
INTRODUCTION
Triple-negative breast cancer (TNBC), which does not express or express low levels of estrogen receptor, progesterone receptor and HER2/neu, is one of the most invasively malignant human cancers and its incidence increases year by year [1, 2]. Currently, there only exist few standard therapies for TNBC [3]. The development of nanoparticles (NPs), as tools of nanomedicine, has made significant advances towards TNBC treatment [4]. Unfortunately, the therapeutic efficiency of NPs is usually unsatisfied [5–7]. Firstly, the targeting ability of conventional NPs is too low to effectively accumulate around tumor sites, reducing the concentration of chemical drugs delivered by NPs and compromising the therapeutic effect. Secondly, after accumulating around tumors, NPs faced vastly diffusional hinderance due to the presence of the compressed intratumoral blood and lymphatic vessels and the dense collagen-rich extracellular matrix (ECM). Therefore, they were always unable to access into the nonvascularized and anoxic regions within deep tumor parenchyma, and the cancerous cells of core area are still aggressively survived, resulting in chemotherapeutic bland and latent crisis for tumor metastasis and regeneration [8, 9]. Based on these considerations, the development of novel NPs for TNBC is still urgently needed and the above two issues should be addressed.
For improving the tumor targeting efficiency of NPs, a variety of ligands are modified on the surface of NPs in favor to actively tumorous accumulation [10–12]. Considering the overexpression of LRP1 on TNBC cells, the corresponding peptide, angiopep-2 was employed to decorate NPs to endow our NPs with active TNBC targeting ability [13]. Furthermore, size-alterable NPs are constructed for overcoming the diffusional hinderance within tumors. Compared with small-sized NPs, NPs with larger size (100–200 nm) usually possessed higher tumor accumulation abilities [14, 15], but their diffusional abilities within tumor were severely impeded by the dense ECM and high tumor interstitial pressure, thus yielding a heterogeneous distribution in solid tumors [16, 17]. In contrast, small-sized NPs displayed superior permeability within tumor tissues but largely restricted by fast clearance in vivo [18–21]. Thus size-reducible NPs were developed for addressing the low tumor accumulation and limited tumor penetration. Taking together, angiopep-2 modified and intelligently size-reducible NPs were developed to effectively deliver drugs to TNBC.
In our study, Angio-DOX-DGL-GNP with core-shell nanostructure, were designed to integrate the active tumor targeting and size-shrinkable property. As shown in Figure 1, the core was composed of gelatin NPs (GNP) degraded by MMP-2, while the shell was made up of DGL linked with DOX and angiopep-2. During blood circulation, Angio-DOX-DGL-GNP could effectively accumulate around in tumor sites through passive and active tumor targeting [22, 23]. Then the core of Angio-DOX-DGL-GNP was dissembled when exposed to MMP-2 overexpressed in TNBC [24–26] and released the small-sized Angio-DOX-DGL-PEG, facilitating the delivery of NPs to the core area to kill more viable cells [24, 27]. Specifically, in consideration of the uniformly small size (4 ~ 7 nm) and structural modifications for specific biomedical applications, DGL was employed as the small-sized carriers linked with angiopep-2 [28–30]. Moreover, with the aim to examine the ability of Angio-DOX-DGL-GNP’s targeting effect and therapeutic effect, DOX was attached to DGL via a pH-sensitive cis-aconitic anhydride bond [31, 32]. In deed, our in vitro and in vivo results indicated that Angio-DOX-DGL-GNP possessed notable tumor accumulation and penetration abilities, thus maximizing antitumor effect.
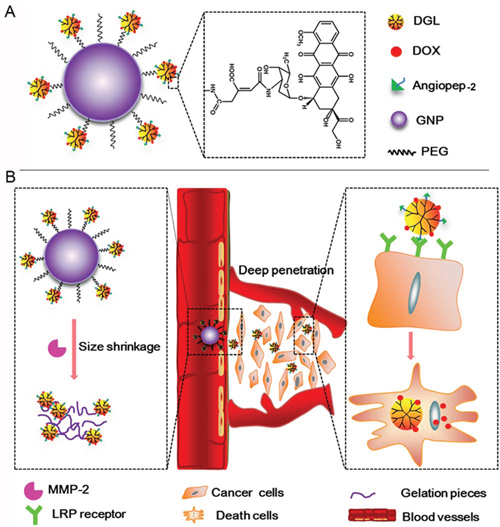
Figure 1: A. The schematic illustration of Angio-DOX-DGL-GNP, the linker between DOX and DGL was acid-sensitive cis-aconitic anhydride bond. B. The schematic illustration of the delivery of Angio-DOX-DGL-GNP in breast cancer cells. Large-sized Angio-DOX-DGL-GNP accumulated around the perivascular sites through active and passive targeting and then underwent large-to-small size transition by MMP-2 overexpressed in TNBC for deep tumor penetration; LRP receptor mediated cellular uptake of Angio-DOX-DGL-PEG and cell apoptosis induced by the released DOX.
RESULTS
Synthesis and characterization of Angio-DOX-DGL-GNP
DOX was conjugated to the residual primary amino groups of Angio-PEG-DGL by cis-aconityl bond. Infrared spectrometry exhibited the absorption band of the primary amino of DOX was 3328 cm−1 and 3523 cm−1, while cis-aconitic anhydride-doxorubicin (CAD) was in 3423 cm−1 consistent with the absorption band of the secondary amino groups, indicating the formation of amide bond and successive synthesis of CAD (Figure S1A and S1B). In NMR spectra of Angio-PEG-DGL, the solvent peak of D2O was found at 4.65 ppm. The repeat units (-O-CH2-CH2-O-) of PEG presented as a sharp peak at 3.4–3.6 ppm (Figure S1C). The methylene protons of branching units of DGL had double peaks between 1 ppm and 2 ppm. The peaks between 6.7 ppm and 7.3 ppm proved the existence of angiopep-2. Finally, the activated carboxyl groups of CAD were grafted on the amino groups of Angio-DGL-PEG via amide linkage to obtain the final product: Angio-DOX-DGL-PEG.
The particle size of Angio-DOX-DGL-PEG and Angio-DOX-DGL-GNP were 35.1 ± 1.7 nm and 185.7 ± 3.2 nm respectively with a narrow distribution. The drug content of Angio-DOX-DGL-PEG and Angio-DOX-DGL-GNP were 10.32 ± 0.3% and 4.82 ± 0.2%, respectively. As determined by MTT (Figure S2), the constructed DGL-GNP was safer than DGL, suggesting the carrier was biocompatible.
In vitro release of DOX
The in vitro release of DOX from Angio-DOX-DGL-GNP was performed at pH 7.4, 6.0, 5.0 and 3.0 (Figure S3). The drug release rate obviously increased as reduction of pH value. Little amount of DOX released at pH 7.4 and the cumulative release amount was less than10% after 24 h, suggesting the stability of cis-aconitic anhydride bond under neutral pH. When the pH decreased to 5.0, the 24 h cumulative release amount was nearly 85%, indicating the acidic sensitivity of cis-aconitic anhydride bond.
Degradation of GNPs triggered by MMP-2
Particle size of Angio-DOX-DGL-GNP notably decreased from 185.7 nm to 55.6 nm in the presence of MMP-2, which was attributed to the degradation of gelatin in Angio-DOX-DGL-GNP (Figure 2). Moreover, the zeta potential of Angio-DOX-DGL-GNP increased from +1.65 mV to +7.48 mV, which may facilitate the cellular uptake in tumor sites. However, the diameter of Angio-DOX-DGL-PEG remained stable and the transmission electron microscope (TEM) image also proved the same shrinkable properties. More importantly, the degradation of the GNP core by MMP-2 facilitated the release of small-sized Angio-DOX-DGL-PEG at tumor sites, which can largely decrease the diffusional hinderance. All the results suggested Angio-DOX-DGL-GNP could accomplish large-to-small shrinkage in response to MMP-2, thereby penetrating into the core area in tumor sites.
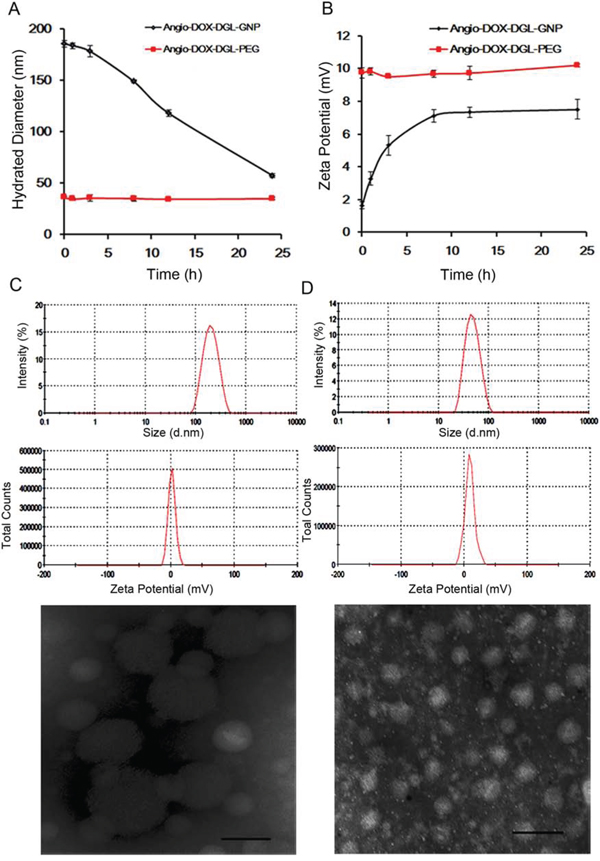
Figure 2: A. Hydrated Diameter of Angio-DOX-DGL-GNP and Angio-DOX-DGL-PEG incubated with MMP-2 during 24 h. B. Zeta Potential of Angio-DOX-DGL-GNP and Angio-DOX-DGL-PEG incubated with MMP-2 during 24 h. C. DLS data (Size distribution of Zeta potential) and TEM images of Angio-DOX-DGL-GNP before incubation with MMP-2, bar represents 100 nm. D. DLS data (Size distribution of Zeta potential) and TEM images of Angio-DOX-DGL-GNP after incubation with MMP-2 for 24 h, bar represents 100 nm.
In vitro penetration efficiency of Angio-DOX-DGL-GNP2
4T1 cells, which are extremely aggressive, highly tumorigenic and metastatic, is selected as a model for TNBC [2, 33, 34]. The multicellular tumor spheroids (MCTs) were employed to evaluate the penetration efficiency of Angio-DOX-DGL-GNP after degradation by MMP-2 because the MCTs’ environment was similar to in vivo tumors, such as poor drug penetration, free of microvessel, changed protein expression and activity and gradients of oxygen tension and nutrients [35, 36]. The distribution of different formulations in superficial sections of 4T1 MCTs was observed. MCTs treated with Angio-DOX-DGL-PEG exhibited higher intensity in all slices than that of Angio-DOX-DGL-GNP, implying that smaller-sized particles possessed better penetrating efficiency. Moreover, Angio-DOX-DGL-GNP (with MMP-2) displayed the similar penetration ability as small-sized Angio-DOX-DGL-PEG, which was largely attributing to the size shrinkable properties of Angio-DOX-DGL-GNP in presence of MMP-2 (Figure 3A). By comparison, Angio-DOX-DGL-GNP was located at the edge of MCTs, while the fluorescence of Angio-DOX-DGL-GNP pre-incubated with MMP-2 distributed more extensively and penetrated much deeper in the distance of 100 μm than that of Angio-DOX-DGL-GNP groups (Figure 3B). For better evaluated the permeability of NPs, the penetrating percentage (fluorescent intensity of inner region to edge region) at 130 μm depth was introduced to determine the penetrating efficiency (Figure 3C). After pre-treatment with MMP-2, the penetrating efficiency of Angio-DOX-DGL-GNP in MCTs was apparently improved. The results confirmed that Angio-DOX-DGL-GNP possessed better penetration ability after incubation with MMP-2 and the reduction of particle size was beneficial for enhancing penetrating efficiency through the tumor spheroids experiments.
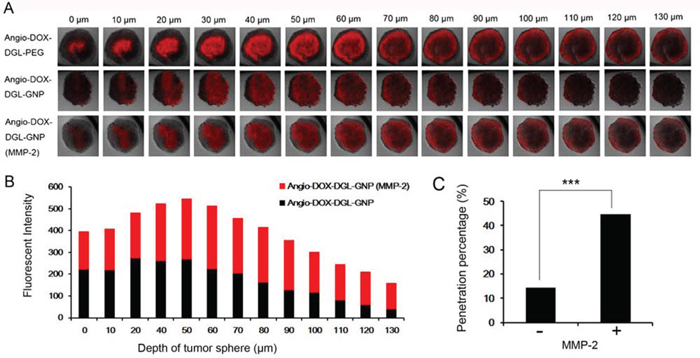
Figure 3: A. Fluorescent images of 4T1 MCTs after incubation with Angio-DOX-DGL-GNP and Angio-DOX-DGL-GNP (MMP-2) for 24 h. B. Semi-quantitative intensity of inner region at different sections of 4T1 tumor spheroids. C. Penetration percentage of Angio-DOX-DGL-GNP at 130 μm.
In vitro cellular uptake study
In order to validate the enhanced cellular uptake of Angio-DOX-DGL-GNP, 4T1 cells were incubated with these NPs for 2 h. The fluorescent intensity of Angio-DOX-DGL-GNP was significantly stronger than that of DOX-DGL-GNP, which confirmed that Angio-DOX-DGL-GNP possessed stronger ability of entering into tumor cells (Figure 4A and 4C).
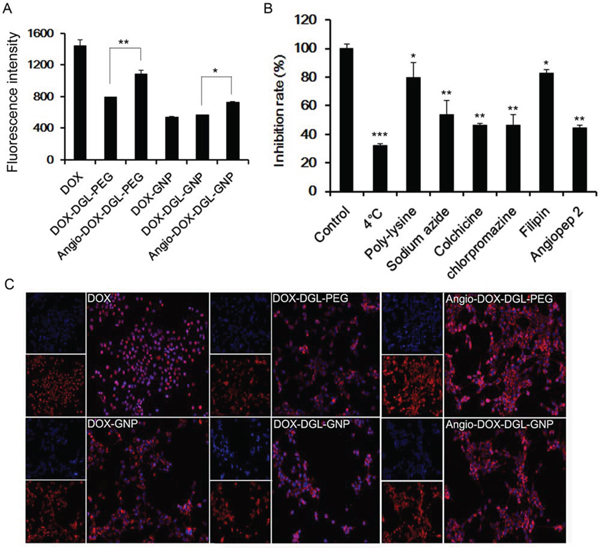
Figure 4: A. Cellular uptake of 4T1 cells after incubation with different formulations for 2 h. Data represent the mean ± SD (n = 3). B. The endocytosis inhibition assay of Angio-DOX-DGL-GNP on 4T1 cells. The inhibition rate (%) is expressed as the ratios of the cellular uptake in the presence of various inhibitors to the uptake in absence of inhibitor. C. Images of 4T1 cells treated with DOX, DOX-DGL-PEG, Angio-DOX-DGL-PEG, DOX-GNP, DOX-DGL-GNP and Angio-DOX-DGL-GNP.
In vitro cellular uptake mechanism
Different inhibitors were utilized to conduct the endocytosis inhibition assay. 4°C and sodium azide were employed to deplete cellular ATP in order to explore the impact of energy on the cellular uptake. Colchicine, chlorpromazine and filipin were chosen to block macropinocytosis-mediated, clathrin-mediated and caveolin-mediated endocytosis respectively and poly-lysine was chosen as positive charge inhibitor [39, 40]. Ligands on the surface of NPs also correlate with the internalization of NPs. Our data exhibited that various mechanisms were involved in the uptake process. The cellular uptake was remarkably reduced under 4°C and sodium azide, implying that the endocytosis of Angio-DOX-DGL-GNP was energy-correlated. Moreover, the rate of uptake was also significantly inhibited by colchicine, chlorpromazine and angiopep-2, indicating that macropinocytosis, clathrin- and receptor-mediated endocytosis were involved in the cellular uptake of Angio-DOX-DGL-GNP.
In vitro cellular apoptosis assay
As demonstrated in Figure 5, the apoptosis of 4T1 cells induced by Angio-DOX-DGL-GNP was higher than that by DOX-DGL-GNP, suggesting angiopep-2 modification could enhance the antitumor effect. However, the apoptosis percentage of Angio-DOX-DGL-GNP was not higher than that of Angio-DOX-DGL-PEG, which was because the particles directly contacted with cells and this study could not reflect the difference between particles with different sizes.
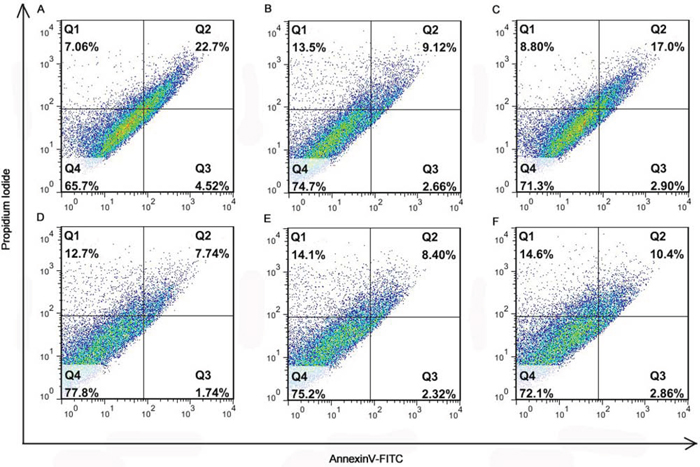
Figure 5: The diagrams of the apoptosis assay of different formulations. A. DOX; B. DOX-DGL-PEG; C. Angio-DOX-DGL-PEG; D. DOX-GNP; E. DOX-DGL-GNP; F. Angio-DOX-DGL-GNP.
In vivo tumor distribution and penetration
To test the biodistribution and tumor targeting of Angio-DOX-DGL-GNP in vivo, a murine 4T1 xenograft model of TNBC was established and a real-time fluorescence imaging technique was applied. At 24 h post-injection, the large-sized Angio-DOX-DGL-GNP exhibited a stronger fluorescent signal at tumor site than small-sized Angio-DOX-DGL-PEG (Figure 6, S4), which attributed to the enhanced tumor retention effect. More importantly, the fluorescent intensity of Angio-DOX-DGL-GNP in tumor region was higher compared with that of other groups, validating the significant tumor targeting effect of angiopep-2 (Figure 7). Accordingly, it was demonstrated that Angio-DOX-DGL-GNP had a high accumulation in tumor sites as a result of a combination of the passive and active targeting mechanisms. To further visualize the distribution of NPs, the tumor slices was stained with CD34 and LRP-1 (Red) and the distribution of DOX (green) was captured (Figure 7–8). The fluorescent intensity of Angio-DOX-DGL-GNP was remarkably higher than other groups, which benefited from better tumor targeting and penetration effect. Although many of Angio-DOX-DGL-GNP colocalized with microvessel, there were also fluorescence distributed in the area free of vessel (Figure 8), suggesting the Angio-DOX-DGL-GNP could penetrate deeper in tumor than other groups.
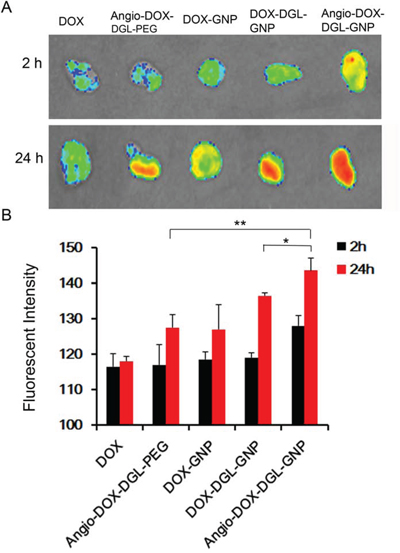
Figure 6: A. Ex vivo fluorescence imaging of the tumor and normal tissues of 4T1 tumor-bearing BALB/C mice after 2 h or 24 h post-injection of different DOX formulations. B. Semi-quantitative analysis of fluorescent intensity of tumor sites.
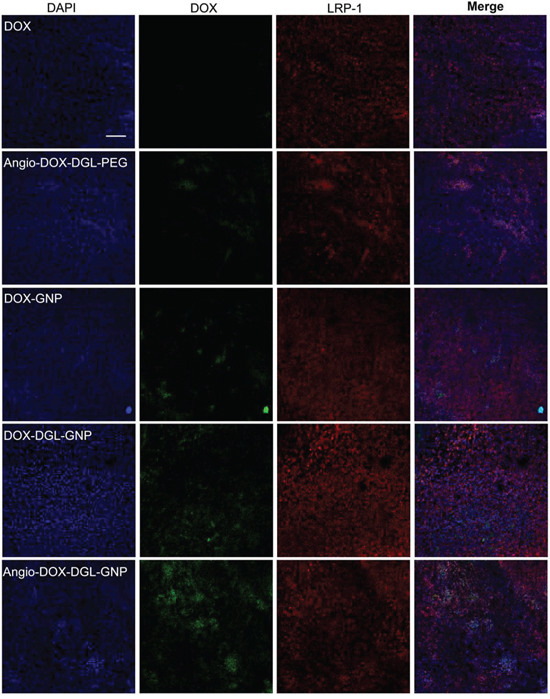
Figure 7: In vivo distribution of DOX, Angio-DOX-DGL-PEG, DOX-GNP, DOX-DGL-GNP and Angio-DOX-DGL-GNP in breast cancer. Blue represents DAPI; Green represents the fluorescence of DOX; Red represents LRP-1. Bar represents 100 μm.
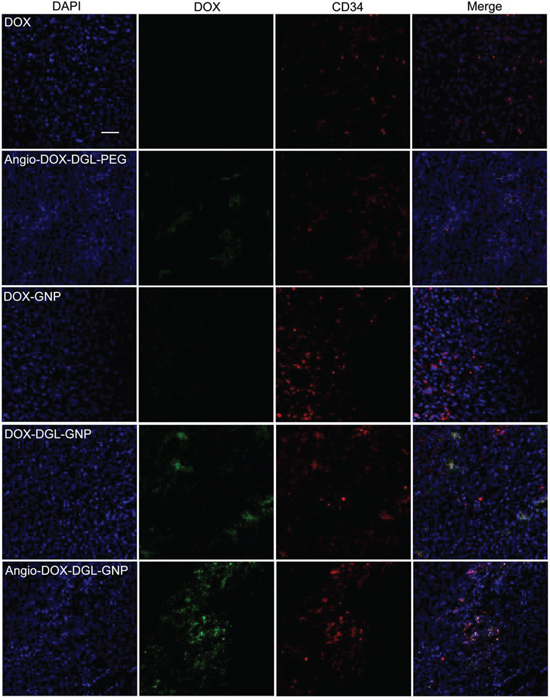
Figure 8: In vivo tumor distribution of DOX, Angio-DOX-DGL-PEG, DOX-GNP, DOX-DGL-GNP and Angio-DOX-DGL-GNP. Blue represents DAPI, green represents the fluorescence of DOX, red represents CD34 labeled blood vessels and bar represents 50 μm.
In vivo therapy studies
To demonstrate the feasibility of Angio-DOX-DGL-GNP for cancer treatment in vivo, the therapeutic efficacy of Angio-DOX-DGL-GNP compared with several other NPs was evaluated in 4T1 tumor bearing mice. The rate of tumor growth was suppressed to different degree after intravenous administration of various formulations compared with the control group (Figure 9). Angio-DOX-DGL-GNP exerted a noticeably higher effect on tumor inhibition compared with non-targeted NP (DOX-GNP, DOX-DGL-GNP) and small-sized Angio-DOX-DGL-PEG, which mainly ascribed to better tumor retention effect combined with the active target ability provided by angiopep-2. More importantly, Angio-DOX-DGL-GNP cannot only accumulate more efficiently at tumor sites via active targeting but also penetrate deeper within the tumor. During the entire treatment period, no noticeable alteration of mice body weight was observed in all groups except DOX group, which was due to the high toxicity of free DOX. Moreover, the images of tumor slices using hematoxylin and eosin (H&E) staining presented a massive cancer cell remission after administration of Angio-DOX-DGL-GNP (Figure 9E), suggesting Angio-DOX-DGL-GNP could induce more apoptosis tumor cells. The histologic images of main tissues stained by H&E showed that DOX induced the necrosis of heart with acute inflammatory cells infiltration (Figure 10). In contrast, no noticeable changes of Angio-DOX-DGL-GNP groups were visualized in comparison with control group, indicating the application of them in vivo was safe. Taking together, the Angio-DOX-DGL-GNP significantly enhanced tumor targeting efficacy and possessed better antitumor efficacy and low side effect.
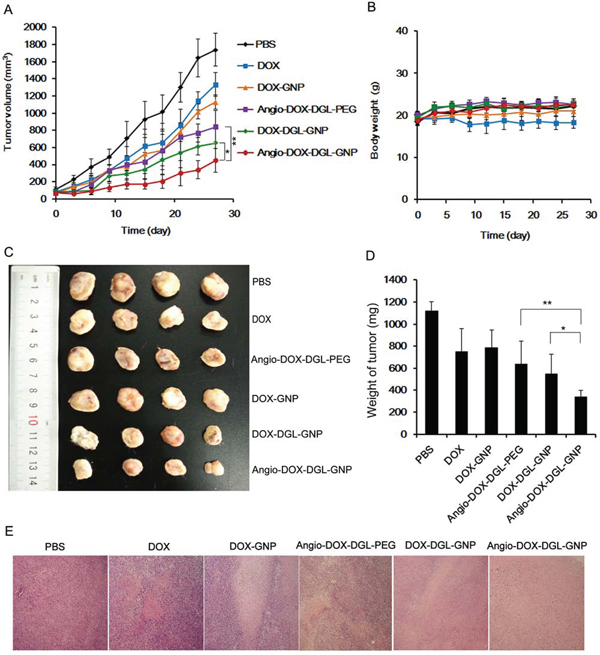
Figure 9: In vivo tumor growth inhibition experiment. A. The 4T1 tumor growth curves after intravenous injection of different formulations of DOX. B. The body weight of mice after administration of different formulations of DOX and PBS up to day 27 (n = 6). C. The images of tumors of mice obtained from sacrificed mice at the end of this experiment. D. The weight of tumor after 27 days treatment (n = 6). E. Histological images of tumors sections stained by H&E (tumors: × 100).
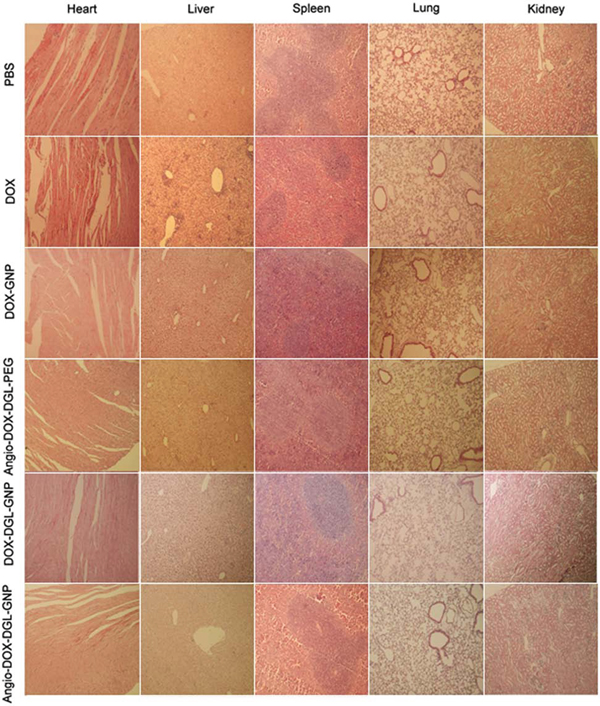
Figure 10: Histological analysis for different organs of 4T1 bearing mice administrated PBS, DOX, Angio-DOX-DGL-PEG, DOX-GNP, DOX-DGL-GNP and Angio-DOX-DGL-GNP (all tissues: × 100).
DISCUSSIONS
Many factors in vivo constitute challenging hurdles for TNBC treatment such as high interstitial fluid pressure, dense stromal tissue, complex interaction with fibroblasts and tumor cells, thus conventional NPs were not homogeneously distributed in tumor sites and just achieved compromising therapeutic effects. Many strategies were addressed to eradicating the bland area of chemotherapy such as size-reduction, depletion of ECM and pulsed high intensity focused ultrasound [16, 27, 37, 38]. Among them, size-reducible NPs hold most attention for its incomparable advantages such as high accumulation and superior tumor penetration [37, 39]. For example, Daniel S. Kohane reported a photo-triggered NPs undergoing large-to-small size shrinkage from 150 nm to 40 nm [27]. In another study, Fukumura designed a QDs-GNP with shrinkable size triggered by MMP-2, thus releasing small-sized QDs for deep tumor penetration [24, 40].
In our study, significantly higher cellular uptake of Angio-DOX-DGL-GNP was observed in 4T1 cells indicating that angiopep-2 modifying facilitated the cellular uptake of Angio-DOX-DGL-GNP. Furthermore, size shrinkage NPs were designed to overcome the hinderance of tumor region, thus maximizing the antitumor effect. In our study the degradability of GNP core was evaluated by monitoring the size change of nanocarrier after incubation with MMP-2. The results showed that the size of Angio-DOX-DGL-GNP could reduce from 185.7 nm to 55.6 nm, indicating GNP could be degraded into small fragments by MMP-2 as previous study showed [41, 42]. In vivo imaging experiment also demonstrated that Angio-DOX-DGL-GNPs accumulated in tumor tissues more efficiently via EPR effect and active targeting in 4T1 tumor bearing mice. Our study expected that after the GNP core were degraded by MMP-2, the released small-sized Angio-DOX-DGL-PEG penetrated into the regions away from the tumor blood vessels to kill more cancer cells. However, the penetration ability of Angio-DOX-DGL-GNP in vivo wasn’t satisfied which may due to the sensitivity to MMP-2 degradation, but in the evaluation of anti-TNBC effect in 4T1 grafted model, Angio-DOX-DGL-GNP exhibited the highest tumor growth inhibition rate than other groups, implying that Angio-DOX-DGL-GNP accomplished our targets to some extent. An ideal enzyme-responsive multistage NPs should been rapidly degraded into small-sized NPs after they accumulated in tumor sites, therefore small-sized NPs penetrated into the core of tumor sites and released the anticancer drug, leading to killing more tumor cells. In fact, when the NPs reached the tumor sites by EPR effect, the multistage NPs were degraded into small NPs in accompany with penetration. The smaller the particle size of NPs shrank, the deeper the NPs penetrated. Certainly, further research was also needed for us to increase the responsiveness of our NPs in the future. More importantly, in the aspect of the in vivo application of safety of Angio-DOX-DGL-GNP, no significant weight change and hematoxylin and eosin staining results proved that these nanocarrier were of biological safety, which was corresponded with other researches [43, 44].
MATERIALS AND METHODS
Materials
Doxorubicin hydrochloride was purchased from Beijing Huafenglianbo Technology Co. Ltd. (Beijing, China). Aconitic anhydride (CA) was purchased from Alfa Aesar Chemical Co. Ltd. (Tianjin, China). DGL-G3 dendrimer was purchased from Colcom (Montpellier Cedex, France). Gelatin type A was purchased from MP Biomedicals Co. Ltd. (California, USA). Glutaraldehyde solution (Grade II, 25%) was purchased from Beijing Solarbio Technology Co. Ltd. (Beijing, China). Angiopep-2 was purchased from Qiangyao Biotechnology Co. Ltd. (Shanghai, China). Amino-PEG5000-succinimidyl carbonate, α-mPEG5000-ω-amino and dicarboxyl-PEG5000 were purchased from Seebio Biotechnology Co. Ltd. (Shanghai, China). 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) and N-hydroxy-succinimide (NHS) were purchased from Keddia Reagent Co. Ltd. (Chengdu, China). 6-Diamidino-2-pheylindole (DAPI) was obtained from Beyotime Insitute Biotechnology (Haimen, China). Anti-CD34 antibody was purchased from Abcam (Hong Kong). Alexa Fluor 594-conjugated AffiniPure donkey anti-rabbit was purchased from Jackson Immuno Research Laboratories Inc (West Grove, USA). Rb mAb to LRP-1 (EPR 3724) was purchased from Abscam (Hong Kong). Cy3-conjugated Affinipure Goat Anti-Rabbit IG (H+L) was purchased from Jackson ImmunoRearch Laboratories, Inc.(West Grove, USA). MMP-2 protein (rat) was purchased from Abcam Ltd. (Hong Kong, China). Purified rabbit mAb to CD34 was purchased from Abcam Ltd. (Hong Kong, China). RPMI1640, 3-(4, 5-dimethylthialzolayl)-2, 5-diphenyltetrazolium bromide (MTT) was purchased from Baoxin Biotechnology Ltd. (Chengdu, China). Mouse mammary breast tumor cell line (4T1) was purchased from Shanghai Institute of Cell Biology (Shanghai, China). All other reagents and solvents were of analytical or HPLC grade and were used without further purification.
Synthesis of Angio-DOX-DGL-PEG
The conjugation of DOX to DGL via cis-aconityl bond was performed as previously reported literature [32, 45]. Briefly, cis-aconitic anhydride (CA) (54 mg) dissolved in 1 mL of 1, 4-dioxane was added dropwise to the doxorubicin solution (30 mg, 51.7 μmol) with continuous stirring in dark at room temperature and the pH value was controlled in the range of 8.4–8.7 by NaOH. Then a great amount of heavy precipitate was produced by slowly adding in HCl (1 M) in ice bath and distributed in 25 mL of ethyl acetate for 4 times. After the organic solvent was removed by the rotary evaporator, the structure of CAD was determined by Infrared Spectrometer (VECTOR22) for FT-IR analysis. Secondly, DGL was reacted with the activated dicarboxyl-PEG5000 at the ratio 1: 8 (molar ratio) in pH 8.0 PBS for 2 h in the presence of EDC and NHS followed by ultrafiltration by using a 10KDa molecular weight cutoff membrane to remove the small molecules. Angiopep-2 was added to the purified mixture at the ratio of 1: 8 (DGL to peptide, molar ratio) for 24 h in the presence of EDC and NHS, finally the reaction mixture was dialyzed against deionized water twice for 12 h. Next the purified reaction was freeze-dried to obtain a white power and analyzed in a 400 MHz spectrometer. At last, a 10-fold molar excess of EDC and NHS was added to CAD in 2 mL of PBS and reacted under the dark condition for 0.5 h. Then the solutions (CAD: Angio-PEG-DGL = 48: 1, molar ratio) were mixed and reacted for another 12 h. At last, the unreacted CAD and other small molecules were removed by ultrafiltration through a 10KDa molecular weight cutoff membrane.
Preparation of GNPs
GNPs were fabricated by a two-step desolvation method as previous reports with minor modification [46, 47]. Briefly, 625 mg of gelatin type A was dissolved in 12.5 mL of deionized water (DI) at 40°C, then 12.5 mL of acetone was added to the gelatin solution under low-speed stirring. Stop stirring exactly 1 min, the supernatant was thrown away and 8 mL of DI was added to the solution. Then acetone was added to the solution (pH 2.7–3.0) until a sustained faint turbidity was visualized, followed by addition of 60 μL of 25% glutaraldehyde solution diluted in 1 mL of acetone to harden the NPs. Finally, the solution was stirred at 40°C and 600 rpm for 7 h and the acetone was removed. 0.2 mL of 1 M glycine solution was added to terminate the cross linking. Finally the GNPs were purified by passage through a Sephadex G-50 column.
Preparation and drug content of Angio-DOX-DGL-GNP
1 mL of GNP solution (20 ~ 25 mg) was activated by an additional solution of EDC (0.8 mg) and sulfo-NHS (0.8 mg) for 30 min, then COOH-PEG5000-NH2 (20 mg, × 2 μmol) was added to the activated GNP solution to obtain GNP-PEG5000-COOH for 2 h. Next the carboxyl groups of GNP-PEG5000-COOH were activated by an additional solution of EDC (2 mg) and sulfo-NHS (2 mg) in 50 μL of DI under pH 6.0 for 30 min. After the pH value of the mixture was adjusted to 8.0 again, Angio-DOX-DGL-PEG (equivalent to 0.5 mg DOX) was added and stirring for another 8 h to obtain Angio-DOX-DGL-GNP. Finally, the solutions were purified by ultrafiltration through a 100KDa molecular weight cutoff membrane at 4500 g for 30 min.
Drug content was quantified by hydrolyzing the glycosidic bond between the doxorubicinone and amino sugar at acidic condition and determining the released doxorubicinone. Different formulations (0.1 mL) was mixed with 2.5 M HCl (0.8 mL) and methanol (1 mL), and the mixture was incubated at 50°C for 1.5 h. The generated doxorubicinone was analyzed by HPLC (WelchromR C18 column (4.6 × 250 mm, 5 mm particle size, Agilent 1200 series), 0.01 M KH2PO4 : acetonitrile : acetic acid = 45 : 55 : 0.27 (v/v/v), 1.0 mL/min, 30°C, 490 nm). A standard curve of doxorubicinone was generated by hydrolyzing free DOX at the same condition, then the amount of DOX conjugation were obtained and the drug loading efficiency were finally calculated.
In vitro release
As the linker between DOX and DGL was a pH-sensitive linker, cis-aconitic anhydride bond, in vitro release profiles of Angio-DOX-DGL-GNP (equivalent to 0.1 mg DOX) were performed in 40 mL of PBS (pH 7.4, 6.0, 5.0 and 3.0) medium at 37°C and 50 rpm. The samples were sealed in a dialysis bag (MW 3500). At predetermined intervals, 1 mL of released media was taken away and the corresponding fresh medium was added. The amount of released DOX was measured by fluorescence spectrophotometer. Each drug release test was performed thrice. Each drug release test was performed thrice.
Degradation of GNP triggered by MMP-2 in vitro
MMPs, especially MMP-2 and MMP-9, are associated with the invasion, progression angiogenesis and metastasis of many human cancers. The expression levels of MMPs were found to be relatively high in breast cancer, liver cancer, liver cancer, lung cancer and ovarian cancer, whereas they are minimally expressed in healthy tissue [48–51]. To investigate the enzyme-sensitivity of Angio-DOX-DGL-GNP, 0.5 mL of Angio-DOX-DGL-GNP (0.4 mg) was incubated with 0.5 mL of MMP-2 in HEPES (460 ng). At prearranged time intervals, the particle size and zeta potential of the NPs were immediately determined by a dynamic light scattering detector (Nano-ZS, Malvern, UK). The morphology of Angio-DOX-DGL-GNP before or after MMP-2 incubation was observed via TEM (JEM-100CX, JEOL, Japan).
Penetration assay using MCTs
4T1 cells were seeded into 96-well plates pre-coated with sterile agarose solution (2%, m/v) at a density of 8 × 103 cells per well. Subsequently, the MCTs were monitored with optical microscope in order to assure that they form intact spheres and ready to use after 3 days. Then MCTs were selected with uniform size and then incubated with different formulations for 12 h. Subsequently, the MCTs were rinsed thrice with cold PBS followed by fixation of 4% paraformaldehyde for 30 min and placed in 96-well plates for confocal microscopy.
In vitro cellular uptake study
The cellular uptake of different formulations was evaluated by fluorescent microscope, 4T1 were seeded at a density of 5 × 104 cells/well on 6-well plates containing square coverships for 24 h. After achieving 70%–80% confluence, various samples (12.5 μg/mL, DOX equal) was added to each well and incubated for 2 h. Then the coverships were washed trice with PBS followed by fixing with 4% paraformaldehyde for 20 min and then staining with DAPI to identify the nuclei for 5 min. Finally, the coverships were visualized and photographed by fluorescent microscope (Nikon, Japan).
4T1 cells were plated in 6-cell plates at a density of 1 × 105 cells per cell and cultured for 24 h. Cells were treated with DOX, DOX-DGL-PEG, Angio-DOX-DGL-PEG, DOX-DGL-GNP and Angio-DOX-DGL-GNP at a final DOX concentration of 12.5 μg/mL. After incubation for 2 h, the cells were washed three times with cold PBS, trypsinized and resuspended in 0.5 mL of PBS. Then the fluorescent intensity was tested by flow cytometry (BD, USA).
Cellular uptake mechanism study
In order to study the uptake mechanism of Angio-DOX-DGL-GNP, 4T1 cells were pre-incubated with various endocytosis inhibitors including poly-lysine (400 μg/mL), sodium azide (0.651 mg/mL), chlorpromazine (10 μg/mL), filipin (5 μg/mL) meanwhile the inhibition of free angiopep-2 (250 μg/mL) for 30 min and the effect of temperature (4°C) was also studied. Then the inhibitors were withdrawn from the wells and the cells were treated with Angio-DOX-DGL-GNP. After 1 h incubation, the solution was discarded and the cells were washed with ice-cold PBS twice and responded in 0.4 mL PBS. The fluorescence intensity was determined by flow cytometer (Cytomics™ FC 500, Beckman Coulter, Miami, FL, USA).
Cell apoptosis assay
The apoptosis of 4T1 cells was determined by using Annexin V-FITC apoptosis detection kit (Doshido, Japan). Briefly, the 4T1 cells were plated in 6-well plates at a density of 1 × 105 cells per cell and cultured for 24 h. Then the cells were treated with different formulations at DOX concentration of 12.5 μg/mL. After incubated at 37°C for 24 h, the cells were harvested, washed by PBS twice and suspended in 500 μL of binding buffer. 5 μL of Annexin V-FITC and 5 μL of propidium iodide (PI) were added into the cell suspension for 15 min incubation, respectively. The cells were immediately analysed by flow cytometry (BD, USA).
In vivo imaging and tumor distribution
Animals were performed in accordance with national regulations and approved by the Institutional Animal Care and Use Committee of Sichuan University. 4T1 tumor-bearing mice were randomly into 5 groups and intravenously administrated with different formulations at a dose of 5 mg/kg DOX (equal) per mouse via the tail vein. 24 hours after administration, the mice were sacrificed and the major organs were further visualized. Images were taken on Bio-Real Quick view 3000 (Geneway International, Australia) and the fluorescence intensities were analyzed by Living Image Software.
After dehydration with sucrose solution, the tumors were embedded and frozen in OCT embedding medium (Leica, Germany), frozen slices of 16 μm thickness were prepared with cryotome Cryostat (Leica, Germany). Subsequently, the slices were stained with rabbit mAb to LRP1 antibody (1:100) overnight, followed by staining with secondary antibody Cy3-conjugated Affinipure Goat Anti-Rabbit IgG(H+L) and 0.5 μg/mL of DAPI at room temperature. After washing with PBS trice, the slices were immediately examined by a confocal microscope at corresponding excitation wavelength (LSM710, Carl Zeiss, Germany). At the same time, the slices were stained with Rab mAb to CD34 antibody (1:100) and Alexa Fluor 594-conjugated Affinipure Donkey Anti-Rabbit IgG secondary antibody with a procedure established previously. Then the distribution was determined by a confocal microscope above at corresponding excitation wavelength.
In vivo therapy studies
The therapy studies were conducted in 4T1 tumor models. Briefly, 4T1 cells (5 × 105) were injected into the right flank of BALB/c mice (20 ± 2). BALB/c mice bearing 4T1 tumor were randomly divided into six groups (n = 6) and treated with different formulations at a dose of 5 mg/kg of DOX and saline via intravenously injection at an interval of 2 days for 4 times, respectively. The tumor size and body weight of mice were recorded at the meantime and the tumor volume was calculated using the formula: Tumor volume = (width2/length)/2. At the end of the experiment, all the mice were killed and the main organs were isolated. Finally the tumors were imaged and weighed. For HE staining, formalin-fixed tissues were embedded in paraffin blocks and observed by optical microscope (Axiovert 40 CFL, Carl Zeiss, Germany).
Statistical analysis
All data were displayed as mean ± SD. Statistical difference between two groups were performed by Students t-test. P value < 0.05 and < 0.01 were considered indications of statistical difference and statistically significant difference respectively.
CONCLUSIONS
In summary, Angio-DOX-DGL-GNP was designed to overcome the limitations of current NPs such as low targeting efficiency and poor penetration ability. Based on the enhanced accumulation of angiopep-2 mediated active targeting and size-reduction of enhanced penetration efficacy, Angio-DOX-DGL-GNP significantly inhibited tumor growth and provided a promising nanoplatform for TNBC treatment.
ACKNOWLEDGEMENTS AND FUNDING
This work was mainly supported by the National Natural Science Foundation of China (81402866, 31571016, 81202926).
CONFLICTS OF INTEREST
There is no conflict of interest.
REFERENCES
1. Bosch A, Eroles P, Zaragoza R, Vina JR, Lluch A. Triple-negative breast cancer: molecular features, pathogenesis, treatment and current lines of research. Cancer Treat Rev. 2010; 36:206–215.
2. Singh M, Ramos I, Asafu-Adjei D, Quispe-Tintaya W, Chandra D, Jahangir A, Zang X, Aggarwal BB, Gravekamp C. Curcumin improves the therapeutic efficacy of Listeria(at)-Mage-b vaccine in correlation with improved T-cell responses in blood of a triple-negative breast cancer model 4T1. Cancer Med. 2013; 2:571–582.
3. Rammohan Devulapally, Narayana M. Sekar, Thillai V. Sekar, Kira Foygel, Tarik F. Massoud, Jürgen K. Willmann, and Ramasamy Paulmurugan*. Polymer Nanoparticles Mediated Codelivery of AntimiR-10b and AntimiR-21 for Achieving Triple Negative Breast Cancer Therapy. ACS Nano. 2015.
4. Tatsiana Y. Rakovich, Omar K. Mahfoud, Bashir M. Mohamed, Adriele Prina-Mello, Kieran Crosbie-Staunton, Tina Van Den Broeck, Line De Kimpe, Alyona Sukhanova, Aliaksandra Rakovich, Stefan A. Maier, Frauke X. Alves, Frans Nauwelaers, Igor Nabiev, Patrick R. Chames and Yuri Volkov. Highly Sensitive Single Domain Antibody Quantum Dot Conjugates for Detection of HER2 Biomarker in Lung and Breast Cancer Cells. ACS Nano. 2014; 8:5682–5695.
5. Bear HD, Tang G, Rastogi P, Geyer CE Jr. Robidoux A, Atkins JN, Baez-Diaz L, Brufsky AM, Mehta RS, Fehrenbacher L, Young JA, Senecal FM, Gaur R, Margolese RG, Adams PT, Gross HM, et al. Bevacizumab added to neoadjuvant chemotherapy for breast cancer. N Engl J Med. 2012; 366:310–320.
6. Beyer I, Li Z, Persson J, Liu Y, van Rensburg R, Yumul R, Zhang XB, Hung MC, Lieber A. Controlled extracellular matrix degradation in breast cancer tumors improves therapy by trastuzumab. Mol Ther. 2011; 19:479–489.
7. Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nat Rev Clin Oncol. 2010; 7:653–664.
8. Shishuai Su, Yanhua Tian, Yiye Li, Yanping Ding, Tianjiao Ji, MeiyuWu, YanWu, Guangjun Nie. “Triple-Punch” Strategy for Triple Negative Breast Cancer Therapy with Minimized Drug Dosage and Improved Antitumor Efficacy. ACS Nano. 2015;.
9. Zan M, Li J, Luo S, Ge Z. Dual pH-triggered multistage drug delivery systems based on host-guest interaction-associated polymeric nanogels. Chem Commun (Camb). 2014; 50:7824–7827.
10. Wang B, Lv L, Wang Z, Zhao Y, Wu L, Fang X, Xu Q, Xin H. Nanoparticles functionalized with Pep-1 as potential glioma targeting delivery system via interleukin 13 receptor alpha2-mediated endocytosis. Biomaterials. 2014; 35:5897–5907.
11. Wen Y, Roudebush SL, Buckholtz GA, Goehring TR, Giannoukakis N, Gawalt ES, Meng WS. Coassembly of amphiphilic peptide EAK16-II with histidinylated analogues and implications for functionalization of beta-sheet fibrils in vivo. Biomaterials. 2014; 35:5196–5205.
12. Wen Y, Liu W, Bagia C, Zhang S, Bai M, Janjic JM, Giannoukakis N, Gawalt ES, Meng WS. Antibody-functionalized peptidic membranes for neutralization of allogeneic skin antigen-presenting cells. Acta biomaterialia. 2014; 10:4759–4767.
13. Pires LA, Hegg R, Freitas FR, Tavares ER, Almeida CP, Baracat EC, Maranhão RC. Effect of neoadjuvant chemotherapy on low-density lipoprotein (LDL) receptor and LDL receptor-related protein 1 (LRP-1) receptor in locally advanced breast cancer. Braz J Med Biol Res. 2012; 45:557–564.
14. Xiangsheng Liu, Yangjun Chen, Huan Li, Nan Huang, Qiao Jin, Kefeng Ren, and Jian Ji*. Enhanced Retention and Cellular Uptake of Nanoparticles in Tumors by Controlling Their Aggregation Behavior. ACS Nano. 2013; 7:6244–6257.
15. She W, Luo K, Zhang C, Wang G, Geng Y, Li L, He B, Gu Z. The potential of self-assembled, pH-responsive nanoparticles of mPEGylated peptide dendron-doxorubicin conjugates for cancer therapy. Biomaterials. 2013; 34:1613–1623.
16. Wang S, Shin IS, Hancock H, Jang BS, Kim HS, Lee SM, Zderic V, Frenkel V, Pastan I, Paik CH, Dreher MR. Pulsed high intensity focused ultrasound increases penetration and therapeutic efficacy of monoclonal antibodies in murine xenograft tumors. J Control Release. 2012; 162:218–224.
17. Alessandro Parodi, X Seth G. Haddix, Nima Taghipour, Shilpa Scaria, Francesca Taraballi, Armando Cevenini, Iman K. Yazdi, Claudia Corbo, Roberto Palomba, Sm Z. Khaled, Jonathan O. Martinez, Brandon S. Brown, Lucas Isenhart, and Ennio Tasciotti*. Bromelain Surface Modification Increases the Diffusion of Silica Nanoparticles in the Tumor. ACS Nano. 2013.
18. Sunoqrot S, Bugno J, Lantvit D, Burdette JE, Hong S. Prolonged blood circulation and enhanced tumor accumulation of folate-targeted dendrimer-polymer hybrid nanoparticles. J Control Release. 2014; 191:115–122.
19. Zhang C, Li C, Liu Y, Zhang J, Bao C, Liang S, Wang Q, Yang Y, Fu H, Wang K, Cui D. Gold Nanoclusters-Based Nanoprobes for Simultaneous Fluorescence Imaging and Targeted Photodynamic Therapy with Superior Penetration and Retention Behavior in Tumors. Advanced Functional Materials. 2015; 25:1314–1325.
20. Popovic Z, Liu W, Chauhan VP, Lee J, Wong C, Greytak AB, Insin N, Nocera DG, Fukumura D, Jain RK, Bawendi MG. A nanoparticle size series for in vivo fluorescence imaging. Angew Chem Int Ed Engl. 2010; 49:8649–8652.
21. Wu W, Driessen W, Jiang X. Oligo(ethylene glycol)-based thermosensitive dendrimers and their tumor accumulation and penetration. J Am Chem Soc. 2014; 136:3145–3155.
22. Huile G, Shuaiqi P, Zhi Y, Shijie C, Chen C, Xinguo J, Shun S, Zhiqing P, Yu H. A cascade targeting strategy for brain neuroglial cells employing nanoparticles modified with angiopep-2 peptide and EGFP-EGF1 protein. Biomaterials. 2011; 32:8669–8675.
23. Ruan S, Yuan M, Zhang L, Hu G, Chen J, Cun X, Zhang Q, Yang Y, He Q, Gao H. Tumor microenvironment sensitive doxorubicin delivery and release to glioma using angiopep-2 decorated gold nanoparticles. Biomaterials. 2015; 37:425–435.
24. Cliff Wonga, Triantafyllos Stylianopoulosb, Jian Cuia, John Martin, Vikash P. Chauhan, Wen Jiang, Zoran Popovi’c, Rakesh K. Jain, Moungi G. Bawendia and Dai Fukumura. Multistage nanoparticle delivery system for deep penetration into tumor tissue. Pro. Acad. Natl. Sci. 2011; 108:2426–2431.
25. Li-Li Li, Jun-Hua Xu, Guo-Bin Qi, Xingzhong Zhao, Faquan Yu and HaoWang. *Core Shell Supramolecular Gelatin Nanoparticles for Adaptive and “On-Demand” Antibiotic Delivery. ACS Nano. 2014.
26. Zou Z, He X, He D, Wang K, Qing Z, Yang X, Wen L, Xiong J, Li L, Cai L. Programmed packaging of mesoporous silica nanocarriers for matrix metalloprotease 2-triggered tumor targeting and release. Biomaterials. 2015; 58:35–45.
27. Tong R, Hemmati HD, Langer R, Kohane DS. Photoswitchable nanoparticles for triggered tissue penetration and drug delivery. J Am Chem Soc. 2012; 134:8848–8855.
28. Han L, Ma H, Guo Y, Kuang Y, He X, Jiang C. pH-controlled delivery of nanoparticles into tumor cells. Adv Healthc Mater. 2013; 2:1435–1439.
29. Han L, Guo Y, Ma H, He X, Kuang Y, Zhang N, Lim E, Zhou W, Jiang C. Acid active receptor-specific peptide ligand for in vivo tumor-targeted delivery. Small. 2013; 9:3647–3658.
30. Yevlampieva N, Dobrodumov A, Nazarova O, Okatova O, Cottet H. Hydrodynamic Behavior of Dendrigraft Polylysines in Water and Dimethylformamide. Polymers. 2012; 4:20–31.
31. Hu FQ, Liu LN, Du YZ, Yuan H. Synthesis and antitumor activity of doxorubicin conjugated stearic acid-g-chitosan oligosaccharide polymeric micelles. Biomaterials. 2009; 30:6955–6963.
32. Yabbarov NG, Posypanova GA, Vorontsov EA, Obydenny SI, Severin ES. A new system for targeted delivery of doxorubicin into tumor cells. J Control Release. 2013; 168:135–141.
33. McRae Page S, Henchey E, Chen X, Schneider S, Emrick T. Efficacy of polyMPC-DOX prodrugs in 4T1 tumor-bearing mice. Mol Pharm. 2014; 11:1715–1720.
34. Tao K, Fang M, Alroy J, Sahagian GG. Imagable 4T1 model for the study of late stage breast cancer. BMC Cancer. 2008; 8:228–246.
35. Gao H, Yang Z, Zhang S, Pang Z, Liu Q, Jiang X. Study and evaluation of mechanisms of dual targeting drug delivery system with tumor microenvironment assays compared with normal assays. Acta biomaterialia. 2014; 10:858–867.
36. Goodman TT, Ng CP, Pun SH. 3-D tissue culture systems for the evaluation and optimization of nanoparticle-based drug carriers. Bioconjug Chem. 2008; 19:1951–1959.
37. Yu Y, Zhang X, Qiu L. The anti-tumor efficacy of curcumin when delivered by size/charge-changing multistage polymeric micelles based on amphiphilic poly(beta-amino ester) derivates. Biomaterials. 2014; 35:3467–3479.
38. Kohli AG, Kivimae S, Tiffany MR, Szoka FC. Improving the distribution of Doxil(R) in the tumor matrix by depletion of tumor hyaluronan. J Control Release. 2014; 191:105–114.
39. Fan Y, Li C, Cao H, Li F, Chen D. The intranuclear release of a potential anticancer drug from small nanoparticles that are derived from intracellular dissociation of large nanoparticles. Biomaterials. 2012; 33:4220–4228.
40. Rong Tong, Homer H. Chiang and Daniel S. Kohane. Photoswitchable nanoparticles for in vivo cancer chemotherapy. Pro. Natl. Acad. Sci. 2013; 110:19048–19053.
41. Xu JH, Gao FP, Liu XF, Zeng Q, Guo SS, Tang ZY, Zhao XZ, Wang H. Supramolecular gelatin nanoparticles as matrix metalloproteinase responsive cancer cell imaging probes. Chem Commun (Camb). 2013; 49:4462–4464.
42. Zhu Q, Jia L, Gao Z, Wang C, Jiang H, Zhang J, Dong L. A tumor environment responsive doxorubicin-loaded nanoparticle for targeted cancer therapy. Mol Pharm. 2014; 11:3269–3278.
43. Lee SJ, Yhee JY, Kim SH, Kwon IC, Kim K. Biocompatible gelatin nanoparticles for tumor-targeted delivery of polymerized siRNA in tumor-bearing mice. J Control Release. 2013; 172:358–366.
44. Santoro M, Tatara AM, Mikos AG. Gelatin carriers for drug and cell delivery in tissue engineering. J Control Release. 2014; 190:210–218.
45. Zhu S, Hong M, Tang G, Qian L, Lin J, Jiang Y, Pei Y. Partly PEGylated polyamidoamine dendrimer for tumor-selective targeting of doxorubicin: the effects of PEGylation degree and drug conjugation style. Biomaterials. 2010; 31:1360–1371.
46. Tatsiana G. Shutava, James A. Cardelli, Shantanu S. Balkundi, Pranitha Vangala* Joshua J. Steffan, D. Patrick O’Neal and Yuri M. Lvov, Rebecca L. Bigelow. Layer-by-Layer-Coated Gelatin Nanoparticles as a Vehicle for Delivery of Natural Polyphenols. ACS Nano. 2009; 3:1877–1885.
47. Li-Li Li, Jun-Hua Xu, Guo-Bin Qi, Xingzhong Zhao, Faquan Yu and HaoWang*. Core-Shell Supramolecular Gelatin Nanoparticles for Adaptive and “On-Demand” Antibiotic Delivery. ACS Nano. 2014.
48. Zhu L, Kate P, Torchilin VP. Matrix Metalloprotease 2-Responsive Multifunctional Liposomal Nanocarrier for Enhanced Tumor Targeting. ACS Nano. 2012; 6:3491–3498.
49. Wang H-X, Yang X-Z, Sun C-Y, Mao C-Q, Zhu Y-H, Wang J. Matrix metalloproteinase 2-responsive micelle for siRNA delivery. Biomaterials. 2014; 35:7622–7634.
50. Terada T, Iwai M, Kawakami S, Yamashita F, Hashida M. Novel PEG-matrix metalloproteinase-2 cleavable peptide-lipid containing galactosylated liposomes for hepatocellular carcinoma-selective targeting. J Control Release. 2006; 111:333–342.
51. Li R, Wu W, Liu Q, Wu P, Xie L, Zhu Z, Yang M, Qian X, Ding Y, Yu L, Jiang X, Guan W, Liu B. Intelligently Targeted Drug Delivery and Enhanced Antitumor Effect by Gelatinase-Responsive Nanoparticles. PLoS One. 2013; 8:e69643.