–13].
Matrix metalloproteinases (MMPs) mediated degradation of the extracellular matrix (ECM) is an initial step for metastasis. MMP-3 has been revealed to remodel ECM [14–15] and has a close correlation with the progression of breast, gastric and lung cancer [16–18]. Meanwhile, MMP-13 was also documented to be activated by MMP-3 and contribute to metastasis [19–21]. The activities of MMP-3/MMP-13 could be increased by TNF-α treatment and mediated TNF-α augmented metastasis [22]. But until now, little is known about the roles of MMP-3 and MMP-13 in IL-6 correlated lung cancer metastasis.
Ataxia-telangiectasia mutated (ATM) is a serine/threonine kinase that is activated by DNA double strand break [23]. The treatment with IL-6 also triggers ATM phosphorylation without apparent DNA damage [11]. The phosphorylation of ATM up-regulates MDR-associated protein expression, and contributes to chemotherapeutic resistance [24–25]. The effect of IL-6 on ATM raises the question of whether ATM activation is involved in IL-6 correlated lung cancer metastasis. Nevertheless, little is known about the role of ATM phosphorylation in IL-6 increased expressions of MMP-3 and MMP-13.
In the present study, we found that the high IL-6 level reveals both the increased expression of MMP-3/MMP-13 and the enhanced migration abilities of lung cancer cells. Then, the inhibition of ATM phosphorylation abolishes IL-6 increased expression of MMP-3/MMP-13, hence abrogates IL-6 correlated lung cancer metastasis both in vitro and in vivo. All these findings demonstrate that IL-6 inducing ATM activation increases the expression of MMP-3/MMP-13, augments the abilities of cell migration and promotes lung cancer metastasis, indicating that ATM is a potential target molecule to overcome IL-6 correlated lung cancer metastasis.
RESULTS
IL-6 level correlates cell migration abilities in lung cancer cells
To explore the effect of IL-6 on cell migration, we firstly determined IL-6 levels in a panel of lung cancer cells. Contrast to NCI-H446 and NCI-H1299 cells, there is a relatively higher IL-6 level in A549, LTEP-a-2 and NCI-H520 cells (Figure 1a–1b). Consistent with the higher IL-6 level, the more migration cells were observed in A549, LTEP-a-2 and NCI-H520 cells (Figure 1c). When NCI-H446 cells were replenished with IL-6, the ability of cell migration increased accordingly (Figure 1d). Meanwhile, the down-regulation of IL-6 in A549, LTEP-a-2 and NCI-H520 cells led to a significant decline of migration cells (Figure 1e). As IL-8 was reported to affect cell migration by mediating angiogenic activity [26], we next detected the IL-8 level in lung cancer cells. No difference was found between NCI-H446 and A549 cells (Supplementary Figure S1a). As the increased migration cells is usually yielded by promoted cell proliferation or augmented cell migration, the observation that IL-6 had no effect on cell proliferation excludes the possibility that IL-6 increases cell migration by promoting proliferation (Supplementary Figure S1b–S1c). The above results demonstrate that the IL-6 level correlates cell migration abilities in lung cancer cells.
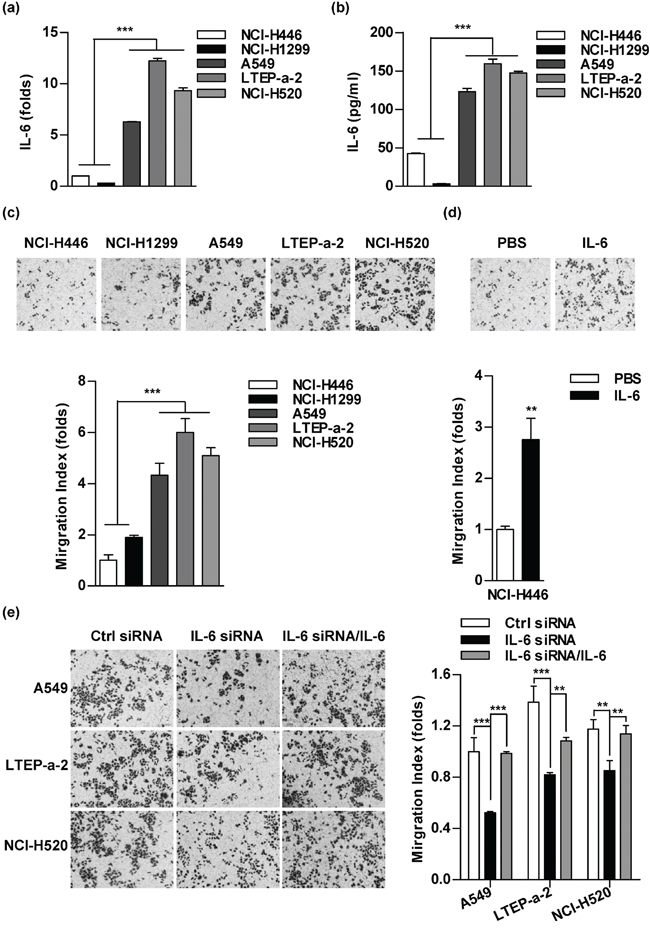
Figure 1: The level of IL-6 correlates to the abilities of cell migration in lung cancer cells. The cellular RNA and supernatant of indicated lung cancer cells were prepared and the expression of IL-6 was determined by RT-qPCR a. and ELISA b. A panel of lung cancer cells was treated with PBS c. IL-6 (5 ng/ml) d. or IL-6 siRNA transfection/IL-6 e. and the cell migration was determined via Transwell migration assay by calculating the number of migrated cells in three visual fields. The data are presented as the mean ± SEM, n = 3. **p < 0.01, ***p < 0.001, Student's t test or one-way ANOVA with post Newman-Keuls test. One representative from three experiments is shown.
MMP-3/MMP-13 is involved in IL-6 increasing cell migration in lung cancer cells
MMPs activities and epithelial-mesenchymal transition (EMT) are the main landmarks of metastasis. To investigate the roles of MMPs in IL-6 increased cell migration, MMPs inhibitors were used and cell migration was determined. Both the broad-spectrum inhibitor (NOB) and the specific inhibitors, such as HYD (MMP-1), SB-3CT (MMP-2), NNGH (MMP-3) and UK-356618 (MMP-13), efficiently abolished the effect of IL-6 on cell migration in NCI-H446 (Figure 2a) and A549 cells (Supplementary Figure S2). As MMP-3/MMP-13 plays the pivotal roles in TNF-α increased metastasis [22], we therefore investigated the role of MMP-3/MMP-13 in IL-6 enhanced lung cancer metastasis. As shown in Figure 2b, the silencing of MMP-3 and MMP-13 abrogated the effect of IL-6 on cell migration in NCI-H446 cells. Together, these data demonstrate that MMP-3 and MMP-13 are the key MMPs in IL-6 promoting lung cancer metastasis.
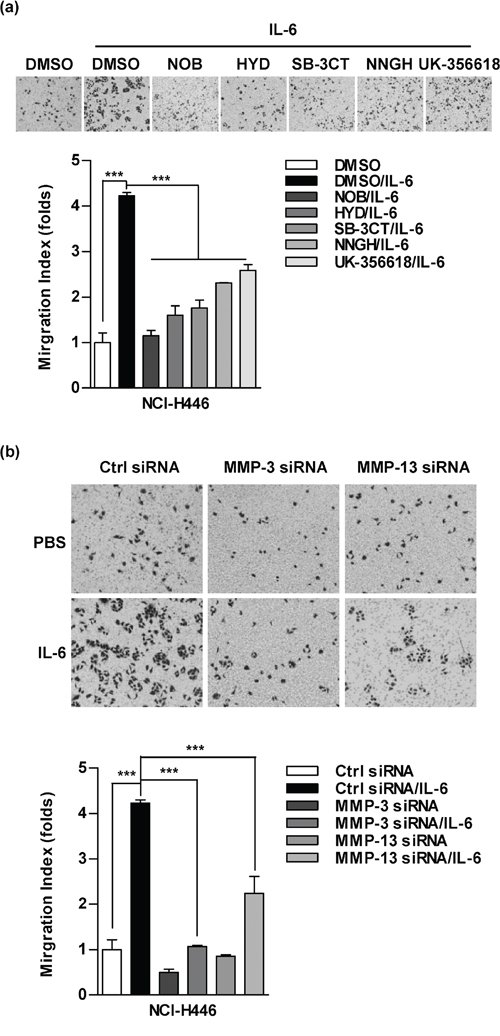
Figure 2: MMPs are involved in interleukin 6 (IL-6) increasing cell migration in lung cancer cells. NCI-H446 a. or A549 b. cells were conferred the treatment with Nobiletin (NOB, 64 μM), N-CBZ-Pro-Leu-Gly hydroxamate (HYD, 50 μM), SB-3CT (10 μM), NNGH (10 μM), UK-356618 (73 nM) (a) or MMP-3/MMP-13 siRNA transfection (b), respectively, which were further stimulated with IL-6 (5 ng/ml). The ability of cell migration was determined by Transwell migration assay. The data are presented as the mean ± SEM, n = 3. ***p < 0.001, One-way ANOVA with post Newman-Keuls test. One representative from three experiments is shown.
IL-6 increases MMP-3/MMP-13 expression and activity in lung cancer cells
As the cells that had higher level of IL-6 (Figure 1) also revealed higher MMP-3/MMP-13 expression (Figure 3a), we next explored the effect of IL-6 on MMPs’ expression by IL-6 replenishment or siRNA silencing. The IL-6 replenishment increases MMP-3/MMP-13 expression not only in protein level (Figure 3b), but also in mRNA level (Figure 3c) in NCI-H446 and NCI-H1299 cells. On the other hand, MMP-3/MMP-13 expression can be decreased by IL-6 silencing in A549, LTEP-a-2 and NCI-H520 cells (Figure 3d). Importantly, the activities of MMP-3/MMP-13 were also increased by IL-6 replenishment (Figure 3e). Apart from MMP-3/MMP-13, the expression of MMP-1/MMP-2 was also found to correlate with the level of IL-6 (Supplementary Figure S3). All these results indicate that IL-6 increases MMPs expressions and activities in lung cancer cells.
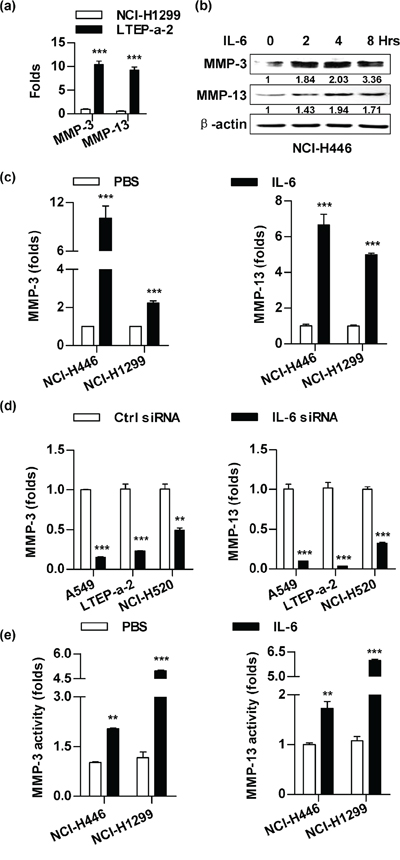
Figure 3: The treatment with IL-6 promotes the expressions and activities of MMP-3/MMP-13 in lung cancer cells. A panel of lung cancer cells was conferred the treatment with PBS a. IL-6 (5 ng/ml) b–c, e. or IL-6 siRNA transfection d. The effects of IL-6 on expression (b-d) and activities (e) of MMP-3/MMP-13 were determined by RT-qPCR (a, c-d), western blot (b) and ELISA (e), respectively. The relative band intensity of MMP-3/MMP-13 in western blot was quantified using IMAGE J software and indicated under each lane. The data are presented as the mean ± SEM, n = 3. **p < 0.01, ***p < 0.001, Student t test. One representative from three experiments is shown.
Inhibition of ATM phosphorylation abrogates the effect of IL-6 on cell migration in lung cancer cells
Despite the effects of IL-6 in ATM phosphorylation [11] and in tumor invasion [9–10] were documented respectively, the exact role of ATM phosphorylation in IL-6 increasing lung cancer metastasis is still unknown. Indeed, both the silencing of ATM/p65 (Figure 4a) and the inhibition of relative kinases (Figure 4b, Supplementary Figure S4) abrogated IL-6’s effect on cell migration in NCI-H446 cells. The silencing of ATM/p65 in A549, LTEP-a-2 and NCI-H520 cells also decreased the ability of cell migration (Figure 4c). As the silencing of these genes had no effect on cell proliferation (Supplementary Figure S5a–S5b) and down-regulation of MMP-3/MMP-13 inhibited cell migration (Figure 4d), the inhibition of ATM phosphorylation abolishing IL-6’s effect on cell migration indicates that the phosphorylation of ATM is involved in IL-6 increasing lung cancer metastasis.
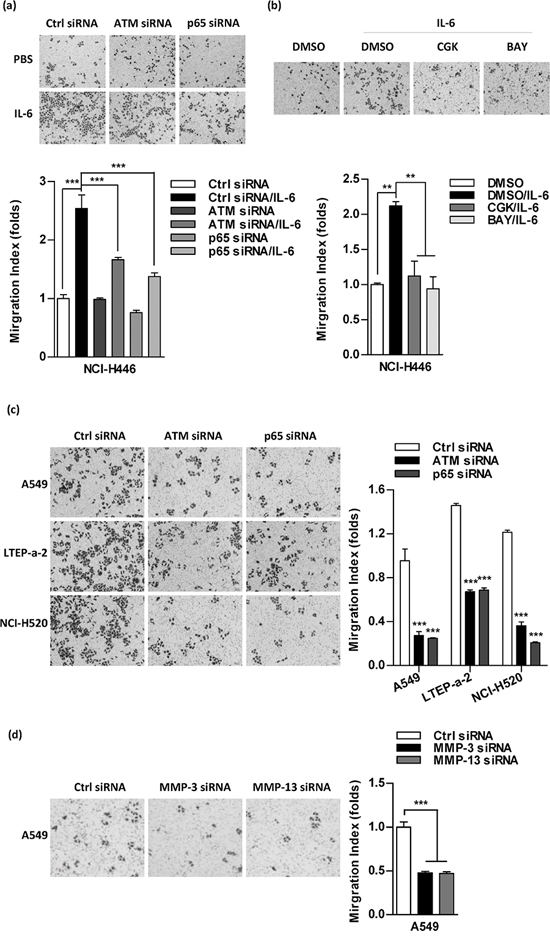
Figure 4: Inhibition of ATM and NF-κB activation abrogates the effect of IL-6 on cell migration in lung cancer cells. To investigate the role of ATM, NF-κB and MMP-3/MMP-13 in IL-6 increasing cell migration, the indicated cells were performed siRNA transfection a, c, d. or kinases inhibitor treatment b. and cell migration was determined by Transwell migration assay. The data are presented as the mean ± SEM, n = 3. ***p < 0.001, One-way ANOVA with post Newman-Keuls test. One representative from three experiments is shown. CGK733 and BAY11–7082 were shown as CGK and BAY, respectively, for limited space in the figure.
Inhibition of ATM phosphorylation abrogates IL-6 increasing MMP-3/MMP-13 expression
To further explore the role of ATM phosphorylation in IL-6 augmenting lung cancer metastasis, a panel of lung cancer cells was conferred the inhibition of ATM/p65 and the MMPs’ expressions were monitored. While the usage of ATM/p65 inhibitors abrogated IL-6 increasing MMP-3/MMP-13 expression in protein level (Figure 5a), the silencing of ATM/p65 abolished IL-6’s effect on MMP-3/MMP-13 expression in mRNA level (Figure 5b). Importantly, the suppression of ATM/p65 in LTEP-a-2 and NCI-H520 cells also decreased high IL-6 level correlating MMP-3/MMP-13 expression (Figure 5c). Apart from MMP-3 and MMP-13, MMP-1/MMP-2 expression was obviously decreased by the inhibition of ATM/p65 (Supplementary Figure S5c–S5d). All together, these data suggest that ATM could be activated by the treatment with IL-6, hence increases the expression of MMP-3/MMP-13 and promotes lung cancer metastasis.
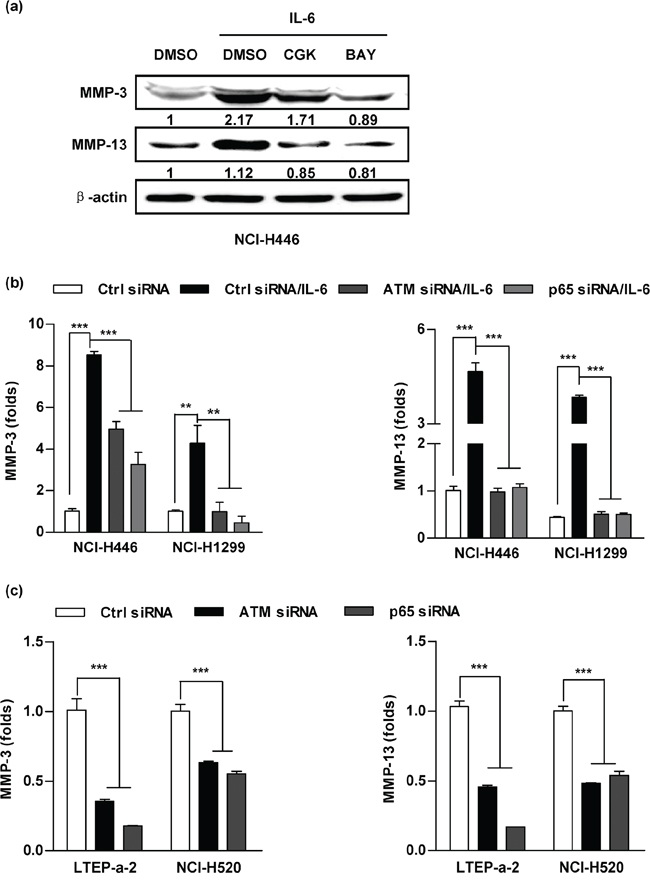
Figure 5: Inhibition of ATM and NF-κB activation abrogates the effect of IL-6 on MMP-3/MMP-13 up-regulation. A panel of lung cancer cells was pretreated with inhibitors of ATM (CGK733, 20 μM), p65 (BAY11–7082, 20 μM) a. or siRNA transfection b–c. prior to (5 ng/ml) IL-6 (a-b) or PBS (c) treatment. The expressions of MMP-3 and MMP-13 were determined by western blot (a) and RT-qPCR (b-c). The relative band intensity of MMP-3/MMP-13 in western blot was quantified using IMAGE J software and indicated under each lane. The data are presented as the mean ± SEM, n = 3. **p < 0.01, ***p < 0.001, One-way ANOVA with post Newman-Keuls test. One representative from three experiments is shown. CGK733 and BAY11–7082 were shown as CGK and BAY, respectively, for limited space in the figure.
Inhibition of ATM activation abrogates IL-6 increasing lung cancer metastasis in vivo
Tail vein injection is a widely accepted method to measure metastatic potential and the ability of cancer cells to attach to blood vessels and survive in the lung [21]. Similar amounts of lung cancer cells suffered from ATM inhibition anchored in the vessels of the lungs (data not shown). Despite IL-6 treatment increased cell retention in the lungs, the inhibition of ATM obviously abrogated IL-6’s effect on lung cancer cell retention (Figure 6a), indicating that ATM phosphorylation facilitates lung cancer cell metastasis. To address this issue, in vivo lung cancer metastasis test was performed. Hematoxylin and eosin sections revealed nest cells with prominent and irregular nuclei (Figure 6b–6c, Figure S6a–S6b). The number of cancer nests in the lung represents the proximal colonization of the cancer cells (Figure 6b–6c), while the metastatic nodes in the liver represent the remote metastasis (Figure S6a–S6b). Importantly, the inhibition of ATM abrogated IL-6’s effect on cancer nests formation in the lung of NCI-H446 or A549 cells transferred recipients (Figure 6b–6c). Moreover, the expressions of MMP-3/MMP-13 in both lung (Figure 6d–6e) and liver (Supplementary Figure S6c–S6d) were also suppressed by the inhibition of ATM. As the silencing of ATM/p65, IL-6, MMP-3/MMP-13 efficiently abrogated ATM phosphorylation (Supplementary Figure S7) and gene expression (Supplementary Figure S8), the above observations demonstrate that high level of ATM phosphorylation contributes to IL-6 correlating MMP-3/MMP-13 expression and lung cancer metastasis in vivo.
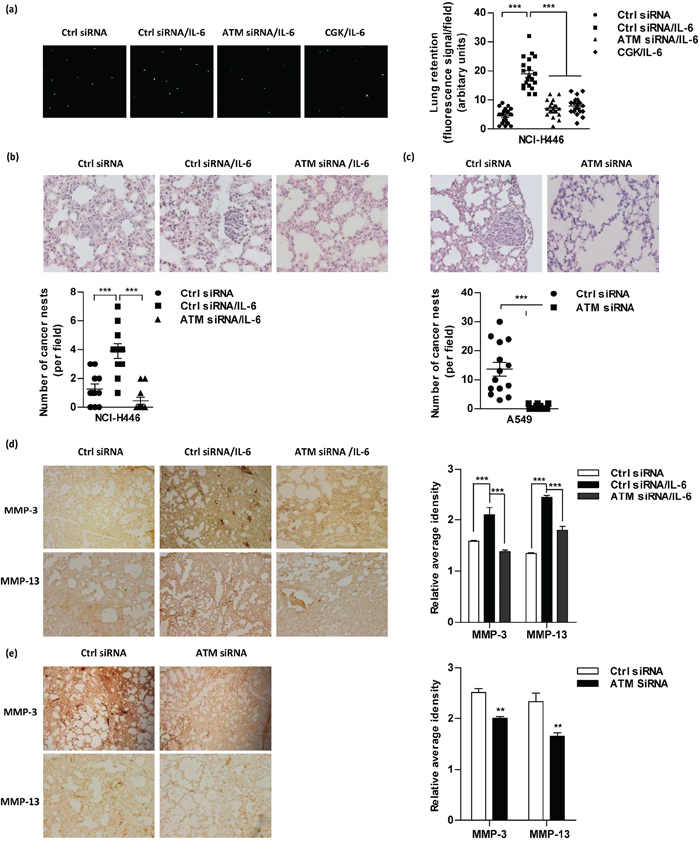
Figure 6: Inhibition of ATM phosphorylation abrogates IL-6 increasing lung cancer metastasis in vivo. a. For lung retention assays, 5 × 105 NCI-H446 cells pretreated with IL-6, siRNA or related kinase inhibitors were labeled with CFSE and then injected to Balb/c nude mice (5–6 weeks old) through the tail vein (n = 4 per group). Lung retention is represented as fluorescence signals (CFSE-Green from lung epithelium cells in left panel). Dots represent NCI-H446 cells retained in the lungs 10 h post-tail vein injection (right panel). 10 fields per mouse were analyzed. b–e. For lung cancer metastasis model, 8 × 105 NCI-H446 (b, d) or A549 (c, e) cells were conferred ATM siRNA transfection and transferred to Balb/c nude mice (5–6 weeks old) through tail vein (n = 4 per group). NCI-H446 cells transferred mice were further subcutaneously conferred IL-6 administration. 2 weeks after adoptive transfer, the lungs were dissociated and performed H&E (b-c) and Immunohistochemistry staining (d-e). The number of cancer nests in lungs was counted; 11–13 slides per condition (b-c). The signals of MMP-3 and MMP-13 were quantified using the Image-Pro Plus software (d-e); 3 fields per condition.
DISCUSSION
Our previous studies demonstrate that ATM phosphorylation [25] and elevated IL-6 level [11] contribute to chemotherapeutic agents-inducing MDR formation in lung cancer. The present study demonstrated that high IL-6 level increases both expressions and activities of MMP-3 and MMP-13, hence augments cell migration abilities of lung cancer (Figures 1–3). Moreover, the inhibition of ATM phosphorylation not only abrogates IL-6 increased, MMP-3/MMP-13 mediated cell migration in vitro (Figure 4–5), but also suppresses IL-6 correlating lung cancer metastasis in vivo (Figure 6).
The degradation of ECM and basement membrane by MMPs is a critical process in tumor invasion and metastasis [27]. Although down-regulation of MMP-3 reduced lung cancer spontaneous metastasis [28], genetic ablation of MMP-3 did not significantly affect breast cancer metastasis [29] was also documented. MMP-13 abrogation was demonstrated to reduce breast cancer metastases by inhibiting osteoclast cells’ differentiation [30] or decreasing stromal MMP-13 expression [31]. While MMP-13 replenishment increased tumor metastasis by promoting angiogenesis [32], MMP-13 inactivation inhibited stromal-promoting melanoma metastasis [33], indicating that the interactions of tumor and stromal cells might be an important issue for MMP-13 participating in metastasis. Here, we investigated the role of ATM phosphorylation in IL-6 increasing MMP-3/MMP-13 expression, but the exact mechanism by which MMP-3/MMP-13 promoting lung cancer metastasis is still to be clarified.
As the MMP family is comprised of more than 25 related zinc-dependent enzymes [34], the findings that MMP-3/MMP-13 is involved in ATM activation increasing cell migration (Figures 2–6) and IL-6 increases MMP-1/MMP-2 expression (Supplementary Figure S2) cannot exclude the possibility that other MMPs might also contribute to inflammation-associated lung cancer metastasis. Meanwhile, we also mention that EMT contributes to tumor progression and metastasis [35–37]. Apart from transforming growth factor beta (TGF-β) [38], IL-6 has been documented to be involved in lung cancer EMT via signal transducer and activator of transcription 3 (STAT3) [39], Notch [40–41] pathway. As EMT could be affected by the composition and structure of ECM [42], the findings that ATM phosphorylation increases MMP-3/MMP-13 expression and promotes lung cancer metastasis indicate that the phosphorylation of ATM might be involved in IL-6 promoting EMT. The exact effects and mechanism of ATM activation on IL-6 increasing EMT require further investigation.
STAT3 has been documented to be involved in tumor growth and metastasis in many types of tumor [43–50]. Once binding to IL-6 receptor (IL-6R) and gp130 receptor, IL-6 could activate STAT3, mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI3K) pathways and increase tumor metastasis [43–44, 51], indicating that IL-6 increasing metastasis is IL-6R/gp130 dependent. Meanwhile, a novel mechanism that IL-6 promoted prostate cancer metastases through a soluble IL-6 receptor (sIL-6R) without activation of STAT1, STAT3 or MAPK was also documented [52]. In the present study, despite ATM phosphorylation was achieved by IL-6 treatment, the exact mechanism that IL-6 inducing ATM activation still needs further investigation.
The migration and invasion characteristics that are related to inflammatory response play important roles in the development of lung cancer. While Erk1/2-NF-κB pathway was reported to be partially involved in inflammatory factors TGF-β1, TNF-α and IL-6 correlated lung cancer invasion [53], p38-NF-κB and STAT3-NF-κB pathways were demonstrated to inhibit miR-365 expression and regulate IL-6 repressing miR-98 levels respectively [54–55]. All these studies indicate that Erk1/2, p38 and STAT3 are up-stream molecules of NF-κB. The activation of ATM-NF-κB increasing MDR associated genes expression [11] indicates that ATM is another up-stream kinase of NF-κB. Hence, it was not surprise to find that the inhibition of NF-κB pathway was more efficient than that of ATM in abolishing IL-6’s effects on MMP-3/MMP-13 expression and cell migration (Figure 4, 5). As the STAT3 phosphorylation at Ser(727) is triggered by active RSK2 or JNK1 in the presence of intracellular phosphorylation process of ATM [56], the exact crosstalk between ATM and other NF-κB upstream molecules in IL-6 correlated lung cancer metastasis still need further exploration.
ATM, which expresses in a variety of tumor, is commonly considered as a tumor suppressor for its role in DNA damage repair machinery [57–63] and in SATB1-induced tumorigenic progression [64]. In the present study, our results reveal that IL-6 inducing ATM phosphorylation increases lung cancer metastasis via up-regulation of MMP-3/MMP-13. Consistent to our study, high phosphorylation level of ATM decrease disease-free survival was also documented [65]. Hence, there is still controversial about the role of ATM, to suppress or activate tumor progression. As an early DNA damage-response kinase, ATM might immediately repair SATB1-induced chromatin remodeling, inhibit genetic alterations, and suppress malignant transformation. Therefore, it is no surprise to find that ATM abundant breast epithelial cells, but not ATM depletion cells, were resistant to SATB1-induced malignant progression [64]. On the other hand, the phosphorylation of ATM can also be induced by IL-6 treatment without obvious DNA double break [11]. Similar to the present study, hypoxia treatment increasing ATM phosphorylation without apparent DNA damage was demonstrated to promote breast cancer metastasis via NF-κB pathway [66]. Thus, the high phosphorylation level of ATM contributing to IL-6 correlated lung cancer metastasis indicates that the functions of ATM might be dependent on cell stress, DNA damage and signaling networks.
Taken together, our results reveal for the first time that the inhibition of ATM phosphorylation efficiently decreases the expressions of MMP-3/MMP-13 and inhibits IL-6 correlated lung cancer metastasis, indicating that the inhibition of ATM phosphorylation is a potential strategy for dealing with inflammation correlating lung cancer metastasis.
MATERIALS AND METHODS
Reagents
Recombinant human or mouse IL-6 and Human IL-6 Module Set ELISA kit were obtained from eBioscience (San Diego, CA, USA). Antibodies for western blot and BAY11–7082 were purchased from Cell Signaling Technology (Beverly, MA, USA) and Abcam (San Francisco, CA, USA). ATM inhibitor CGK733, MMPs inhibitor Z-Pro-Leu-Gly-hydroxamate (HYD), 3B-3CT and NNGH was acquired from Sigma (Shanghai, China). MMP-3 inhibitor UK-356618 was from Toronto Research Chemicals (North York, Canada). The siRNA of IL-6, ATM, p65 and controls was purchased from Santa Cruz Biotechnology (Dallas, TX, USA). SYBR Premix Ex Taq, Trizol and Prime-Script Reverse Transcriptase were obtained from TaKaRa Biotechnology (Dalian, China). Transwell compartment was from Corning (New York city, NY, USA). RPMI-1640, DMEM and fetal bovine serums were acquired from Hyclone (Logan, UT, USA). UltraSensitiveTM SP IHC Kit was from Maixin Biotech (Fuzhou, China). Human MMP-3 and MMP-13 ELISA kits were from Shanghai Huiying Biological Technology (Shanghai, China). Lipofectamine 2000 and carboxyfluorescein diacetate succinimidyl ester (CFSE) were bought from Invitrogen (Eugene, OR, USA).
Cell culture, cell lines and siRNA transfection
Human small cell lung cancer (SCLC) NCI-H446 cells, non-small cell lung cancer (NSCLC) NCI-H1299 cells, lung adenocarcinoma LTEP-a-2 cells and squamous cell carcinoma NCI-H520 cells were obtained from Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). Human lung carcinoma A549 cells were kindly provided by Professor GH. Jin (Xiamen University). All the cells were grown in RPMI-1640 or DMEM medium containing 10% FBS. For siRNA transfection, 30–50% confluent cells were transfected with siRNA using Lipofectamine 2000. The cells were harvested 48 h after transfection. The final concentration for siRNA is 100 nM. The silence effects of indicative siRNA in NCI-H446 cells were validated in Supplementary Figure S8.
Animals
Pathogen-free BALB/c nude mice (5–6 weeks old, female) were bought from the Shanghai Laboratory Animal Center of Chinese Academy of Sciences and kept at the Animal Center of Xiamen University. The protocol was approved by the Committee on the Ethics of Animal Experiments of the Xiamen University.
RT-qPCR
Endogenous IL-6 expression of lung cancer cell lines and MMPs expression were investigated by RT-qPCR analysis. Briefly, the cellular RNA was extracted, and reverse transcription was performed using PrimeScript Reverse Transcriptase. Quantitative PCR analysis was performed using ABI 7000 Sequence Detection System in the presence of SYBR Green. The cycling parameters were 95°C for 3 min, followed by 40 cycles of 95°C for 5 s, 60°C for 30 s, and 72°C for 60 s, with a final extension at 72°C for 10 min; a melting curve analysis was subsequently conducted. Each assay was performed in triplicate, and the relative expression levels (defined as fold changes) of the target genes were normalized to the folds of corresponding control cells. The PCR primer sequences are listed in Table 1.
Table 1: Real-time PCR primer sequence
Genes |
F/R |
Sequence |
|---|---|---|
β-actin |
F |
5′-TCAAGATCATTGCTCCTCCTG-3′ |
β-actin |
R |
5′-CTGCTTGCTGATCCACATCTG-3′ |
IL-6 (F) |
F |
5′-CCACACAGACAGCCACTCACC-3′ |
IL-6 (F) |
R |
5′-CTACATTTGCCGAAGAGCCCT-3′ |
MMP-3 |
F |
5′-CCTGCTTTGTCCTTTGATGC-3′ |
MMP-3 |
R |
5′-TGAGTCAATCCCTGGAAAGTC-3′ |
MMP-13 |
F |
5′-TTGTTGCTGCGCATGAGTTCG-3′ |
MMP-13 |
R |
5′-GGGTGCTCATATGCAGCATCA-3′ |
ELISA
To detect the release of IL-6 in corresponding lung cancer cell lines and determine MMP-3/MMP-13 activity, 1 × 105 cells plated in 24-well plates were treated with PBS or IL-6 (5 ng/ml) for 24 h and the supernatant was collected. The IL-6 concentration or the MMP-3/MMP-13 activity was determined by enzyme double-antibody indirect immunoassays with respective ELISA kits in accordance with manufacturer protocol [67].
Western blot
NCI-H446 cells were treated with IL-6 (5 ng/ml) for indicated periods or pretreated with inhibitors prior to IL-6 48 h stimulation. The expressions of MMP-3 and MMP-13 were determined via western blot analysis. The expression of ß-actin was used as a loading control. The relative band intensity of western blot was quantified using IMAGE J software and indicated under each lane.
Transwell migration assay
Cell migration ability was determined via transwell migration assay [22]. Briefly, 4 × 103 cells pretreated with IL-6, siRNA or related kinase inhibitors were seeded onto the top chamber of Transwell inserts (24-well insert, 8 μm pore size; Corning) in serum-free-medium. Culture medium with 10% fetal bovine serum in the lower compartment was used as a chemo attractant. After 18 h incubation, the cells on the upper surface of the membrane were wiped out using a cotton swab. Then the cells that migrated to the lower surface were fixed and stained. Cell migration ability was determined by calculating the number of migrated cells in three visual fields per well by microscopy with 100 × magnification.
In vivo lung retention assays
Early migration ability was determined via lung retention assays [21]. Briefly, 5 × 105 NCI-H446 cells pretreated with IL-6, siRNA or related kinase inhibitors were labeled with CFSE (1:1000 dilution in PBS), trypsinized and injected into BALB/c nude mice through the tail vein in 200 μl saline. Mice were sacrificed after 1 h (to show the early arrive at the lung) and 10 h. The lungs were washed, fixed and examined for fluorescently labeled cells under fluorescence microscope. Lung retention is represented as fluorescence dots per field, and 10 fields per mouse were analyzed. Four mice per condition were conducted.
In vivo lung cancer metastasis test
Late migration ability was determined via murine lung cancer metastasis test [21]. Briefly, NCI-H446 or A549 cells were transfected with ATM siRNA. 48 h post-transfection, 8 × 105 cells per mouse were transferred to BALB/c nude mice in 100 μl saline through the tail vein. The mice were further subcutaneously conferred IL-6 administration, once a day. The dose of IL-6 was 1.5 μg/kg for first 3 days, then 2.5 μg/kg throughout next 3 days and reached 5 μg/kg over next 7 days. 2 days after the last injection, the lungs and livers were dissociated and preserved for further studies. Each experiment had four mice per condition.
H&E and immunohistochemistry staining
In vivo cell migration ability and the expression of MMP-3/MMP-13 in lung were determined via H&E and Immunohistochemistry staining [68]. Briefly, free-floating lung sections (4 μM) were obtained using a slicing system (Leica RM2135). For H&E staining, the sections were stained with hematoxylin, then counterstaining with eosin. For immunohistochemistry staining, endogenous peroxidase activity was quenched in H2O2, and 90% formic acid min was used to expose epitope. The primary antibody was applied overnight at 4°C and then biotinylated secondary antibody at room temperature. After a final wash, the slides were developed with diaminobenzidine substrate by using the avidin-biotin HRP system. The signals were quantified using the Image-Pro Plus software.
Statistical analysis
All experiments were repeated at least three times to confirm the similar results. Data were presented as the mean ± SEM. Student's t test or one-way ANOVA with the post Newman-Keuls test was applied. Statistical differences were considered to be significant at p < 0.05.
ACKNOWLEDGMENTS AND FUNDING
We thank Prof. GH. Jin (Medical College of Xiamen University) for kindly providing A549 cell line.
CONFLICTS OF INTEREST
The authors have no conflicts of interests to declare.
GRANT SUPPORT
This work was supported by grants from the State Key Laboratory of Oncogenes and Related Genes (No. 90-14-05 to FG. Gao), grants from the National Natural Science Foundation of China (No. 81273203 to FG. Gao; No. 81201275 to Q. Li) and grants from Natural Science Foundation of Fujian Province of China (No. 2015J01353 to Q. Li).
Authorship
FG. Gao designed the research and copy-edited the paper. YN. Jiang conducted the transwell migration, histochemestry and immunohistochemestry analyses; HQ. Yan conducted the real-time PCR and transwell migration analyses; XB. Huang conducted the western blot; YN. Wang contributed to important reagents; and Q. Li wrote the manuscript.
REFERENCES
1. Wang R, Zhang J, Chen SF, Lu M, Luo X, Yao S, Liu S, Qin Y, Chen H. Tumor-associated macrophages provide a suitable microenvironment for non-small lung cancer invasion and progression. Lung Cancer. 2011; 74:188–96.
2. Wang TH, Chan YH, Chen CW, Kung WH, Lee YS, Wang ST, Chang TC, Wang HS. Paclitaxel (Taxol) upregulates expression of functional interleukin-6 in human ovarian cancer cells through multiple signaling pathways. Oncogene. 2006; 25:4857–66.
3. Poth KJ, Guminski AD, Thomas GP, Leo PJ, Jabbar IA, Saunders NA. Cisplatin treatment induces a transient increase in tumorigenic potential associated with high Interleukin-6 expression in head and neck squamous cell carcinoma. Mol Cancer Ther. 2010; 9:2430–9.
4. Duan Z, Lamendola DE, Penson RT, Kronish KM, Seiden MV. Overexpression of IL-6 but not IL-8 increases paclitaxel resistance of U-2OS human osteosarcoma cells. Cytokine. 2002; 17:234–42.
5. Hong DS, Angelo LS, Kurzrock R. Interleukin-6 and its receptor in cancer: implications for translational therapeutics. Cancer. 2007; 110:1911–28.
6. Shirabe K, Mano Y, Muto J, Matono R, Motomura T, Toshima T, Takeishi K, Uchiyama H, Yoshizumi T, Taketomi A, Morita M, Tsujitani S, Sakaguchi Y, et al. Role of tumor-associated macrophages in the progression of hepatocellular carcinoma. Surg Today. 2012; 42:1–7.
7. Wang YY, Lo GH, Lai KH, Cheng JS, Lin CK, Hsu PI. Increased serum concentrations of tumor necrosis factor-α are associated with disease progression and malnutrition in hepatocellular carcinoma. J Chin Med Assoc. 2003; 66:593–8.
8. Ding J, Huang S, Wang Y Tian Q, Zha R, Shi H, Wang Q, Ge C, Chen T, Zhao Y, Liang L, Li J, He X. Genome-wide screening reveals that miR-195 targets the TNF-α/NF-κB pathway by down-regulating IκB kinase alpha and TAB3 in hepatocellular carcinoma. Hepatology. 2013; 58:654–66.
9. Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006; 124:263–6.
10. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010; 140:883–99.
11. Yan HQ, Huang XB, Ke SZ, Jiang YN, Zhang YH, Wang YN, Li J, Gao FG. Interleukin 6 augments lung cancer chemotherapeutic resistance via ataxia-telangiectasia mutated/NF-KappaB pathway activation. Cancer Sci. 2014; 105:1220–7.
12. Sansone P, Storci G, Tavolari S, Guarnieri T, Giovannini C, Taffurelli M, Ceccarelli C, Santini D, Paterini P, Marcu KB, Chieco P, Bonafè M. IL-6 triggers malignant features in mammospheres from human ductal breast carcinoma and normal mammary gland. J Clin Invest. 2007; 117:3988–4002.
13. Bellone G, Smirne C, Mauri FA, Tonel E, Carbone A, Buffolino A, Dughera L, Robecchi A, Pirisi M, Emanuelli G. Cytokine expression profile in human pancreatic carcinoma cells and in surgical specimens: implications for survival. Cancer Immunol Immunother. 2006; 55:684–98.
14. Wang X, Wang H, Yang H, Li J, Cai Q, Shapiro IM, Risbud MV. Tumor necrosis factor-α- and interleukin-1β-dependent matrix metalloproteinase-3 expression in nucleus pulposus cells requires cooperative signaling via syndecan 4 and mitogen-activated protein kinase-NF-κB axis: implications in inflammatory disc disease. Am J Pathol. 2014; 184:2560–72.
15. Sternlicht MD, Bissell MJ, Werb Z. The matrix metalloproteinase stromelysin-1 acts as a natural mammary tumor promoter. Oncogene. 2000; 19:1102–13.
16. Slattery ML, John E, Torres-Mejia G, Stern M, Lundgreen A, Hines L, Giuliano A, Baumgartner K, Herrick J, Wolff RK. Matrix metalloproteinase genes are associated with breast cancer risk and survival: the Breast Cancer Health Disparities Study. PLoS One. 2013; 8:e63165.
17. Shantha Kumara HM, Gaita DJ, Miyagaki H, Yan X, Herath SA, Cekic V, Whelan RL. Minimally invasive colorectal resection is associated with significantly elevated levels of plasma matrix metalloproteinase 3 (MMP-3) during the first month after surgery which may promote the growth of residual metastases. Surg Endosc. 2014; 28:3322–8.
18. Fakhoury HM, Noureddine S, Tamim H, Chmaisse H, Makki R. Association of MMP3-1171(5A>6A) polymorphism with lung cancer in Lebanon. Genet Test Mol Biomarkers. 2012; 16:988–90.
19. Leeman MF, Curran S, Murray GI. The structure, regulation, and function of human matrix metalloproteinase-13. Crit Rev Biochem Mol Biol. 2002; 37:149–66.
20. Hsu CP, Shen GH, Ko JL. Matrix metalloproteinase-13 expression is associated with bone marrow microinvolvement and prognosis in non-small cell lung cancer. Lung Cancer. 2006; 52:349–57.
21. Orgaz JL, Pandya P, Dalmeida R, Karagiannis P, Sanchez-Laorden B, Viros A, Albrengues J, Nestle FO, Ridley AJ, Gaggioli C, Marais R, Karagiannis SN, Sanz-Moreno V. Diverse matrix metalloproteinase functions regulate cancer amoeboid migration. Nat Commun. 2014; 5:4255.
22. Zhang YH, Yan HQ, Wang F, Jiang YN, Wang YN, Gao FG. TIPE2 inhibits TNF-α-induced hepatocellular carcinoma cell metastasis via Erk1/2 downregulation and NF-κB activation. Int J Oncol. 2015; 46:254–64.
23. Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol. 2013; 14:197–210.
24. Svirnovski AI, Serhiyenka TF, Kustanovich AM, lebko PV, Fedosenko VV, Taras IB, Bakun AV. DNA-PK, ATM, and MDR proteins inhibitors in overcoming fludarabine resistance in CLL cells. Exp Oncol. 2010; 32:258–62.
25. Ke SZ, Ni XY, Zhang YH, Wang YN, Wu B, Gao FG. Camptothecin and cisplatin upregulate ABCG2 and MRP2 expression by activating the ATM/NF-κB pathway in lung cancer cells. Int J Oncol. 2013; 42:1289–96.
26. Arenberg DA, Kunkel SL, Polverini PJ, Glass M, Burdick MD, Strieter RM. Inhibition of interleukin-8 reduces tumorigenesis of human non-small cell lung cancer in SCID mice. J Clin Invest. 1996; 97:2792–802.
27. Martin Del Campo SE, Latchana N, Levine KM, Grignol VP, Fairchild ET, Jaime-Ramirez AC, Dao TV, Karpa VI, Carson M, Ganju A, Chan AN, Carson WE 3rd. MiR-21 enhances melanoma invasiveness via inhibition of tissue inhibitor of metalloproteinases 3 expression: in vivo effects of MiR-21 inhibitor. PLoS One. 2015; 10:e0115919.
28. Banik D, Netherby CS, Bogner PN, Abrams SI. MMP3-mediated tumor progression is controlled transcriptionally by a novel IRF8-MMP3 interaction. Oncotarget. 2015; 6:15164–79.
29. Juncker-Jensen A, Rømer J, Pennington CJ, Lund LR, Almholt K. Spontaneous metastasis in matrix metalloproteinase 3-deficient mice. Mol Carcinog. 2009; 48:618–25.
30. Pivetta E, Scapolan M, Pecolo M, Wassermann B, Abu-Rumeileh I, Balestreri L, Borsatti E, Tripodo C, Colombatti A, Spessotto P. MMP-13 stimulates osteoclast differentiation and activation in tumour breast bone metastases. Breast Cancer Res. 2011; 13:R105.
31. Perry SW, Schueckler JM, Burke K, Arcuri GL, Brown EB. Stromal matrix metalloprotease-13 knockout alters Collagen I structure at the tumor-host interface and increases lung metastasis of C57BL/6 syngeneic E0771 mammary tumor cells. BMC Cancer. 2013; 13:411.
32. Kudo Y, Iizuka S, Yoshida M, Tsunematsu T, Kondo T, Subarnbhesaj A, Deraz EM, Siriwardena SB, Tahara H, Ishimaru N, Ogawa I, Takata T. Matrix metalloproteinase-13 (MMP-13) directly and indirectly promotes tumor angiogenesis. J Biol Chem. 2012; 287:38716–28.
33. Zigrino P, Kuhn I, Bäuerle T, Zamek J, Fox JW, Neumann S, Licht A, Schorpp-Kistner M, Angel P, Mauch C. Stromal expression of MMP-13 is required for melanoma invasion and metastasis. J Invest Dermatol. 2009; 129:2686–93.
34. Deryugina EI, Quigley JP. Matrix metalloproteinases and tumor metastasis. Cancer Metastasis. 2006; 25:9–34.
35. Kan H, Guo W, Huang Y, Liu D. MicroRNA-520g induces epithelial-mesenchymal transition and promotes metastasis of hepatocellular carcinoma by targeting SMAD7. FEBS Lett. 2015; 589:102–9.
36. Okada T, Sinha S, Esposito I, López-Lago MA, Su W, Pratilas CA, Abele C, Hernandez JM, Ohara M, Okada M, Viale A, Heguy A, Socci ND, et al. The Rho GTPase Rnd1 suppresses mammary tumorigenesis and EMT by restraining Ras-MAPK signalling. Nat Cell Biol. 2015; 17:81–94.
37. Qu B, Yu W, Huang Y, Cai B, Du L, Liu F. 6-OH-BDE-47 promotes human lung cancer cells epithelial mesenchymal transition via the AKT/Snail signal pathway. Environ Toxicol Pharmacol. 2014; 39:271–279.
38. Yu H, Shen Y, Hong J, Xia Q, Zhou F, Liu X. The contribution of TGF-β in Epithelial-Mesenchymal Transition (EMT): Down-regulation of E-cadherin via snail. Neoplasma. 2015; 62:1–15.
39. Zhao Z, Cheng X, Wang Y, Han R, Li L, Xiang T, He L, Long H, Zhu B, He Y. Metformin inhibits the IL-6-induced epithelial-mesenchymal transition and lung adenocarcinoma growth and metastasis. PLoS One. 2014; 9:e95884.
40. Hassan WA, Yoshida R, Kudoh S, Hasegawa K, Niimori-Kita K, Ito T. Notch1 controls cell invasion and metastasis in small cell lung carcinoma cell lines. Lung Cancer. 2014; 86:304–10.
41. Liu L, Chen X, Wang Y, Qu Z, Lu Q, Zhao J, Yan X, Zhang H, Zhou Y. Notch3 is important for TGF-β-induced epithelial-mesenchymal transition in non-small cell lung cancerbone metastasis by regulating ZEB-1. Cancer Gene Ther. 2014; 21:364–72.
42. Guan J, Zhang H, Wen Z, Gu Y, Cheng Y, Sun Y, Zhang T, Jia C, Lu Z, Chen J. Retinoic acid inhibits pancreatic cancer cell migration and EMT through the downregulation of IL-6 in cancer associated fibroblast cells. Cancer Lett. 2014; 345:132–9.
43. Tawara K, Oxford JT, Jorcyk CL. Clinical significance of interleukin (IL)-6 in cancer metastasis to bone: potential of anti-IL-6 therapies. Cancer Manag Res. 2011; 3:177–89.
44. Chang Q, Bournazou E, Sansone P, Berishaj M, Gao SP, Daly L, Wels J, Theilen T, Granitto S, Zhang X, Cotari J, Alpaugh ML, de Stanchina E, et al. The IL-6/JAK/Stat3 feed-forward loop drives tumorigenesis and metastasis. Neoplasia. 2013; 15:848–62.
45. Snyder M, Huang XY, Zhang JJ. Signal transducers and activators of transcription 3 (STAT3) directly regulates cytokine-induced fascin expression and is required for breast cancer cell migration. J Biol Chem. 2011; 286:38886–93.
46. Studebaker AW, Storci G, Werbeck JL, Sansone P, Sasser AK, Tavolari S, Huang T, Chan MW, Marini FC, Rosol TJ, Bonafé M, Hall BM. Fibroblasts isolated from common sites of breast cancer metastasis enhance cancer cell growth rates and invasiveness in an interleukin-6-dependent manner. Cancer Res. 2008; 68:9087–95.
47. Zhao Y, Yao J, Wu XP, Zhao L, Zhou YX, Zhang Y, You QD, Guo QL, Lu N. Wogonin suppresses human alveolar adenocarcinoma cell A549 migration in inflammatory microenvironment by modulating the IL-6/STAT3 signaling pathway. Mol Carcinog. 2015; 54:E81–93.
48. Sanford DC, DeWille JW. C/EBP delta is a downstream mediator of IL-6 induced growth inhibition of prostate cancer cells. Prostate. 2005; 63:143–54.
49. Liu H, Ren G, Wang T, Chen Y, Gong C, Bai Y, Wang B, Qi H, Shen J, Zhu L, Qian C, Lai M, Shao J. Aberrantly expressed Fra-1 by IL-6/STAT3 transactivation promotes colorectal cancer aggressiveness through epithelial-mesenchymal transition. Carcinogenesis. 2015; 36:459–68.
50. Yang X, Liang L, Zhang XF, Jia HL, Qin Y, Zhu XC, Gao XM, Qiao P, Zheng Y, Sheng YY, Wei JW, Zhou HJ, Ren N, et al. MicroRNA-26a suppresses tumor growth and metastasis of human hepatocellular carcinoma by targetinginterleukin-6-Stat3 pathway. Hepatology. 2013; 58:158–70.
51. Junk DJ, Bryson BL, Jackson MW. HiJAK’d signaling; the STAT3 paradox in senescence and cancer progression. Cancers (Basel). 2014; 6:741–55.
52. Santer FR, Malinowska K, Culig Z, Cavarretta IT. Interleukin-6 trans-signalling differentially regulates proliferation, migration, adhesion and maspin expression in human prostate cancer cells. Endocr Relat Cancer. 2010; 17:241–53.
53. Cheng X, Gu J, Zhang M, Yuan J, Zhao B, Jiang J, Jia X. Astragaloside IV inhibits migration and invasion in human lung cancer A549 cells via regulating PKC-α-ERK1/2-NF-κB pathway. Int Immunopharmacol. 2014; 23:304–13.
54. Hong B, Li H, Zhang M, Xu J, Lu Y, Zheng Y, Qian J, Chang JT, Yang J, Yi Q. p38 MAPK inhibits breast cancer metastasis through regulation of stromal expansion. Int J Cancer. 2015; 136:34–43.
55. Li F, Li XJ, Qiao L, Shi F, Liu W, Li Y, Dang YP, Gu WJ, Wang XG, Liu W. miR-98 suppresses melanoma metastasis through a negative feedback loop with its target gene IL-6. Exp Mol Med. 2014; 46:e116.
56. Zhang Y, Cho YY, Petersen BL, Bode AM, Zhu F, Dong Z. Ataxia telangiectasia mutated proteins, MAPKs, and RSK2 are involved in the phosphorylation of STAT3. J Biol Chem. 2003; 278:12650–9.
57. Bhattacharya N, Mukherjee N, Singh RK, Sinha S, Alam N, Roy A, Roychoudhury S, Panda CK. Frequent alterations of MCPH1 and ATM are associated with primary breast carcinoma: clinical and prognostic implications. Ann Surg Oncol. 2013; 20:S424–S432.
58. Angèle S, Falconer A, Edwards SM, Dörk T, Bremer M, Moullan N, Chapot B, Muir K, Houlston R, Norman AR, Bullock S, Hope Q, Meitz J, et al. ATM polymorphisms as risk factors for prostate cancer development. Br J Cancer. 2004; 91:783–7.
59. Kang B, Guo RF, Tan XH, Zhao M, Tang ZB, Lu YY. Expression status of ataxia-telangiectasia-mutated gene correlated with prognosis in advanced gastric cancer. Mutat Res. 2008; 638:17–25.
60. Grabsch H, Dattani M, Barker L, Maughan N, Maude K, Hansen O, Gabbert HE, Quirke P, Mueller W. Expression of DNA double-strand break repair proteins ATM and BRCA1 predicts survival in colorectal cancer. Clin Cancer Res. 2006; 12:1494–500.
61. Hong YS, Hong SW, Kim SM, Jin DH, Shin JS, Yoon DH, Kim KP, Lee JL, Heo DS, Lee JS, Kim TW. Bortezomib induces G2-M arrest in human colon cancer cells through ROS-inducible phosphorylation of ATM-CHK1. Int J Oncol. 2012; 41:76–82.
62. Li DH, Frazier M, Evans DB, Hess KR, Crane CH, Jiao L, Abbruzzese JL. Single nucleotide polymorphisms of RecQ1, RAD54L and ATM genes are associated with reduced survival of pancreatic cancer. J Clin Oncol. 2006; 24:1720–28.
63. Lee KW, Tsai YS, Chiang FY, Huang JL, Ho KY, Yang YH, Kuo WR, Chen MK, Lin CS. Lower ataxia telangiectasia mutated (ATM) mRNA expression is correlated with poor outcome of laryngeal and pharyngeal cancer patients. Ann Oncol. 2011; 22:1088–93.
64. Ordinario E, Han HJ, Furuta S, Heiser LM, Jakkula LR, Rodier F, Spellman PT, Campisi J, Gray JW, Bissell MJ, Kohwi Y, Kohwi-Shigematsu T. ATM suppresses SATB1-induced malignant progression in breast epithelial cells. PLoS One. 2012; 7:e51786.
65. Roossink F, Wieringa HW, Noordhuis MG, ten Hoor KA, Kok M, Slagter-Menkema L, Hollema H, de Bock GH, Pras E, de Vries EG, de Jong S, van der Zee AG, Schuuring E, et al. The role of ATM and 53BP1 as predictive markers in cervical cancer. Int J Cancer. 2012; 131:2056–66.
66. Chen WT, Ebelt ND, Stracker TH, Xhemalce B, Van Den Berg CL, Miller KM. ATM regulation of IL-8 links oxidative stress to cancer cell migration and invasion. Elife. 2015; 4:e07270.
67. Gao FG, Li HT, Li ZJ, Gu JR. Nicotine stimulated dendritic cells could achieve anti-tumor effects in mouse lung and liver cancer. J Clin Immunol. 2011; 31:80–8.
68. Xue MQ, Liu XX, Zhang YL, Gao FG. Nicotine exerts neuroprotective effects against β-amyloid-induced neurotoxicity in SH-SY5Y cells through the Erk1/2-p38-JNK-dependent signaling pathway. Int J Mol Med. 2014; 33:925–33.