
Introduction
Multidrug resistance (MDR) can influence cancer clinical outcomes and ~90% of cancer mortality is correlated to primary or acquired MDR and consequent metastases [1]. Thus, elucidating underlying molecular mechanisms of MDR can improve clinical outcomes for cancer patients. Currently three major mechanisms of MDR have been proposed: decreased chemotherapeutic uptake, absorption and distribution; aberrant drug metabolism that diminishes therapeutic efficacy, such as target alterations, DNA damage repair and cell death signaling pathway dysregulation; and finally, increased energy-dependent efflux of hydrophobic drugs by ATP-binding cassette (ABC) transporters [2].
Of these mechanisms, drug efflux by ABC transporters may explain more MDR. ABC transporters are encoded by 48 genes and classified into seven subfamilies (ABCA-ABCG) based on sequence homology and domain organization [3]. To date, sixteen members have been identified, but only three major ABC drug transporters, including ABCB1 (P-glycoprotein), ABCC1 (multidrug resistance protein 1, MRP1) and ABCG2 (BCRP), are thought to affect cancer chemosensitivity [4]. Numerous natural or synthetic inhibitors targeting ABC transporters were developed to reverse MDR. However, neither strong evidence nor clinical trial data are available to confirm the feasibility of these inhibitors for improving chemotherapeutic bioavailability or restoring drug sensitivity in resistant cancer patients [5]. Insufficient specificity, potency and intrinsic toxicity obstruct identifying ideal inhibitors to suppress ABC transporter activity. Also, substantial overlapping specificities exist among major ABC transporters, and some ABC proteins are expressed on vital organs for protective functions [6]. Thus, finding novel membrane transporters related to MDR is required for designing better drugs.
Caveolin-1(Cav-1) is an essential constituent protein of specialized membrane invaginations, referred to as caveolae. Research indicates that Cav-1 is a molecular hub, integrating transduction of multiple signals including Src, EGFR, HER2, and the mitogen-activated protein kinase (MAPK) cascade [7]. Cav-1 silencing was reported to result in activation of these survival signals and promote cancer transformation and initiation [7]. Meanwhile, loss of Cav-1 was frequently observed in various types of malignancies such as breast and colon cancers and ovarian carcinomas [8]. Therefore, Cav-1 may be a tumor suppressor gene. Recent work also indicates that Cav-1 may be a stress-related molecule mediating cancer drug resistance and metastasis. Cav-1 was up-regulated in MDR colon cancer cells, adriamycin-resistant breast cancer cells and taxol- and gemcitabine-resistant lung cancer cells [9]. Research indicated that Cav-1 expression was positively correlated to ABCB1 in various cancers such as acute myeloid leukemia [10-12]. Clinical findings also suggested that cancer patients with high Cav-1 expression had a worse chemotherapeutic response and worse progression-free survival or overall survival [13]. Moreover, Cav-1 was highly expressed on cancer stem cells (CSCs) and affected CSCs’ chemosensitivity [14]. Also, Cav-1 was found to be a critical regulator of aerobic glycolysis, a key metabolic switch influencing cancer chemosensitivity [15]. Finally, Cav-1 inhibition was shown to be correlated to autophagic induction, an important regulatory mechanism to protect cells from stress. However, autophagy overactivation can cause type II programmed cell death [16]. These data suggest that Cav-1 might be a stress-response protein and involved in the modulation of cancer chemosensitivity. Thus, targeting Cav-1 may offer a novel strategy for preventing cancer drug resistance and improving clinical outcomes.
Here, we summarize current evidence of a relationship between Cav-1 and cancer drug resistance, discuss pathophysiological implications of this pathway and propose a targeted therapeutic strategy. A greater understanding of Cav-1 and its role in modulating drug resistance will help us understand cancer therapy and discover better chemotherapeutics.
Cav-1 signaling
Caveolins are 21-24 KDa membrane-associated proteins highly enriched in caveolae [17]. Each caveolae contains approximately 100 to 200 caveolin molecules formed by three principle members, Cav-1, -2, and -3 [18]. Cav-1 and -2 are ubiquitously co-expressed on epithelial and endothelial cells, fibroblasts, smooth muscle cells, adipocytes and pneumocytes, while Cav-3 expression is mainly restricted to muscle and glia cells [19]. The CAV1 gene consists of three exons and maps to 7q31.1, where is close to a known fragile site (FRA7G) frequently deleted in cancer. CAV1 encodes a 21- to 24-kDa integral membrane protein, and the topology of Cav-1 can be divided into three domains including a C-terminal spanning domain (residues 135-150), a transmembrane domain (residues 102-134), an N-terminal scaffolding domain (residues 82-101) and an oligomerization domain (residues 61-101). Both the C- and N-termini face the cytoplasm (Figure 1) [20]. Notably, the Cav-1 scaffolding domain (CSD) is a region that can mediate protein-protein interactions, such as Src-family tyrosine kinases, H-Ras, HER2, estrogen receptor, MAPK and G protein-coupled receptors (Figure 1) [7, 19]. Moreover, due to alternative splicing or initiation, Cav-1 exists in two isoforms, α or β. Cav-1β is distinct in that it has a 31 amino acid residue deletion at the amino terminus [7].
Cav-1 was initially described as a prevalent target for tyrosine phosphorylation in Rous sarcoma virus transformed chicken fibroblasts. Upon stimulation by agonists including insulin, epidermal growth factor (EGF), platelet-derived growth factor (PDGF), mechanical stress or oxidative stress, tyrosine 14 site of Cav-1 can be phosphorylated, subsequently transmitting extracellular signals via intracellular pathways [21]. Co-localization and co-fractionation bioassays demonstrated that Cav-1 not only interacted with many signal transduction proteins, such as Ras-p44/42 MAPK, Her-2, src family kinases and eNOS, but also inhibited their catalytic activity [22]. Many components of the Ras-p44/42 MAPK cascade localize within caveolae, including EGFR, PDGFR, H-Ras, Raf kinase, ERK1/2 kinases, Shc and Grb [23-25]. Suppression of Ras-p42/44 MAPK activity by treatment of PD98059 (a MAPK kinase inhibitor) was found to upregulate Cav-1 expression in Ras-transformed cells [26]. In contrast, Cav-1 overexpression inhibited Ras-p42/44 MAPK signaling by acting as an endogenous inhibitor of EGFR, MEK-1 and ERK1/2 in vitro and in vivo via the CSD domain [27]. Similar reciprocal regulation was also observed between Cav-1 and c-ErbB2, a proto-oncogene encoding Her-2 in human breast carcinomas. Cav-1 expression is significantly reduced in mammary tumors of c-ErbB2 transgenic mice [28]. Conversely, Cav-1 upregulation blocks Her-2 mediated signal transduction in vivo by directly inhibiting the Her-2 autophosphorylation via its CSD domain [27]. In addition, Cav-1 is known to bind and sequester Src family kinases in an inactive configuration [29, 30]. Accordingly, Cav-1 deprivation leads to a marked increase in Src family tyrosine kinase activity, and therefore significantly contributes to the migration and anchorage-dependent growth of osteosarcoma cells [31]. Meanwhile, Cav-1 loss is suggested to trigger the phosphatidylinositol 3-kinase (PI3K)/Akt pathway and induce cell transformation in mammary epithelial cell line MCF-10ACE [32]. Furthermore, Cav-1 was shown to participate in the formation of a multi-protein complex, which includes E-cadherin/β-catenin and helps sequester β-catenin to the membrane, thereby precluding β-catenin/Tcf-Lef-dependent transcription of genes such as survivin, COX-2 and cyclin D1 [33]. Notably, Cav-1 was also shown to inhibit eNOS enzymatic activity and nitric oxide production in a Ca2+/calmodulin-dependent fashion [34]. Overall, Cav-1 was reported to have a central role in regulating cell survival, proliferation, and apoptosis. Elucidation of Cav-1 in cancer development and progression may be significant for improving patient prognosis and preventing tumor onset.
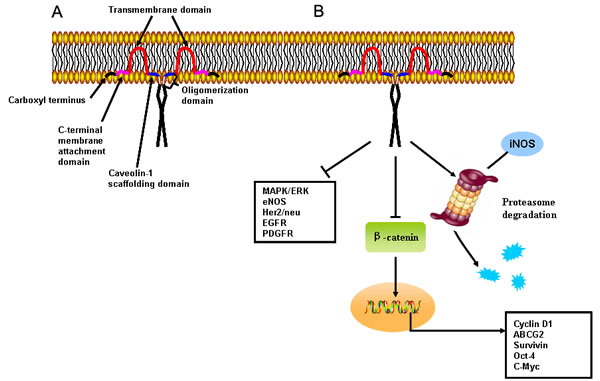
Figure 1: Primary structure and cellular signaling of Cav-1. A. Topology of membranous Cav-1. Cav-1 has a C-terminal spanning, transmembrane, N-terminal scaffolding, and oligomerization domains. Both C- and N-termini face the cytoplasm. B. The Cav-1 scaffolding domain interacts with and inhibits activity of well-known signaling regulators including G-protein coupled and tyrosine-kinase receptors, and eNOS and mitogen-activated protein kinases. In addition, the scaffolding domain may mediate proteasome degradation of iNOS and its inhibitory effects on transcriptional activity of β-catenin were noted.
Cav-1 and cancer development
Despite advancements in understanding Cav-1 structure and function, the relationship between dysfunctional Cav-1 and tumorigenesis is unclear. Downregulation of Cav-1 and its tumor suppression function has been validated in breast, colon, and ovarian cancer and soft-tissue sarcomas. First, studies show that Cav-1 is negatively associated with cancer cells’ transformation. Xie’s group found that Cav-1 expression was significantly down-regulated in 3-phosphoinositide-dependent protein kinase-1 (PDK1)-mediated transformation of mammary epithelial cells [35]. Another study revealed that stable expression of HPV E6 viral oncoprotein in NIH 3T3 cells increased Cav-1 protein. In contrast, adenoviral-mediated overexpression of Cav-1 in SiHa cells, a human cervical squamous carcinoma cell line, abrogated their anchorage-independent growth in soft agar [36]. Similar findings were also validated in alveolar rhabdomyosarcomas. The Cav-1 expression was found either undetectable or very low in alveolar rhabdomyosarcomas cell lines and tumor samples, whereas Cav-1 reintroduction impaired cells clonogenic capacity and promoted features of muscular differentiation [37]. Next, Cav-1 associates with known tumor suppressors. In MCF-7 cells, transfection of murine Cav-1 cDNA led to increased BRCA1 transcription and protein overexpression in a P53 dependent manner [38]. BRCA1 was also found to activate the Cav-1 promoter region and induce redistribution of Cav-1 from the cytoplasm to the cell membrane [39]. Several studies indicate that Cav-1 expression was positively correlated with PTEN in mammalian cells, and Cav-1 restoration in fibroblasts could elevate membrane PTEN, inhibit Akt phosphorylation, and suppress proliferation [40]. In addition, a series of tumor suppressors including deleted in liver cancer 1 (DLC-1), INK4a and RB, were revealed to be closely correlated to Cav-1 status [41-43]. Finally, in vivo studies confirmed that the absence of Cav-1 resulted in mammary ductal hyperplasia, precocious lobuloalveolar development and gestational lactation [44]. Meanwhile, hybridization of Cav-1 knockout mice with tumor prone transgenic mice (MMTV-PyVT) decreased tumor development latency, doubled tumor burden and lung metastatic lesions [28, 45]. Accumulating evidence indicates that Cav-1 was significantly reduced or absent in mammary tumors from MMTV-c-Myc, -Her2, -Src, -Ha-Ras and p53 null transgenic mice [46]. Notably, clinical studies with microarray analysis and immunohistochemistry revealed that Cav-1 expression was equally decreased in either mammary invasive lobular carcinomas or invasive ductal carcinomas [47]. Similar findings were also confirmed in other malignancies including colon and ovarian cancers and osteosarcomas [47].
Intriguingly, opposing evidence supports a role for Cav-1 as a tumor-promoting protein. Cav-1 in pancreatic ductal adenocarcinoma is positively associated with tumor size, grade and stage and Cav-1 expression is correlated to Ki67, P53 in tumor tissues and serum CA 19-9 serum [48]. Other work suggested that Cav-1 expression was significantly correlated to tumor diameter, histological grade and poor prognosis in pancreatic cancer, and indicated that Cav-1 might be an independent negative predictor of survival [49]. Similar findings were confirmed in renal cancer. Campbell’s group suggested that high Cav-1 expression was closely correlated to larger and higher grade tumors, as well as microvessel density, and survival analysis confirmed that increased Cav-1 staining portended shorter survival [50]. In hepatocellular carcinoma, Cav-1 expression was also markedly upregulated in specimens or cell lines, and Cav-1 upregulation positively correlated with histological differentiation, venous invasion, intrahepatic metastasis and VEGF expression [51]. Even in breast cancer, a strong association was found between elevated Cav-1 and a basal-like phenotype [52]. Cav-1 expression was upregulated in inflammatory breast cancer cells and tissues [53] and Cav-1 overexpression significantly contributed to cancer metastasis. With using proteomic identification, Cav-1 was found to be elevated in metastatic hepatocellular cancer cell lines [54]. A microarray analysis also supported Cav-1 as a metastasis-related gene by comparing gene expression profiles between weakly and highly invasive breast cancer cells [55]. Specifically, Cav-1 silencing was effective for reducing metastatic activity of prostate cancer cells in vitro and in vivo, whereas Cav-1 restoration was shown to enhance the motility of tumor cells in lung cancer, hepatocellular carcinoma and melanoma [56]. In addition, as early as 2001, Hayashi’s group reported that the Cav-1 mutant form P132L was identified in 16% of primary breast cancers and mutation-positive cases were mostly invasive scirrhous carcinomas [57]. Subsequent studies revealed that Cav-1 (P132L) behaves in a dominant-negative manner, causing mislocalization and intracellular retention of WT Cav-1 [58]. Mechanistic investigation further verified that Cav-1 (P132L) upregulated the expression of estrogen receptor-α and significantly increased cell migration, invasion and experimental metastasis. Gene profile analysis demonstrated that Cav-1 (P132L) mutation was associated with stem cell/metastasis-associated gene signature, including Cdcp1, Cyr61, Fox1a, Krt14, Pif, Tacstd1, Tnc and Wnt10a [59]. However, several studies also claimed that the P132L mutation was not identified in breast cancer samples, either by direct DNA sequencing or restriction fragment length polymorphism analysis [60-63]. Thus, reevaluating the existence and frequency of P132L mutations in breast cancer is needed. Here, we summarize conflicting evidence regarding Cav-1 during cancer development (Figure 2). Cav-1 may be a duplex signal during cancer progression, and its expression status might be closely correlated to tumor stage. During cancer transformation and initiation, Cav-1 acted as a tumor suppressor gene in breast, lung, cervical, gastric, glioma and pancreatic malignancies [64-69]. Cav-1 loss not only inactivate tumor suppressors BRCA1, P53 and PTEN but also promoted cell cycle progression and activated pro-survival signaling such as PI3K/Akt and MAPK [36, 38, 70]. However, with cancer advanced progression, Cav-1 re-expression was observed in various cancers including breast, lung, prostate, liver, ovarian, pancreas, melanoma, thyroid, colorectal, gastric, renal and pleomorphic malignancies [51, 54, 57, 71-79]. Cav-1 overexpression was found in metastatic lesions of breast cancer and was closely correlated with tumor stage and clinical prognosis [80]. Meanwhile, Cav-1 positively correlated with CSC self-renewal and the tumor biomarker CA19-9 in cancer patients [81, 82]. These data suggest that validating Cav-1 in cancer development with time point-guided detection and genetic intervention tools to investigate the function of Cav-1 in different cancer stages are needed. Although the underlying mechanisms of Cav-1 fluctuation during cancer progression are unclear, studies to better understand the therapeutic and prognostic values of Cav-1 for cancer—especially for chemosensitivity and chemoresistance—are underway.
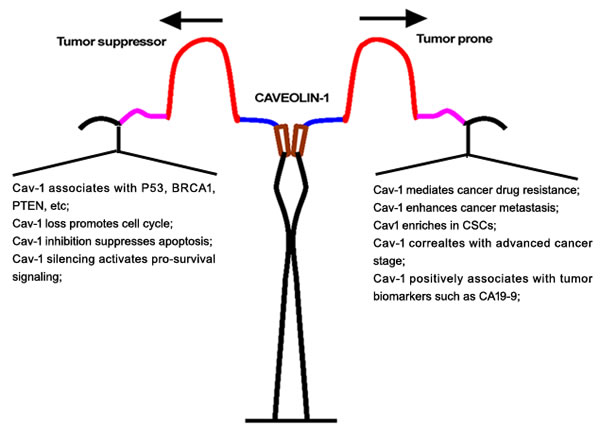
Figure 2: Duplex role of Cav-1 in cancer origination and metastasis.
Implication of Cav-1 in cancer drug resistance
Studies suggest that Cav-1 is a chemotherapy-response gene independent of cancer type. Cav-1 expression was significantly upregulated in a series of drug-resistant cancer cells. In a taxol-resistant (9-fold resistant to taxol) lung cancer cell line A549, Cav-1 expression increased 3.4-folds compared to the parental cell line. Increasing resistance taxol to 17-fold, increased Cav-1 expression 9.5-fold [83]. Meanwhile, increased Cav-1 expression has been reported in other drug-resistant cancer cells, such as SKVLB1 cells resistant to vinblastine, HT-29 cells resistant to colchicine and MCF-7 cells resistant to adriamycin [83, 84]. In addition, Cav-1 expression is significantly induced after treating lung cancer cells with etoposide or bleomycin, indicating that Cav-1 might be a stress-related protein [11]. Thus, genetic and pharmacological studies confirmed a correlation between Cav-1 and drug resistance. Cav-1 silencing increased chemosensitivity or radiosensitivity in diverse cancer cells such as Ewing’s sarcoma cells A4573, Madin-Darby canine kidney cells MDCKII and the pancreatic cancer cell line MiaPaCa2 [84-86]. Cav-1 silencing increased susceptibility of human renal carcinoma cells to doxorubicin-induced apoptosis and inhibited lung metastasis. In contrast, Cav-1 restoration promoted resistance to chemotherapy-induced apoptosis in Ewing’s sarcoma cells [87]. Cav-1 knockout mice had less survival compared with the Cav-1 wide-type mice after whole body irradiation [88]. In a prostate cancer animal model, combined dasatinib and anti-Cav1 antibody treatment or sunitinib and anti-Cav1 antibody produced greater tumor regression than either treatment alone [89]. More importantly, a clinical study revealed that Cav-1 expression was correlated to chemotherapy response in non-small lung cancer. Patients who had tumors samples with strong Cav-1 staining had less response to therapy and worse progression-free and overall survival [90]. Similar findings were also confirmed on gastric cancer. Cav-1 was positively associated with tumor stage and nodal status, and was inversely correlated with the response to ECF (epirubicin, cisplatin and 5-fluorouracil) chemotherapy administrated to gastric cancer patients [91]. Another study validated the value of Cav-1 in predicting chemoresponse. A comprehensive methylation microarray analysis of 49 patients with advanced non-small cell lung cancer (NSCLC), suggested that Cav-1 was a powerful predictor for therapeutic response after platinum-taxane based treatment [92]. Cav-1 methylation was significantly associated with improved overall survival [92]. Therefore, the underlying mechanisms between Cav-1 and cancer chemosensitivity modulation warrant study.
Cav-1 and ABC transporters
Since the discovery of the first ABC family member, ABCB1, in multidrug resistant ovarian cancer cells in 1976, research to identify drug transporters contributing to cancer drug resistance has been underway. Currently, at least seven ABC subfamilies (A-G) encoded by 48 genes have been identified [4]. Among them, ABCB1 is the most well-studied target to regulate cancer drug resistance and develop chemosensitizing agents. Numerous studies show that ABCB1 can extrude hydrophobic compounds central to most chemotherapeutic regimens, including vinca alkaloids, anthracyclines, taxanes and epipodophyllotoxins [93]. In addition, some tyrosine kinase inhibitors such as imatinib, nilotinib and erlotinib, can be exported by ABCB1 [94]. ABCB1 is normally expressed in the transport epithelium of the liver, kidney and gastrointestinal tract so it is difficult to justify the role of ABCB1 for predicting chemosensitivity of tumors originating from these sites. However, clinical studies revealed a positive correlation between ABCB1 expression and poor prognosis in breast cancer, sarcoma and certain types of leukemia, because ABCB1-positive biopsies from these cancers can be compared with ABCB1-negative patients [95]. Interestingly, a close association has been observed between Cav-1 and ABCB1. Both proteins are predominantly located in the Lubrol-based detergent-insoluble glycosphingolipid-enriched membrane domains and co-immunoprecipitation assays confirm that Cav-1 interacts with ABCB1 in brain capillaries, rat brain endothelial, Chinese hamster ovary and breast cancer cells [96]. Sucrose gradient fractionation revealed that Cav-1 co-localized with ABCB1 in lipid rafts. The interaction seems to be mediated by a Cav-1-binding motif in the N-terminal portion of ABCB1 (37-FSMFRYSNW-45) [97]. Clinical studies indicated a positive correlation between Cav-1 and ABCB1 mRNA in acute myeloid leukemia independent of tumor stage [9]. However, studies suggested that Cav-1-regulated chemosensitivity was ABCB1 independent. In ABCB1 deficient taxol-resistant A549 cancer cells, Cav-1 upregulation was observed [83] and Cav-1 overexpression correlated with little ABCB1 protein expression in human ovarian cancer cells, and ABCB1 overexpression was not localized in caveolae [98]. Although accumulating evidence suggested a potential molecular interaction between Cav-1 and ABCB1, detailed signaling pathways between the two molecules are unknown, and more studies of Cav-1 to understand the transcriptional and translational expression of ABCB1 are urgently needed.
Other than ABCB1, ABCG2 is a high-capacity transporter widely expressed in drug resistant cancer cells. ABCG2 substrates include organic anion conjugates, nucleoside analogs, tyrosine kinase inhibitors, organic dyes, cytotoxic drugs, methotrexate and food toxins or carcinogens. Compared with ABCB1, ABCG2 is a mediator of MDR in breast, colon, small cell lung, ovarian, gastric and intestinal cancers and melanomas [99]. Several reports also indicated a potential correlation between Cav-1 and ABCG2. Cav-1 silencing in MDCKII-BCRP cells led to a reduction of ABCG2 activity by 35% [86]. Co-immunoprecipitation experiments demonstrated a physical interaction between Cav-1 and ABCG2, which were co-localized in detergent-resistant membranes [11]. Another study indicated that Cav-1 was highly expressed on breast cancer stem cells or side-population cells, which were enriched with ABCG2. Furthermore, Cav-1 silencing may decrease ABCG2 expression via accelerating β-catenin proteasomal degradation [14]. Because CSCs are considered a root of drug resistance, the study also suggested a role for Cav-1 in regulating chemosensitivity mediated by stem cells.
Cav-1 and CSCs
The first compelling evidence of CSCs came from acute myeloid leukemia in 1997 which indicated that only CD34+CD38- cells could passage hematopoietic malignancy in NOD/SCID mice after serial dilution [100]. Similar phenomena were subsequently validated in various types of cancers, such as breast cancer, hepatocellular carcinoma, gastric and lung cancers. Based on accumulating evidence, the American Association for Cancer Research (AACR) defined CSCs as “a cell within a tumor that possesses the capacity to self-renew and to cause heterogeneous lineages of cancer cells that comprise the tumor” [101]. Therefore, self-renewal and multi-differentiation abilities are considered basic characteristics of CSCs. Research with CSCs suggests that they are relatively resistant to chemotherapy and radiotherapy. High expressions of ABCG2, ABCB1, or ABCC1 were validated in CSCs in various cancers, and the ability of CSCs to extrude Hochest 33342 by ABCG2 was used to develop the “side population” method, which is frequently used to isolate stem-like cells from primary tumors or cancer cell lines [102]. Clinical studies also revealed a positive correlation between CSCs population size and poor prognosis in cancer patients. Neumeister’s group applied CD44 and aldehyde dehydrogenase 1 (ALDH1) biomarkers to define CSCs in formalin-fixed paraffin-embedded breast cancer tissue. Through a retrospective analysis of 642 patients, they found that specimens with high expression of both markers predicted worse outcomes (P = 0.0003), and multivariate analysis confirmed that the marker combination is independent of tumor size, histological grade, nodal status, ER, PR, and HER2 status [103]. Meanwhile, another study reported that during neoadjuvant chemotherapy of breast cancer patients, ALDH1-positive tumors were significantly associated with a low pathological complete response. More importantly, the proportion of ALDH1-positive cancer cells increased dramatically (P < 0.001) after neoadjuvant chemotherapy, indicating that CSCs were relatively chemoresistant [104]. Similar to breast cancer, a meta-analysis of hepatocellular carcinoma patients revealed that the presence of CSCs was significantly associated with a poor histological grade (OR = 3.16, P = 0.003), overall survival (HR = 1.62, P < 0.00001) and disease-free survival (HR = 1.85, P < 0.00001). Notably, there were no significant correlation between the populations of CSCS and tumor size, tumor stage, hepatitis and cirrhosis, indicating that CSCs subset might be an independent factor influencing the clinical outcomes of cancer patients [105]. At present, mechanisms underlying therapeutic resistance of CSCs have not been fully elucidated. However, several mechanisms have been suggested, including: (i) stem cells remain quiescent, making them resistant to cell-cycle active drugs; (ii) high DNA repair ability; (iii) high expression of drug efflux proteins such as ABCG2; and (iv) most current therapies do not target signaling pathways regulating CSCs self-renewal. Elucidation of molecular networks regulating drug sensitivity of CSCs might be essential for eliminating cancer cells.
In recent years, increasing interests have been paid to explore the possible link between Cav-1 and stem cells. It was found that in human mesenchymal stem cells (MSCs), Cav-1 expression was significantly increased when cells were induced to senescence, while Cav-1 lost remarkably increased the proliferation of MSCs, indicating that Cav-1 might be closely associated with the maintenance and self-renewal abilities of stem cells [106]. Cav-1 has positive effects on mediating estradiol-17β 0or fibronectin-induced mouse embryonic stem cell proliferation and DNA synthesis via RhoA-PI3K/Akt-ERK1/2 pathway [107]. With regard to the multi-differentiation ability of stem cells, several reports also indicated that Cav-1 knockdown could enhance human MSC osteogenesis, and bone marrow MSCs from the Cav-1 knockout mice had greater osteogenic potential [106]. Cav-1 null mouse mammary gland also showed dramatically accelerated lobuloalveolar development, early milk production and a premature lactation phenotype during lactation, indicating that Cav-1 lost might favor the differentiation process of mammary epithelial cells [108]. Mechanistic studies suggest that Cav-1 expression and caveolar structure help maintain pluripotency marker expression (Oct4, Sox2, FoxD3, Rex1) in mouse embryonic stem cells [109]. All of these findings suggested that Cav-1 may influence self-renewal and multi-differentiation of stem cells. With regard to CSCs, it was found that nitric oxide administration promoted CSC-like phenotype via elevating Cav-1 expression in lung cancer cells [110]. Also, through genome-wide transcription profile analysis, Cav-1 was identified as a key protein for determining the formation of lung CSCs in a carbon nantotube-induced carcinogenesis animal model. Cav-1 was confirmed to be enriched in lung CSCs and significantly promoted tumorigenesis and metastasis through P53 dysregulation [81]. Emerging studies suggested that Cav-1 may regulate chemoresistance of CSCs. Yuan’s group found that epirubicin increased the activity of the human Wnt6 promoter through Cav1-dependent binding of β-catenin to the proximal Wnt6 promoter in gastric cancer, which is considered an important regulator of CSCs [91]. Another study of Wang’s group confirmed that Cav-1 was upregulated in breast CSCs compared with non-CSCs. In addition, Cav-1 silencing sensitized breast CSCs by limiting their self-renewal ability but promoting differentiation through β-catenin regulation. Meanwhile, in vivo animal models indicated that Cav-1 silencing significantly impaired tumorigenicity and chemoresistance of breast CSCs [14]. Thus, current research for Cav-1 in mediating therapeutic resistance of CSCs is limited to a few cancer types, and a more comprehensive approach is needed to validate a critical role of Cav-1 for determining the therapeutic response of CSCs in cancers, and to clarify the physiological function of Cav-1 in maintaining stem cell survival and its molecular network. The hybridization of Cav-1-null mice with a spontaneous cancer model may be helpful.
Cav-1 and Warburg effect
Cancer cells can be distinguished from normal cells by several hallmarks; one is that cancer cells have a fundamentally different bioenergetic metabolism from non-neoplastic cells. In normal cells, energetic metabolism mostly relies upon mitochondrial oxidative phosphorylation, which consumes glucose that is finally metabolized to carbon dioxide, water and ATP. In contrast, cancer cells have developed altered metabolism that permits greater proliferation rates. Cancer cells could predominantly produce energy by glycolysis followed by lactic acid fermentation, even in the presence of oxygen—known as the “Warburg Effect” [111]. Currently, the phenomenon has been exploited clinically to detect tumors by fluorodeoxyglucose positron emission tomography (FDG-PET). Some key enzymes involved in the glycolysis pathway were also developed as promising targets for cancer therapy and drug development. LDH-A is one such attractive target in the glycolytic pathway because its expression is largely confined to skeletal muscle. Moreover, human subjects with LDH-A deficiency only experience myoglobinuria under intense anaerobic exercise, and individuals with a complete lack of the LDH-A subunit have been documented to have no apparent increase in hemolysis [112]. Numerous studies confirm that overexpression of LDH-A in various types of cancer, and LDH-A inhibition in cancer cells could result in significant stimulation of mitochondrial respiration, decreased mitochondrial membrane potential and finally lead to cancer cell death [113]. Interestingly, recent reports also revealed a close association between the Warburg effect and development of cancer drug resistance. Zhou’s group studied a parental breast cancer cell line MDA-MB-435 and reported a significant elevation of LDH-A protein in taxol-resistant 435R1 and 435TRP cell lines. Meanwhile, taxol treatment led to a dose-dependent increase of LDH-A expression in MDA-MB-435 cells, and LDH-A stability appeared to be elevated after taxol administration, indicating that LDH-A might be biomarker for detecting chemosensitivity [114]. In addition, LDH-A silencing or the use of a glycolysis inhibitor oxamate significantly re-sensitized the taxol-resistant cells to taxol treatment. Notably, combination treatment with LDH-A siRNA and oxamate increased sensitivity in taxol-resistant cell viability compared with either agent alone [115]. Also, compared to gemcitabine-sensitive AsPC-1 cells, gemcitabine-resistant cells Panc-1 and Pan-1GemR had more mRNA for many glycolytic enzymes including hexokinase II, glucose phosphate isomerase, 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 4, aldolase, pyruvate kinase M2, and pyruvate dehydrogenase kinase 3. Upon exposure to glucose transporter (GLUT) inhibitor CG-5, the proliferation of Panc-1 and Pan-1GemR was greatly limited, and was accompanied with suppression of Akt-mTOR signaling, activation of amp-activated protein kinase (AMPK), and suppressed expression of a series of oncogenic proteins, such as cyclin D1, Sp1, and Skp2 [15]. Interestingly, glycolytic activity of tumor stromal cells was also reported to contribute to cancer drug resistance. Breast cancer MCF-7 cells are is preliminarily sensitive to tamoxifen. However, when co-culturing breast cancer cells MCF-7 with cancer-associated fibroblasts, MCF-7 cells gained resistance to tamoxifen. Mechanistic studies indicated that glycolytic fibroblasts produced lactate that was sufficient to provide nutrients to support cancer cell survival [116]. Thus, the Warburg effect is correlated to the development of chemoresistance and tumor progression, and elucidation of key modulators involved in the Warburg effect may offer strategies for treating cancer drug resistance.
Considering the significance of Cav-1 in nutrient delivery and cancer development, emerging studies are underscoring the role of Cav-1 in regulating cancer metabolism. Raikar’s group reported that Cav-1 affects expression of glycolytic enzymes phosphofructokinase and aldolase and that co-localization of Cav-1 and phosphofructokinase or aldolase occurred throughout A7r5 cells. Overexpression of Cav-1 shifted phosphofructokinase and aldolase towards the plasma membrane, indicating that Cav-1 may influence activity and cellular distribution of glycolytic enzymes [117]. Also, Cav-1 loss significantly downregulated glucose uptake, intracellular ATP and lactate accumulation in colon cancer cells, whereas Cav-1 overexpression increased glucose uptake and ATP production by stimulating HMGA1-medaited transcription of the glucose transporter SLC2A3/GLUT3, which was reported to play a significant role in the development of drug resistance [118]. Similar findings were also observed in prostate cancer for which Cav-1 overexpression significantly elevated glucose uptake and lactate production, and this was accompanied with upregulation of glucose transporter 3 and hexokinase 2. However, these events did not alter enolase1, LDHA, pyruvate kinase M2, and pyruvate dehydrogenase kinase 1 protein. Mechanistic studies indicated that Cav-1-induced aerobic glycolysis was mediated through the Akt-mTORC1 pathway and independent of β-catenin signaling [119]. Recently, evidence suggests a function for Cav-1 in tumor stroma. Immunohistochemical studies of human breast cancer specimens revealed that biopsies lacking stroma Cav-1 overexpressed key glycolytic enzymes, such as PKM2 and LDH. Meanwhile, transcriptional profiles of Cav-1-negative (-) tumor stroma revealed up-regulation of 238 genes and downregulation of 232 genes compared to Cav-1 positive (+) breast cancer stroma. Notably, a significant gene upregulation of hypoxic target genes (65 transcripts), glycolysis/pyruvate metabolism (15 transcripts), and autophagy (22 transcripts) were involved in the Cav-1-negative (-) tumor stroma [120]. In a Cav-1 knockout mice model, bone-marrow-derived stroma cells upregulated both myofibroblast markers and glycolytic enzymes [121]. Accelerated glycolytic activity of stroma fibroblasts provided nutrients to support cancer cell survival and growth—there was high expression of monocarboxylate transporter 4 (MCT4, for lactate extrusion) in cancer associated fibroblasts, whereas adjacent breast cancer cells expressed MCT1 (for lactate uptake), suggesting metabolic coupling occurred between cancer cells and stromal fibroblasts [116]. Lactate secreted from fibroblasts could be reabsorbed by cancer cells to enter the tricarboxylic acid cycle, and be metabolized into ATP and substrates for DNA or protein synthesis. This hypothesis was validated by increased tumor growth observed after co-injecting breast cancer cells and CL4 fibroblasts into nude mice cancer xenograft [122]. In addition, several studies suggest that stromal Cav-1 loss is an independent factor associated with poor clinical prognosis of breast cancer patients, indicating that stromal Cav-1 loss may assist cancer metastasis or drug resistance [82, 123, 124]. Thus, the effects of Cav-1 on aerobic glycolysis may be opposite in cancer cells and tumor stroma. Combined targeted therapy should be considered when designing novel treatment strategies based on Cav-1 to overcome cancer metastasis or drug resistance. In addition to direct silencing of Cav-1 to target cancer cells, it is necessary to protect Cav-1 from downregulation in tumor stroma. Thus, it is more prudent to combine Cav-1-targeting agents with antibodies against tumor specific antigens, such as EGFR, HER2, and mucins, etc. On the other hand, combining Cav-1 silencing agents with glycolysis inhibitors to prevent glycolytic activation in tumor stroma may also avoid the risk of fueling cancer cell growth.
Cav-1 and autophagy
Autophagy is emerging as a crucial response to metabolic and therapeutic stresses, attempting to maintain cellular homeostasis via eliminating excessive or unnecessary proteins and damaged organelles through lysosome degradation [125]. However, persistent stress can promote extensive autophagy, inducing type II programmed cell death. Increasing evidence suggests that cytotoxic anti-cancer agents including other drugs, tyrosine kinase inhibitors imatinib and cetuximab, tamoxifen and cyclooxygenase inhibitors may induce autophagy in cell culture and animal models [126]. Interestingly, autophagic inhibition opposed camptothecin-induced cytotoxicity in osteosarcoma, whereas autophagic induction by roscovitine assisted cell killing by doxorubicin, indicating that might become a novel strategy to enhance cancer chemosensitivity [127, 128]. Additionally, autophagic induction may overcome drug resistance during cancer therapy. The constitutive expression of ABCB1 in hepatocellular cancer cells was positively linked to Bcl2 and mTOR overexpression, rendering these cells resistant to autophagy [129]. In contrast, autophagic induction by vocamine activated autophagic cell death in doxorubicin-resistant osteosarcoma cells, which was accompanied by decreased ABCB1 expression [130]. Thus, autophagy is important for regulation of cancer drug resistance and molecules that can induce autophagic cell death may be potential targets for regulating chemosensitivity.
At present, genetic screens in yeast have identified a large family of core autophagy-related genes (ATG), such as Atg1, Atg4, LC3/Atg8, and BECN1 (Beclin-1) [131]. Moreover, mTOR also serves as the main regulator of autophagy. Under nutrient deprivation, hypoxia, genomic instability or other stress, mTOR suppression triggers the autophagic cascade and inhibits cell proliferation [132]. Mechanistic work revealed that mTORC1 activation can negatively regulate autophagy by phosphorylating a complex of autophagic proteins such the Unc51-like kinases (ULK1/2), which interfere with the formation of autophagosomes [133]. Furthermore, as a central sensor of cellular nutrient status or energy, AMPK is involved in autophagic regulation and may serve as an upstream regulator of mTORC1 [134]. AMPK activation can subsequently trigger TSC2 to repress mTORC1 and activate autophagy. In addition, research suggests that AMPK may directly phosphorylate multiple sites in ULK1 (S317, S467, S555, T575, S637, and S777) and promote ULK1 function in autophagy. Even with molecular elucidation of autophagic regulation, more detailed signaling networks await exploration.
Intriguingly, recent work indicates a potential correlation between Cav-1 and autophagy. Lay reported that in Cav-1 knockout adipocytes, autophagy marker LC3-II/LC3-I ratios were significantly elevated and this was validated in Cav-1 null mice using electron microscopy and LC3II/LC3-I ratios in adipose tissue [135]. Similar work confirmed that mammary pads of Cav-1 null mice generated a lethal tumor microenvironment and constitutively underwent autophagy by increased oxidative stress and inflammatory cytokines, which provides recycled nutrients to feed cancer cells [136]. Shi’s group also demonstrated that Cav-1 deficiency could induce autophagy via enhancing lysosomal function and autophagosome-lysosome fusion in human breast cancer cells [16]. A recent mechanistic study confirmed that Cav-1 can directly interact with and regulate expression of ATG12-ATG5, an ubiquitin-like conjugation system crucial for autophagosome formation, in lung epithelial Beas-2B cells [137]. Cav-1 deletion increased basal and starvation-induced ATG12-ATG5 and autophagy, whereas mutation of the Cav-1 binding motif on ATG12 disrupted this interaction and augmented autophagy [138]. Thus, Cav-1 loss may cause autophagic flux. Current evidence also explains why Cav-1 expression is restored in metastatic or drug-resistant cancer cells. Thus, Cav-1 re-expression can cause autophagy suppression, which can inhibit lysosomal degradation of ABC transporters, but also block autophagic cell death. Finally Cav-1 enriched cancer cells acquired drug-resistant properties and stem-like characteristics [9, 14]. Although indirect evidence indicates that Cav-1 is positively associated with expression of ABC transporters and a population of stem cells, the role of autophagy in mediating these molecular events requires verification. Meanwhile, autophagic activation by Cav-1 silencing may be a novel strategy for overcoming cancer drug resistance.
Perspective
After drug treatment, cancer cells must activate self-protection systems for survival. Cav-1, as a key membrane-associated protein hub guiding signal transduction and nutrient delivery, may be a first-line response to chemotherapy. Studies indicate an elevation of Cav-1 in cancer cells after chemotherapeutic exposure [47]. Meanwhile, exogenous elevation or knockdown of Cav-1 expression confirmed a critical role for Cav-1 in mediating cancer chemosensitivity [9]. Other than this drug-induced response, Cav-1 may be active after radiotherapy, fluid shear stress, oxidative stress, high osmolarity, and ultraviolet exposure [139, 140]. Therefore, Cav-1 may be a stress-related molecule. Studies indicate that Cav-1 is closely associated with stress signals such as AMPK. Cav-1 silencing could induce autophagy in an AMPK-dependent pathway. Additionally, the AMPK activator metformin required Cav-1 to induce AMPK phosphorylation and an AMP/ATP ratio increase in non-small-cell lung cancer [78, 141]. Notably, Cav-1-null mice had lower body temperatures after fasting or fasting/cold treatment and this was accompanied by a significant reduction of AMPK expression [142]. Moreover, the lifespan of Cav-1-null mice was reduced, indicating that a stress-response for these mice may be weaker after Cav-1 knockout [143]. Therefore, Cav-1 might be a key stress-related molecule protecting cells from hazardous stimulus. This explains Cav-1’s behavior in cancer development. During cancer initiation, malignant transformation may be accelerated due to Cav-1 loss and this would sensitize normal cells to oncogenic events. In contrast, when cancer progress to later stage and is treated, expression of Cav-1 would be upregulated to protect cancer cells escape death by speeding aerobic glycolysis, increasing stem cell populations or overexpressing ABC transporters (Figure 3) [46,144,145,146]. Nevertheless, this model may be only restricted to Cav-1 enriched normal cells such as mammary and lung epithelial cells. Since not all normal cells express Cav-1 and not all cells with low/no Cav-1 expression are primary cancer cells, Cav-1 might be just one of critical molecules responsible for assisting cells defending harmful stress. Beside Cav-1, a number of molecules including glucose regulated proteins (GRP78, GRP94, GRP170) [147], some miRNAs (miRNA-25, miRNA-30c, miRNA-23a, miRNA-200b, etc) [148] or LncRNAs (UCA1, H19, MRUL, etc) [149], were also recorded as stress-response targets. Therefore, the value of Cav-1 as a diagnostic or therapeutic target to improve chemoresistance is still needed to be validated on independent cancer type. On the other hand, molecular networks underlying Cav-1 and other stress signals must be understood by using conditional knockout models and xenografts to study its precise role in carcinogenesis or chemosensitization process in the future.
Taken together, an improved understanding of Cav-1 regulation and its role in chemosensitivity modulation is required to improve cancer prevention and therapy. Drugs targeting Cav-1 are expected to be developed and thereby provide powerful tools to explore Cav-1 molecular biology. Furthermore, larger clinical studies are needed to confirm the pathological significance of Cav-1 in cancer onset and response to conventional therapy.
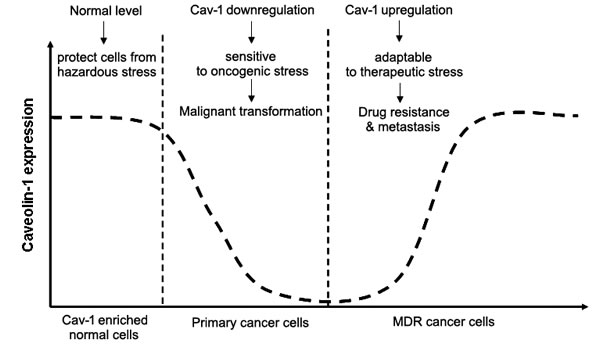
Figure 3: Cav-1 may act as a stress-response signaling during cancer development in Cav-1 enriched cells. When Cav-1 is maintained at the normalized level, it protects Cav-1 enriched normal cells from hazardous damage. However, when Cav-1 declines below the threshold, cells will become sensitive to oncogenic stress and initiate malignant transformation. With cancer progression to the advanced stage or is treated with cytotoxic agents, Cav-1 may re-overexpress to help cancer cells escape death, causing drug resistance and metastasis.
Abbreviations
MDR: multidrug resistance; abc: atp binding cassette; mapk: mitogen-activated protein kinase; cscs: cancer stem cells; cav-1: caveolin-1; csd: cav-1 scaffolding domain; egf: epidermal growth factor; pdgf: platelet-derived growth factor; pi3k: phosphatidylinositol 3-kinase; pdk1: 3-phosphoinositide-dependent protein kinase-1; dlc-1: deleted in liver cancer 1; nsclc: non-small cell lung cancer; aacr: American association for cancer research; aldh1: aldehyde dehydrogenase 1; mscs: mesenchymal stem cells; fdg-pet: fluorodeoxyglucose positron emission tomography; glut: glucose transporter; mct: monocarboxylate transporter; ampk: amp-activated protein kinase;
Acknowledgments
This work was supported by the National Natural science Foundation of China (81402173) and Pearl River S&T Nova Program of Guangzhou (201506010098).
Conflicts of Interests
No potential conflicts of interest exist.
References
1. Wilson TR, Longley DB, Johnston PG. Chemoresistance in solid tumours. Ann Oncol 2006; 17 Suppl 10: x315-24.
2. Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer 2013; 13(10):714-26.
3. Dean M. ABC transporters, drug resistance, and cancer stem cells. J Mammary Gland Biol Neoplasia 2009; 14(1):3-9.
4. Szakács G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov 2006; 5(3): 219-34.
5. Gillet JP, Gottesman MM. Advances in the molecular detection of ABC transporters involved in multidrug resistance in cancer. Curr Pharm Biotechnol 2011; 12(4):686-92.
6. Choi YH, Yu AM. ABC transporters in multidrug resistance and pharmacokinetics, and strategies for drug development. Curr Pharm Des 2014; 20(5):793-807.
7. Boscher C, Nabi IR. Cav-1: role in cell signaling. Adv Exp Med Biol 2012; 729:29-50.
8. Williams TM, Lisanti MP. Caveolin-1 in oncogenic transformation, cancer and metastasis. Am J Physiol Cell Physio 2005; 288:494-506.
9. Shatz M, Liscovitch M. Cav-1 and cancer multidrug resistance: coordinate regulation of pro-survival proteins? Leuk Res 2004; 28(9):907-8.
10. Pang A, Au WY, Kwong YL. Cav-1 gene is coordinately regulated with the multidrug resistance 1 gene in normal and leukemic bone marrow. Leuk Res 2004; 28(9):973-7.
11. Bélanger MM, Gaudreau M, Roussel E, Couet J. Role of caveolin-1 in etoposide resistance development in A549 lung cancer cells. Cancer Biol Ther 2004; 3(10):954-9.
12. Cai C, Chen J. Overexpression of caveolin-1 induces alteration of multidrug resistance in Hs578T breast adenocarcinoma cells. Int J Cancer, 2004; 111(4):522-9.
13. Shatz M, Liscovitch M. Cav-1: a tumor-promoting role in human cancer. Int J Radiat Biol 2008; 84(3):177-89.
14. Wang Z, Wang N, Li W, Liu P, Chen Q, Situ H, Zhong S, Guo L, Lin Y, Shen J, Chen J. Cav-1 mediates chemoresistance in breast cancer stem cells via β-catenin/ABCG2 signaling pathway. Carcinogenesis 2014; 35(10):2346-56.
15. Martinez-Outschoorn UE, Lin Z, Ko YH, Goldberg AF, Flomenberg N, Wang C, Pavlides S, Pestell RG, Howell A, Sotgia F, Lisanti MP. Understanding the metabolic basis of drug resistance: therapeutic induction of the Warburg effect kills cancer cells. Cell Cycle 2011; 10(15):2521-8.
16. Shi Y, Tan SH, Ng S, Zhou J, Yang ND, Koo GB, Mcmahon KA, Parton RG, Hill MM, Del Pozo MA, Kim YS, Shen HM. Critical role of CAV1/caveolin-1 in cell stress responses in human breast cancer cells via modulation of lysosomal function and autophagy. Autophagy 2015; 11(5):769-84.
17. Sargiacomo M, Scherer PE, Tang Z, Kubler E, Song KS, Sanders MC, Lisanti MP. Oligomeric structure of caveolin: implications for caveolae membrane organization. Proc Natl Acad Sci U S A 1995; 92(20):9407-11.
18. Fujimoto T, Kogo H, Nomura R, Une T. Isoforms of Cav-1 and caveolar structure. J Cell Sci 2000; 113 Pt 19:3509-17.
19. Fridolfsson HN, Roth DM, Insel PA, Patel HH. Regulation of intracellular signaling and function by caveolin. FASEB J 2014; pii: fj.14-252320.
20. Lee J, Glover KJ. The transmembrane domain of Cav-1 exhibits a helix-break-helix structure. Biochim Biophys Acta 2012; 1818(5):1158-64.
21. Nomura R, Fujimoto T. Tyrosine-phosphorylated Cav-1: immunolocalization and molecular characterization. Mol Biol Cell 1999; 10(4):975-86.
22. Pike LJ. Growth factor receptors, lipid rafts and caveolae: an evolving story. Biochim Biophys Acta 2005; 1746(3):260-73.
23. Song KS, Li Shengwen, Okamoto T, Quilliam LA, Sargiacomo M, Lisanti MP. Co-purification and direct interaction of Ras with caveolin, an integral membrane protein of caveolae microdomains. Detergent-free purification of caveolae microdomains. J Biol Chem 1996; 271(16):9690-7.
24. Liu P, Ying Y, Ko YG, Anderson RG. Localization of platelet-derived growth factor-stimulated phosphorylation cascade to caveolae. J Biol Chem 1996; 271(17):10299-303.
25. Mineo C, James GL, Smart EJ, Anderson RG. Localization of epidermal growth factor-stimulated Ras/Raf-1 interaction to caveolae membrane. J Biol Chem 1996; 271(20):11930-5.
26. Engelman JA, Wykoff CC, Yasuhara S, Song KS, Okamoto T, Lisanti MP. Recombinant expression of caveolin-1 in oncogenically transformed cells abrogates anchorage-independent growth. J Biol Chem 1997; 272(26):16374-81.
27. Engelman JA, Chu C, Lin A, Jo H, Ikezu T, Okamoto T, Kohtz DS, Lisanti MP. Caveolin-mediated regulation of signaling along the p42/44 MAP kinase cascade in vivo. A role for the caveolin-scaffolding domain. FEBS Lett 1998; 428(3):205-11.
28. Engelman JA, Lee RJ, Karnezis A, Bearss DJ, Webster M, Siegel P, Muller WJ, Windle JJ, Pestell RG, Lisanti MP. Reciprocal regulation of neu tyrosine kinase activity and caveolin-1 protein expression in vitro and in vivo. Implications for human breast cancer. J Biol Chem 1998; 273(32):20448-55.
29. Li S, Couet J, Lisanti MP. Src tyrosine kinases, Gasubunits, and H-Ras share a common membraneanchored scaffolding protein, caveolin. Caveolin binding negatively regulates the auto-activation of Src tyrosine kinases. J Biol Chem 1996; 271:29182–90.
30. Wei Y, Yang X, Liu Q, Wilkins JA, Chapman HA. A role for caveolin and the urokinase receptor in integrinmediated adhesion and signaling. J Cell Biol 1999;144: 1285–94.
31. Cantiani L, Manara MC, Zucchini C, De Sanctis P, Zuntini M, Valvassori L, Serra M, Olivero M, Di Renzo MF, Colombo MP, Picci P, Scotland K. Caveolin-1 reduces osteosarcoma metastasis by inhibiting c-Src activity and met signaling. Cancer Res 2007; 67(16):7675-85.
32. Shack S, Wang XT, Kokkonen GC, Gorospe M, Longo DL, Holbrook NJ. Caveolin-induced activation of the phosphatidylinositol 3-kinase/Akt pathway increases arsenite cytotoxicity. Mol Cell Biol 2003; 23(7):2407-14.
33. Torres VA, Tapia JC, Rodriguez DA, Lladser A, Arredondo C, Leyton L, Quest AF. E-cadherin is required for caveolin-1-mediated down-regulation of the inhibitor of apoptosis protein survivin via reduced beta-catenin-Tcf/Lef-dependent transcription. Mol Cell Biol 2007; 27(21):7703-17.
34. Karuppiah K, Druhan LJ, Chen CA, Smith T, Zwejer JL, Sessa WC, Cardounel AJ. Suppression of eNOS-derived superoxide by caveolin-1: a biopterin-dependent mechanism. Am J Physiol Heart Circ Physiol 2011; 301(3):H903-11.
35. Xie Z, Zeng X, Waldman T, Glazer RI. Transformation of mammary epithelial cells by 3-phosphoinositide- dependent protein kinase-1 activates beta-catenin and c-Myc, and down-regulates caveolin-1. Cancer Res 2003; 63(17):5370-5.
36. Razani B, Altschuler Y, Zhu L, Pestell RG, Mostov KE, Lisanti MP. Caveolin-1 expression is down-regulated in cells transformed by the human papilloma virus in a p53-dependent manner. Replacement of caveolin-1 expression suppresses HPV-mediated cell transformation. Biochemistry 2000; 39(45):13916-24.
37. Huertas-Martinez J, Rello-Varona S, Herrero-Martin D, Barrau I, García-Monclús S, Sáinz-Jaspeado M, Lagares-Tena L, Núñez-Álvarez Y, Mateo-Lozano S, Mora J, Roma J, Toran N, Moran S, et al. Caveolin-1 is down-regulated in alveolar rhabdomyosarcomas and negatively regualtes tumor growth. Oncotarget 2014; 5(20):9744-55.
38. Glait C, Ravid D, Lee SW, Liscovitch M, Werner H. Caveolin-1 controls BRCA1 gene expression and cellular localization in human breast cancer cells. FEBS Lett 2006; 580(22):5268-74.
39. Wang Y, Yu J, Zhan Q. BRCA1 regulates caveolin-1 expression and inhibits cell invasiveness. Biochem Biophys Res Commun 2008; 370(2):201-6.
40. Xia H, Khalil W, Kahm J, Jessurun J, Kleidon J, Henke CA. Pathologic caveolin-1 regulation of PTEN in idiopathic pulmonary fibrosis. Am J Pathol 2010; 176(6):2626-37.
41. Williams TM, Lee H, Cheung MW, Cohen AW, Razani B, Iyengar P, Scherer PE, Pestell RG, Lisanti MP. Combined loss of INK4a and caveolin-1 synergistically enhances cell proliferation and oncogene-induced tumorigenesis: role of INK4a/CAV-1 in mammary epithelial cell hyperplasia. J Biol Chem 2004; 279(23):24745-56.
42. Du X, Qian X, Papageorge A, Schetter AJ, Vass WC, Liu X, Braverman R, Robles AI, Lowy DR.. Functional interaction of tumor suppressor DLC1 and caveolin-1 in cancer cells. Cancer Res 2012; 72(17):4405-16.
43. Mercier I, Casimiro MC, Wang C, Rosenberg AL, Quong J, Minkeu A, Allen KG, Danilo C, Sotgia F, Bonuccelli G, Jasmin JF, Xu H, Bosco E, et al. Human breast cancer-associated fibroblasts (CAFs) show caveolin-1 downregulation and RB tumor suppressor functional inactivation: Implications for the response to hormonal therapy. Cancer Biol Ther 2008; 7(8):1212-25.
44. Williams TM, Cheung MW, Park DS, Razani B, Cohen AW, Muller WJ, Di Vizio D, Chopra NG, Pestell RG, Lisanti MP. Loss of caveolin-1 gene expression accelerates the development of dysplastic mammary lesions in tumor-prone transgenic mice. Mol Biol Cell 2003;14(3):1027-42.
45. Williams TM, Medina F, Badano I, Hazan RB, Hutchinson J, Muller WJ, Chopra NG, Scherer PE, Pestell RG, Lisanti MP. Caveolin-1 gene disruption promotes mammary tumorigenesis and dramatically enhances lung metastasis in vivo. Role of Cav-1 in cell invasiveness and matrix metalloproteinase (MMP-2/9) secretion. J Biol Chem 2004; 279(49):51630-46.
46. Mercier I, Lisanti MP. Caveolin-1 and breast cancer: a new clinical perspective. Adv Exp Med Biol 2012; 729:83-94.
47. Quest AF, Gutierrez-Pajares JL, Torres VA. Caveolin-1: an ambiguous partner in cell signalling and cancer. J Cell Mol Med 2008; 12(4):1130-50.
48. Tanase CP, Dima S, Mihai M, Raducan E, Nicolescu MI, Albulescu L, Voiculescu B, Dumitrascu T, Cruceru LM, Leabu M, Popescu I, Hinescu ME. Caveolin-1 overexpression correlates with tumour progression markers in pancreatic ductal adenocarcinoma. J Mol Histol 2009; 40(1):23-9.
49. Suzuoki M, Miyamoto M, Kato K, Hiraoka K, Oshikiri T, Nakakubo Y, Fukunaga A, Shichinohe T, Shinohara T, Itoh T, Kondo S, Katoh H. Impact of caveolin-1 expression on prognosis of pancreatic ductal adenocarcinoma. Br J Cancer 2002; 87(10):1140-4.
50. Campbell L, Al-Jayyoussi G, Gutteridge R, Gumbleton N, Griffiths R, Gumbleton S, Smith MW, Griffiths DF, Gumbleton M. Caveolin-1 in renal cell carcinoma promotes tumour cell invasion, and in co-operation with pERK predicts metastases in patients with clinically confined disease. Transl Med 2013; 11:255.
51. Tang Y, Zeng X, He F, Liao Y, Qian N, Toi M. Caveolin-1 is related to invasion, survival, and poor prognosis in hepatocellular cancer. Med Oncol 2012; 29(2):977-84.
52. Pinilla SM, Honrado E, Hardisson D, Benítez J, Palacios J. Caveolin-1 expression is associated with a basal-like phenotype in sporadic and hereditary breast cancer. Breast Cancer Res Treat 2006; 99(1):85-90.
53. Van den Eynden GG, Van Laere SJ, Van der Auwera I, Merajver SD, Van Marck EA, van Dam P, Vermeulen PB, Dirix LY, van Golen KL. Overexpression of caveolin-1 and -2 in cell lines and in human samples of inflammatory breast cancer. Breast Cancer Res Treat 2006; 95(3):219-28.
54. Hu MY, Lam CT, Liu KD, Xu Z, Fatima S, Su YC, Tsang F, Chen J, Pang JZ, Qin LX, Luk JM. Proteomic identification of a monoclonal antibody recognizing caveolin-1 in hepatocellular carcinoma with metastatic potential. Protein Pept Lett 2009; 16(5):479-85.
55. Zajchowski DA, Bartholdi MF, Gong Y, Webster L, Liu HL, Munishkin A, Beauheim C, Harvey S, Ethier SP, Johnson PH. Identification of gene expression profiles that predict the aggressive behavior of breast cancer cells. Cancer Res 2001; 61(13):5168-78.
56. Williams TM, Hassan GS, Li J, Cohen AW, Medina F, Frank PG, Pestell RG, Di Vizio D, Loda M, Lisanti MP. Caveolin-1 promotes tumor progression in an autochthonous mouse model of prostate cancer: genetic ablation of Cav-1 delays advanced prostate tumor development in tramp mice. J Biol Chem 2005; 280(26):25134-45.
57. Hayashi K, Matsuda S, Machida K, Yamamoto T, Fukuda Y, Nimura Y, Hayakawa T, Hamaguchi M. Invasion activating Cav-1 mutation in human scirrhous breast cancers. Cancer Res 2001; 61(6):2361-4.
58. Lee H, Park DS, Razani B, Russell RG, Pestell RG, Lisanti MP. Cav-1 mutations (P132L and null) and the pathogenesis of breast cancer: Cav-1 (P132L) behaves in a dominant-negative manner and Cav-1 (-/-) null mice show mammary epithelial cell hyperplasia. Am J Pathol 2002; 161(4):1357-69.
59. Bonuccelli G, Casimiro MC, Sotgia F, Wang C, Liu M, Katiyar S, Zhou J, Dew E, Capozza F, Daumer KM, Minetti C, Milliman JN, Alpy F, et al. Cav-1 (P132L), a common breast cancer mutation, confers mammary cell invasiveness and defines a novel stem cell/metastasis-associated gene signature. Am J Pathol 2009; 174(5):1650-62.
60. Chen ST, Lin SY, Yeh KT, Chang JY, Lin TH, Chen JC, Yuang SS, Chang JG.. Mutational, epigenetic and expressional analyses of Cav-1 gene in breast cancers. Int J Mol Med 2004; 14(4):577-82.
61. Koike S, Kodera Y, Nakao A, Iwata H, Yatabe Y. Absence of the Cav-1 P132L mutation in cancers of the breast and other organs. J Mol Diagn 2010; 12(5):712-7.
62. Patani N, Lambros MB, Natrajan R, Dedes KJ, Geyer FC, Ward E, Martin LA, Dowsett M, Reis-Filho JS. Non-existence of Cav-1 gene mutations in human breast cancer. Breast Cancer Res Treat 2012; 131(1):307-10.
63. Ferraldeschi R, Latif A, Clarke RB, Spence K, Ashton G, O’Sullivan J, Evans DG, Howell A, Newman WG. Lack of Cav-1 (P132L) somatic mutations in breast cancer. Breast Cancer Res Treat 2012; 132(3):1185-6.
64. Fiucci G, Ravid D, Reich R, Liscovitch M. Caveolin-1 inhibits anchorage-independent growth, anoikis and invasiveness in MCF-7 human breast cancer cells. Oncogene 2002; 21(15):2365-75.
65. Feng S, Wang Y, Wang X, Wang Z, Cui Y, Liu J, Zhao C, Jin M, Zou W. Caveolin-1 gene silencing promotes the activation of PI3K/AKT dependent on Eralpha36 and the transformation of MCF10ACE. Sci China Life Sci 2010; 53(5):598-605.
66. Racine C, Bélanger M, Hirabayashi H, Boucher M, Chakir J, Couet J. Reduction of caveolin 1 gene expression in lung carcinoma cell lines. Biochem Biophys Res Commun 1999; 255(3):580-6.
67. Gao X, Sun Y, Huang L, Chen XY, Zhang KL, Kong QY, Liu J, Li H. Down-regulation of caveolin-1 in gastric carcinoma and its clinical biological significance. Ai Zheng 2005; 24(3):311-6.
68. Barresi V, Buttarelli FR, Vitarelli EE, Arcella A, Antonelli M, Giangaspero F. Caveolin-1 expression in diffuse gliomas: correlation with the proliferation index, epidermal growth factor receptor, p53, and 1p/19q status. Hum Pathol 2009; 40(12):1738-46.
69. Han F, Gu D, Chen Q, Zhu H. Caveolin-1 acts as a tumor suppressor by down-regulating epidermal growth factor receptor-mitogen-activated protein kinase signaling pathway in pancreatic carcinoma cell lines. Pancreas 2009; 38(7):766-74.
70. Galbiati F, Volonté D, Liu J, Capozza F, Frank PG, Zhu L, Pestell RG, Lisanti MP. Caveolin-1 expression negatively regulates cell cycle progression by inducing G(0)/G(1) arrest via a p53/p21(WAF1/Cip1)-dependent mechanism. Mol Biol Cell 2001; 12(8):2229-44.
71. Ho CC, Huang PH, Huang HY, Chen YH, Yang PC, Hsu SM. Up-regulated caveolin-1 accentuates the metastasis capability of lung adenocarcinoma by inducing filopodia formation. Am J Pathol 2002; 161(5):1647-56.
72. Zhou H, Jia L, Wang S, Wang H, Chu H, Hu Y, Cao J, Zhang J. Divergent expression and roles for caveolin-1 in mouse hepatocarcinoma cell lines with varying invasive ability. Biochem Biophys Res Commun 2006; 345(1):486-94.
73. Davidson B, Nesland JM, Goldberg I, Kopolovic J, Gotlieb WH, Bryne M, Ben-Baruch G, Berner A, Reich R. Caveolin-1 expression in advanced-stage ovarian carcinoma--a clinicopathologic study. Gynecol Oncol 2001; 81(2):166-71.
74. Moon KC, Lee GK, Yoo SH, Jeon YK, Chung JH, Han J, Chung DH. Expression of caveolin-1 in pleomorphic carcinoma of the lung is correlated with a poor prognosis. Anticancer Res 2005; 25(6C):4631-7.
75. Felicetti F, Parolini I, Bottero L, Fecchi K, Errico MC, Raggi C, Biffoni M, Spadaro F, Lisanti MP, Sargiacomo M, Carè A. Caveolin-1 tumor-promoting role in human melanoma. Int J Cancer 2009; 125(7):1514-22.
76. Nohata N, Hanazawa T, Kikkawa N, Mutallip M, Fujimura L, Yoshino H, Kawakami K, Chiyomaru T, Enokida H, Nakagawa M, Okamoto Y, Seki N. Caveolin-1 mediates tumor cell migration and invasion and its regulation by miR-133a in head and neck squamous cell carcinoma. Int J Oncol 2011; 38(1):209-17.
77. Shankar J, Wiseman SM, Meng F, Kasaian K, Strugnell S, Mofid A, Gown A, Jones SJ, Nabi IR. Coordinated expression of galectin-3 and caveolin-1 in thyroid cancer. J Pathol 2012; 228(1):56-66.
78. Ha TK, Her NG, Lee MG, Ryu BK, Lee JH, Han J, Jeong SI, Kang MJ, Kim NH, Kim HJ, Chi SG. Caveolin-1 increases aerobic glycolysis in colorectal cancers by stimulating HMGA1-mediated GLUT3 transcription. Cancer Res 2012; 72(16):4097-109.
79. Nam KH, Lee BL, Park JH, Kim J, Han N, Lee HE, Kim MA, Lee HS, Kim WH. Caveolin 1 expression correlates with poor prognosis and focal adhesion kinase expression in gastric cancer. Pathobiology 2013; 80(2):87-94.
80. Zhang W, Razani B, Altschuler Y, Bouzahzah B, Mostov KE, Pestell RG, Lisanti MP.. Caveolin-1 inhibits epidermal growth factor-stimulated lamellipod extension and cell migration in metastatic mammary adenocarcinoma cells (MTLn3). Transformation suppressor effects of adenovirus-mediated gene delivery of caveolin-1. J Biol Chem 2000; 275(27):20717-25.
81. Luanpitpong S, Wang L, Stueckle TA, Tse W, Chen YC, Rojanasakul Y. Caveolin-1 regulates lung cancer stem-like cell induction and p53 inactivation in carbon nanotube-driven tumorigenesis. Oncotarget 2014; 5(11):3541-54.
82. Shan T, Lu H, Ji H, Li Y, Guo J, Chen X, Wu T. Loss of stromal Cav-1 expression: a novel tumor microenvironment biomarker that can predict poor clinical outcomes for pancreatic cancer. PLoS One 2014; 9(6):e97239.
83. Yang CP, Galbiati F, Volonte D, Horwitz SB, Lisanti MP. Upregulation of caveolin-1 and caveolae organelles in Taxol-resistant A549 cells. FEBS Lett 1998; 439(3):368-72.
84. Lavie Y, Fiucci G, Liscovitch M. Up-regulation of caveolae and caveolar constituents in multidrug-resistant cancer cells. J Biol Chem 1998; 273(49):32380-3.
85. Tirado OM, MacCarthy CM, Fatima N, Villar J, Mateo-Lozano S, Notario V. Caveolin-1 promotes resistance to chemotherapy-induced apoptosis in Ewing’s sarcoma cells by modulating PKCalpha phosphorylation. Int J Cancer 2010; 126(2):426-36.
86. Herzog M, Storch CH, Gut P, Kotlyar D, Füllekrug J, Ehehalt R, Haefeli WE, Weiss J. Knockdown of caveolin-1 decreases activity of breast cancer resistance protein (BCRP/ABCG2) and increases chemotherapeutic sensitivity. Naunyn Schmiedebergs Arch Pharmacol 2011; 383(1):1-11.
87. Sáinz-Jaspeado M, Lagares-Tena L, Lasheras J, Navid F, Rodriguez-Galindo C, Mateo-Lozano S, Notario V, Sanjuan X, Garcia Del Muro X, Fabra A, Tirado OM. Caveolin-1 modulates the ability of Ewing’s sarcoma to metastasize. Mol Cancer Res 2010; 8(11):1489-500.
88. Li J, Hassan GS, Williams TM, Minetti C, Pestell RG, Tanowitz HB, Frank PG, Sotgia F, Lisanti MP. Loss of caveolin-1 causes the hyper-proliferation of intestinal crypt stem cells, with increased sensitivity to whole body gamma-radiation. Cell Cycle 2005; 4(12):1817-25.
89. Tahir SA, Kurosaka S, Tanimoto R, Goltsov AA, Park S, Thompson TC. Serum caveolin-1, a biomarker of drug response and therapeutic target in prostate cancer models. Cancer Biol Ther 2013; 14(2):117-26.
90. Chen D, Shen C, Du H, Zhou Y, Che G. Duplex value of caveolin-1 in non-small cell lung cancer: a meta analysis. Fam Cancer 2014; 13(3):449-57.
91. Yuan G, Regel I, Lian F, Friedrich T, Hitkova I, Hofheinz RD, Ströbel P, Langer R, Keller G, Röcken C, Zimmermann W, Schmid RM, Ebert MP. WNT6 is a novel target gene of caveolin-1 promoting chemoresistance to epirubicin in human gastric cancer cells. Oncogene 2013; 32(3):375-87.
92. Brodie SA, Lombardo C, Li G, Kowalski J, Gandhi K, You S, Khuri FR, Marcus A, Vertino PM, Brandes JC. Aberrant promoter methylation of caveolin-1 is associated with favorable response to taxane-platinum combination chemotherapy in advanced NSCLC. PLoS One 2014; 9(9):e107124.
93. Hodges LM, Markova SM, Chinn LW, Gow JM, Kroetz DL, Klein TE, Altman RB. Very important pharmacogene summary: ABCB1 (MDR1, P-glycoprotein). Pharmacogenet Genomics 2011; 21(3):152-61.
94. Dohse M, Scharenberg C, Shukla S, Robey RW, Volkmann T, Deeken JF, Brendel C, Ambudkar SV, Neubauer A, Bates SE. Comparison of ATP-binding cassette transporter interactions with the tyrosine kinase inhibitors imatinib, nilotinib, and dasatinib. Drug Metab Dispos 2010; 38(8):1371-80.
95. Leonard GD, Fojo T, Bates SE. The role of ABC transporters in clinical practice. Oncologist 2003; 8(5):411-24.
96. Hinrichs JW, Klappe K, Hummel I, Kok JW. ATP-binding cassette transporters are enriched in non-caveolar detergent-insoluble glycosphingolipid-enriched membrane domains (DIGs) in human multidrug-resistant cancer cells. J Biol Chem 2004; 279(7):5734-8.
97. Radeva G, Perabo J, Sharom FJ. P-Glycoprotein is localized in intermediate-density membrane microdomains distinct from classical lipid rafts and caveolar domains. FEBS J 2005; 272(19):4924-37.
98. Davidson B, Goldberg I, Givant-Horwitz V, Nesland JM, Berner A, Bryne M, Risberg B, Kopolovic J, Kristensen GB, Tropé CG, van de Putte G, Reich R. Caveolin-1 expression in ovarian carcinoma is MDR1 independent. Am J Clin Pathol 2002; 117(2):225-34.
99. Ni Z, Bikadi Z, Rosenberg MF, Mao Q. Structure and function of the human breast cancer resistance protein (BCRP/ABCG2). Curr Drug Metab 2010; 11(7):603-17.
100. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 1997; 3(7):730-7.
101. Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, Visvader J, Weissman IL, Wahl GM. Cancer stem cells--perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res 2006; 66(19):9339-44.
102. Bunting KD. ABC transporters as phenotypic markers and functional regulators of stem cells. Stem Cells 2002; 20(1):11-20.
103. Neumeister V, Agarwal S, Bordeaux J, Camp RL, Rimm DL. In situ identification of putative cancer stem cells by multiplexing ALDH1, CD44, and cytokeratin identifies breast cancer patients with poor prognosis. Am J Pathol 2010; 176(5):2131-8.
104. Tanei T, Morimoto K, Shimazu K, Kim SJ, Tanji Y, Taguchi T, Tamaki Y, Noguchi S.. Association of breast cancer stem cells identified by aldehyde dehydrogenase 1 expression with resistance to sequential Paclitaxel and epirubicin-based chemotherapy for breast cancers. Clin Cancer Res 2009; 15(12):4234-41.
105. Ma YC, Yang JY, Yan LN. Relevant markers of cancer stem cells indicate a poor prognosis in hepatocellular carcinoma patients: a meta-analysis. Eur J Gastroenterol Hepatol 2013; 25(9):1007-16.
106. Baker N, Zhang G, You Y, Tuan RS. Caveolin-1 regulates proliferation and osteogenic differentiation of human mesenchymal stem cells. J Cell Biochem 2012; 113(12):3773-87.
107. Park JH, Lee MY, Han HJ. A potential role for caveolin-1 in estradiol-17beta-induced proliferation of mouse embryonic stem cells: involvement of Src, PI3K/Akt, and MAPKs pathways. Int J Biochem Cell Biol 2009; 41(3):659-65
108. Park DS, Lee H, Frank PG, Razani B, Nguyen AV, Parlow AF, Russell RG, Hulit J, Pestell RG, Lisanti MP. Caveolin-1-deficient mice show accelerated mammary gland development during pregnancy, premature lactation, and hyperactivation of the Jak-2/STAT5a signaling cascade. Mol Biol Cell 2002; 13(10):3416-30.
109. Lee MY, Ryu JM, Lee SH, Park JH, Han HJ. Lipid rafts play an important role for maintenance of embryonic stem cell self-renewal. J Lipid Res 2010; 51(8):2082-9.
110. Yongsanguanchai N, Pongrakhananon V, Mutirangura A, Rojanasakul Y, Chanvorachote P. Nitric oxide induces cancer stem cell-like phenotypes in human lung cancer cells. Am J Physiol Cell Physiol 2015; 308(2):C89-100.
111. Kim JW, Dang CV. Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res 2006; 66:8927-30.
112. Xie H, Valera VA, Merino MJ, Amato AM, Signoretti S, Linehan WM, Sukhatme VP, Seth P. LDH-A inhibition, a therapeutic strategy for treatment of hereditary leiomyomatosis and renal cell cancer. Mol Cancer Ther 2009; 8(3):626-35.
113. Wang ZY, Loo TY, Shen JG, Wang N, Wang DM, Yang DP, Mo SL, Guan XY, Chen JP.. LDH-A silencing suppresses breast cancer tumorigenicity through induction of oxidative stress mediated mitochondrial pathway apoptosis. Breast Cancer Res Treat 2012; 131(3):791-800.
114. Zhou M, Zhao Y, Ding Y, Liu H, Liu Z, Fodstad O, Riker AI, Kamarajugadda S, Lu J, Owen LB, Ledoux SP, Tan M. Warburg effect in chemosensitivity: targeting lactate dehydrogenase-A re-sensitizes taxol-resistant cancer cells to taxol. Mol Cancer 2010; 9:33.
115. Lai IL, Chou CC, Lai PT, Fang CS, Shirley LA, Yan R, Mo X, Bloomston M, Kulp SK, Bekaii-Saab T, Chen CS. Targeting the Warburg effect with a novel glucose transporter inhibitor to overcome gemcitabine resistance in pancreatic cancer cells. Carcinogenesis 2014; 35(10):2203-13.
116. Witkiewicz AK, Whitaker-Menezes D, Dasgupta A, Philp NJ, Lin Z, Gandara R, Sneddon S, Martinez-Outschoorn UE, Sotgia F, Lisanti MP. Using the “reverse Warburg effect” to identify high-risk breast cancer patients: stromal MCT4 predicts poor clinical outcome in triple-negative breast cancers. Cell Cycle 2012; 11(6):1108-17.
117. Raikar LS, Vallejo J, Lloyd PG, Hardin CD. Overexpression of caveolin-1 results in increased plasma membrane targeting of glycolytic enzymes: the structural basis for a membrane associated metabolic compartment. J Cell Biochem 2006; 98(4):861-71.
118. Ha TK, Chi SG. CAV1/caveolin 1 enhances aerobic glycolysis in colon cancer cells via activation of SLC2A3/GLUT3 transcription. Autophagy 2012; 8(11):1684-5.
119. Tahir SA, Yang G, Goltsov A, Song KD, Ren C, Wang J, Chang W, Thompson TC. Caveolin-1-LRP6 signaling module stimulates aerobic glycolysis in prostate cancer. Cancer Res 2013; 73(6):1900-11.
120. Witkiewicz AK, Kline J, Queenan M, Brody JR, Tsirigos A, Bilal E, Pavlides S, Ertel A, Sotgia F, Lisanti MP. Molecular profiling of a lethal tumor microenvironment, as defined by stromal caveolin-1 status in breast cancers. Cell Cycle 2011; 10(11):1794-809.
121. Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, Casimiro MC, Wang C, Fortina P, Addya S, Pestell RG, Martinez-Outschoorn UE, Sotgia F, et al. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009; 8:3984–4001.
122. Sotgia F, Martinez-Outschoorn UE, Pavlides S, Howell A, Pestell RG, Lisanti MP. Understanding the Warburg effect and the prognostic value of stromal caveolin-1 as a marker of a lethal tumor microenvironment. Breast Cancer Res 2011; 13(4):213.
123. Zhao Z, Han FH, Yang SB, Hua LX, Wu JH, Zhan WH. Loss of stromal caveolin-1 expression in colorectal cancer predicts poor survival. World J Gastroenterol 2015; 21(4):1140-7.
124. Wu KN, Queenan M, Brody JR, Potoczek M, Sotgia F, Lisanti MP, Witkiewicz AK.. Loss of stromal caveolin-1 expression in malignant melanoma metastases predicts poor survival. Cell Cycle 2011; 10(24):4250-5.
125. White E. The role for autophagy in cancer. J Clin Invest 2015; 125(1):42-6.
126. Sui X, Chen R, Wang Z, Huang Z, Kong N, Zhang M, Han W, Lou F, Yang J, Zhang Q, Wang X, He C, Pan H. Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death Dis 2013; 4: e838.
127. Hollomon MG, Gordon N, Santiago-O’Farrill JM, Kleinerman ES. Knockdown of autophagy-related protein 5, ATG5, decreases oxidative stress and has an opposing effect on camptothecin-induced cytotoxicity in osteosarcoma cells. BMC Cancer 2013; 13:500.
128. Lambert LA, Qiao N, Hunt KK, Lambert DH, Mills GB, Meijer L, Keyomarsi K. Autophagy: a novel mechanism of synergistic cytotoxicity between doxorubicin and roscovitine in a sarcomamodel. Cancer Res 2008; 68(19):7966-74.
129. Mazzanti R, Platini F, Bottini C, Fantappie O, Solazzo M, Tessitore L. Down-regulation of the HGF/MET autocrine loop induced by celecoxib and mediated by P-gp in MDR-positive human hepatocellular carcinoma cell line. Biochem Pharmacol 2009; 78: 21-32.
130. Meschini S, Condello M, Marra M, Formisano G, Federici E, Arancia G. Autophagy-mediated chemosensitizing effect of the plant alkaloid voacamine on multidrug resistant cells. Toxicol Vitro 2007; 21: 197-203.
131. Frankel LB, Lund AH. MicroRNA regulation of autophagy. Carcinogenesis 2012; 33(11): 2018-25.
132. Chen S, Rehman SK, Zhang W, Wen A, Yao L, Zhang J. Autophagy is a therapeutic target in anticancer drug resistance. Biochim Biophys Acta 2010; 1806(2): 220-9.
133. Chan EY. mTORC1 phosphorylates the ULK1-mAtg13-FIP200 autophagy regulatory complex. Sci Signal 2009; 2(84): pe51.
134. Sui X, Chen R, Wang Z, Huang Z, Kong N, Zhang M, Han W, Lou F, Yang J, Zhang Q, Wang X, He C, Pan H. Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death Dis 2013; 4: e838.
135. Le Lay S, Briand N, Blouin CM, Chateau D, Prado C, Lasnier F, Le Liepvre X, Hajduch E, Dugail I. The lipoatrophic caveolin-1 deficient mouse model reveals autophagy in mature adipocytes. Autophagy 2010; 6(6):754-63.
136. Martinez-Outschoorn UE, Whitaker-Menezes D, Lin Z, Flomenberg N, Howell A, Pestell RG, Lisanti MP, Sotgia F. Cytokine production and inflammation drive autophagy in the tumor microenvironment: role of stromal caveolin-1 as a key regulator. Cell Cycle 2011; 10(11):1784-93.
137. Chen ZH, Cao JF, Zhou JS, Liu H, Che LQ, Mizumura K, Li W, Choi AM, Shen HH. Interaction of caveolin-1 with ATG12-ATG5 system suppresses autophagy in lung epithelial cells. Am J Physiol Lung Cell Mol Physiol 2014; 306(11):L1016-25.
138. Shiroto T, Romero N, Sugiyama T, Sartoretto JL, Kalwa H, Yan Z, Shimokawa H, Michel T. Caveolin-1 is a critical determinant of autophagy, metabolic switching, and oxidative stress in vascular endothelium. PLoS One 2014; 9(2):e87871.
139. Frank PG, Lisanti MP. Role of caveolin-1 in the regulation of the vascular shear stress response. J Clin Invest 2006; 116(5):1222-5.
140. Volonté D, Galbiati F, Pestell RG, Lisanti MP. Cellular stress induces the tyrosine phosphorylation of caveolin-1 (Tyr(14)) via activation of p38 mitogen-activated protein kinase and c-Src kinase. Evidence for caveolae, the actin cytoskeleton, and focal adhesions as mechanical sensors of osmotic stress. J Biol Chem 2001; 276(11):8094-103.
141. Salani B, Maffioli S, Hamoudane M, Parodi A, Ravera S, Passalacqua M, Alama A, Nhiri M, Cordera R, Maggi D. Caveolin-1 is essential for metformin inhibitory effect on IGF1 action in non-small-cell lung cancer cells. FASEB J 2012; 26(2): 788-98.
142. Cohen AW, Schubert W, Brasaemle DL, Scherer PE, Lisanti MP. Caveolin-1 expression is essential for proper nonshivering thermogenesis in brown adipose tissue. Diabetes 2005; 54(3):679-86.
143. Park DS, Cohen AW, Frank PG, Razani B, Lee H, Williams TM, Chandra M, Shirani J, De Souza AP, Tang B, Jelicks LA, Factor SM, Weiss LM, et al. Caveolin-1 null (-/-) mice show dramatic reductions in life span. Biochemistry 2003; 42(51):15124-31.
144. Hehlgans S, Cordes N. Cav-1: an essential modulator of cancer cell radio-and chemoresistance. Am J Cancer Res 2011; 1(4):521-30.
145. Burgermeister E, Liscovitch M, Röcken C, Schmid RM, Ebert MP. Caveats of Cav-1 in cancer progression. Cancer Lett 2008; 268(2):187-201.
146. Sainz-Jaspeado M, Martin-Liberal J, Lagares-Tena L, Mateo-Lozano S, Garcia del Muro X, Tirado OM. Caveolin-1 in sarcomas: freind or foe? Oncotarget 2011; 2(4):305-12.
147. Lee AS. Glucose-regulated proteins in cancer: molecular mechanisms and therapeutic potential. Nat Rev Cancer 2014; 14(4):263-76.
148. Haenisch S, Cascorbi I. miRNAs as mediators of drug resistance. Epigenomics 2012; 4(4):369-81.
149. Malek E, Jagannathan S, Driscoll JJ. Correlation of long non-coding RNA expression with metastasis, drug resistance and clinical outcome in cancer. Oncotarget 2014; 5(18):8027-38.