
INTRODUCTION
Acute myeloid leukemia (AML) is a highly heterogeneous malignancy, especially in terms of the molecular and phenotypic characteristics. Heterogeneity is also observed within the same genetic subgroup of AML tumors. For example, within nucleophosmin 1 (NPM1) mutant subgroup, some patients have concomitant mutations in fms-like-tyrosine kinase 3 (FLT3), isocitrate dehydrogenase (IDH) 1 and 2, DNA methyltransferase 3A (DNMT3A) or ten-elven translocation 2 (TET2) genes. A multi-hit model of leukemogenesis, in which class I mutations confer the proliferation or survival advantages of blast cells and class II mutations block myeloid differentiation, has been observed in most cases of AML [1]. Recently, other studies have also reported the epigenetic effects of class III mutations on AML [2, 3]. However, the exact roles of each alteration in leukemogenesis and the mechanisms of disease progression remain largely unkown, especially with respect to recent data on molecular intraclonal heterogeneity. Recent studies have shown that minimal residual disease (MRD) in AML patients, during or after treatment, has prognostic value [4–9]. However, there are many questions regarding the clinical assessment of MRD in AML patients. First, which of the potential molecular and/or cellular markers should be assessed? Second, what type of biological sample should be analyzed? Third, where should the sensitivity threshold be set, and what are the relevant time-points to consider for MRD assessment? One study found that IDH1/2 gene mutations persisted in patients who were in complete remission (CR), although other molecular markers were not analyzed at the time of AML diagnosis [10]. These mutations may be attributed to a preleukemic clone that acquires additional mutations promoting proliferation and differentiation block, which eventually results in leukemia. This preleukemic clone may be able to survive initial chemotherapy treatments. DNMT3A mutations, which occur in 20% of de novo AML cases, lead to abnormal DNA methylation patterns, which is likely to alter the expression of various target genes [11]. The prognostic impact of DNMT3A mutations seems to be unfavorable [12, 13], but their applicability MRD monitoring remains unclear [14].
Recent technological advances in next-generation sequencing (NGS) have provided new opportunities for MRD monitoring in AML patients and the possibility to simultaneously analyze multiple biomarkers and to detect subclonal populations.
In this study, we used NGS technology to monitor MRD using IDH1/2 and DNMT3A mutations in a cohort of NPM1 mutated AML patients. Our objective was to evaluate the suitability of IDH1/2 and DNMT3A mutations as a target for MRD detection by NGS and to compare the data with NPM1 mutation-based MRD assessed by RTqPCR
RESULTS
Of the 31 NPM1 mutated AML patients, 8 patients harbored an IDH1 mutation, 9 an IDH2 mutation and 15 DNMT3A mutation. Sequencing data showed sufficient sequencing depth with a median of 2,012,459 reads for IDH1/2 (range: 102,657 to 5,160,118 reads) and a median of 966,298 reads for DNMT3A (range: 565,152 to 2,700,349 reads). This coverage allowed the detection of MRD with a sensitivity of approximately 0.001%. Despite such an extensive coverage, a median of 520 reads were positive for mutations in the negative controls, reflecting cross-contamination due to the multiple steps involved in the preparation of the gene libraries (i.e., in the intra-run steps, including preliminary PCR, barcode purity, and adaptors/barcodes ligation, and the inter-run steps, including OT2 and clonal amplification). Thus, the detection limit was 0.07% for IDH1/2 mutation analysis (0.002 - 0.097, p < 0.01, Fisher’s exact test) and 0.11% for DNMT3A mutation analysis (0.001–0.426, p < 0,01, Fisher’s exact test).
MRD level was evaluated at the following time points: post induction (MRD1), post first consolidation course (MRD2) and post second consolidation courses (MRD3). The median clinical followup of the cohort was 673 days (range: 131–2637 days). NPM1 mutation and IDH1/2 mutation MRD levels and NPM1 mutation and DNMT3A mutation MRD levels were highly correlated (r = 0,68183, p < 0,0001; r = 0,55514, p < 0,0001, respectively). Of the 17 IDH1/2 mutation-positive patients, we found concordant MRD results between IDH1/2 and NPM1 mutation levels by RTqPCR in 13 cases. Four patients who relapsed were positive for both IDH1/2 and NPM1 mutations, and 9 patients who remained in complete remission were negative for both NPM1 and IDH1/2 mutations. In the 4 remaining patients, we observed a discrepancy between the NPM1 and IDH1/2 mutation levels. These patients presented with one or more MRD time-points with undetectable NPM1 mutation, whereas the IDH1/2 mutation levels ranged from 0.5% to 47% (Table 1). Three of the patients relapsed after 504, 395 and 158 days, and all of these patients harbored similar NPM1 mutation levels at diagnosis (Figure 1). The patient who did not relapse developed a NPM1-negative myelodysplastic syndrome. In the 15 DNMT3A mutant, we found concordant MRD results between DNMT3A mutation rates and NPM1 mutation levels in 9 cases. Eight out of these 9 patients were positive for both NPM1 and DNMT3A mutations and relapsed, while the remaining case, who remained in persistent CR, was negative for both NPM1 and DNMT3A mutations. In the 6 discordant cases, NPM1 mutations were undetectable in most MRD time-points, whereas DNMT3A mutation levels ranged from 5% to 45% during MRD follow-up. All of these patients remained in first complete remission after a median follow-up of 4 years (Table 2). DNMT3A mutations were detected at different time-points during follow-up (i.e., post-induction, post consolidation 1, and later), whereas other markers studied were undetectable (Figure 2). These results are consistent with clonal heterogeneity in AML, particularly on the molecular level. Samples from 3 of these patients were further examined to investigate the origin of the discrepancy using cell sorting analysis.
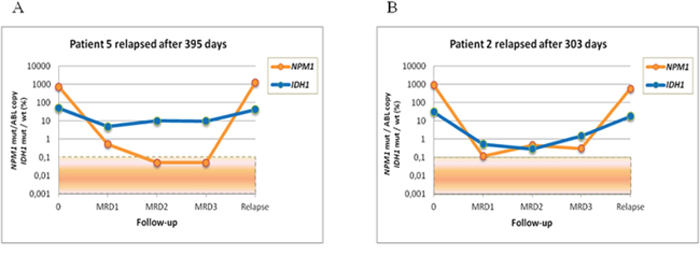
Figure 1: MRD monitoring in AML patients using NGS to analyze IDH1 mutations and using RTqPCR to analyze NPM1 mutations. A. Discrepancy between IDH1 and NPM1 mutations according to the MRD stages in patient 5. B. Correlation between IDH1 and NPM1 mutations according to the MRD stages in patient 2.
Table 1: NGS results for the 4 AML patients with discordant MRD levels between IDH1/IDH2 mutations and NPM1 mutation
Patients |
Age |
Time-point |
IDH1 R132 wild type (wt) |
IDH1 R132C mutated |
%IDH1 R132C mutated |
% RTqPCR NPM1 |
Status |
|---|---|---|---|---|---|---|---|
Patient 4 |
60 |
diagnosis |
1221691 |
918285 |
42.91 |
636 |
Relapse 504 days after diagnosis |
post induction (MRD1) |
2178994 |
152743 |
6.55 |
0.13 |
|||
post consolidation 1 (MRD2) |
2165327 |
202244 |
8.54 |
0.02 |
|||
Patient 8 |
61 |
diagnosis |
684823 |
600112 |
46.70 |
2155 |
Relapse 158 days after diagnosis |
MRD2 |
1236604 |
765254 |
38.22 |
0.01 |
|||
Patients |
Age |
Time-point |
IDH1 R132 wt |
IDH1 R132H mutated |
%IDH1 R132H mutated |
%RTqPCR NPM1 |
Status |
Patient 5 |
62 |
diagnosis |
976395 |
929710 |
48.77 |
734 |
Relapse 395 days after diagnosis |
MRD1 |
1712656 |
84303 |
4.69 |
0.52 |
|||
MRD2 |
1546662 |
166760 |
9.73 |
0.01 |
|||
post consolidation 2 (MRD3) |
1377611 |
147303 |
9.65 |
0.01 |
|||
relapse |
1822544 |
1249478 |
40.67 |
1252 |
|||
Patients |
Age |
Time-point |
IDH2 R140 wt |
IDH2 R140Q mutated |
%IDH2 R140Q mutated |
%RTqPCR NPM1 |
Status |
Patient 17 |
54 |
diagnosis |
512569 |
372370 |
42.07 |
425.07 |
Evolution to myelodysplastic syndrome |
MRD1 |
3848257 |
243415 |
5.94 |
0.24 |
|||
MRD2 |
524595 |
473055 |
47.41 |
0.01 |
Table 2: NGS results in the 6 AML patients who had discordant DNMT3A mutations compared with the results of NPM1 mutation
Patients |
Age |
Time-point |
DNMT3A R882 wild type (wt) |
DNMT3A R882C mutated |
%DNMT3A R882C mutated |
% RTqPCR NPM1 |
Status |
|---|---|---|---|---|---|---|---|
Patient 14 |
52 |
diagnosis |
719399 |
619232 |
46.25 |
797 |
complete remission (CR) at 73 months |
post induction (MRD1) |
1275523 |
416971 |
24.63 |
0.01 |
|||
post consolidation 1 (MRD2) |
925043 |
414427 |
30.93 |
0.1 |
|||
post consolidation 2 (MRD3) |
1406882 |
775928 |
35.54 |
0.01 |
|||
Patient 29 |
59 |
diagnosis |
1596027 |
1284406 |
44.59 |
5077.40 |
CR at 41 months |
MRD1 |
3920773 |
228372 |
5.50 |
0.26 |
|||
MRD3 |
3134982 |
579300 |
15.59 |
0.01 |
|||
Patient 31 |
23 |
diagnosis |
523504 |
388290 |
42.58 |
727.22 |
CR at 47 months |
MRD1 |
2302664 |
226061 |
8.93 |
0.30 |
|||
MRD2 |
2326575 |
391473 |
14.40 |
0.01 |
|||
MRD3 |
2452633 |
733293 |
23.01 |
0.01 |
|||
Patients |
Age |
Time-point |
DNMT3A R882 wt |
DNMT3A R882H mutated |
%DNMT3A R882H mutated |
%RTqPCR NPM1 |
Status |
Patient 20 |
57 |
diagnosis |
677880 |
446557 |
39.71 |
284 |
CR at 87 months |
MRD1 |
787783 |
341300 |
30.23 |
0.34 |
|||
Patient 30 |
56 |
diagnosis |
1028857 |
681492 |
39.85 |
706.29 |
CR at 46 months |
MRD1 |
1283146 |
1283146 |
38.46 |
0.54 |
|||
MRD3 |
1081738 |
1081738 |
45.25 |
0.04 |
|||
Patients |
Age |
Time-point |
DNMT3A Q886 wt |
DNMT3A Q886E mutated |
%DNMT3A Q886E mutated |
%RTqPCR NPM1 |
Status |
Patient 26 |
62 |
diagnosis |
821493 |
559728 |
40.52 |
284 |
CR at 49 months |
MRD2 |
1477600 |
555883 |
27.33 |
0,34 |
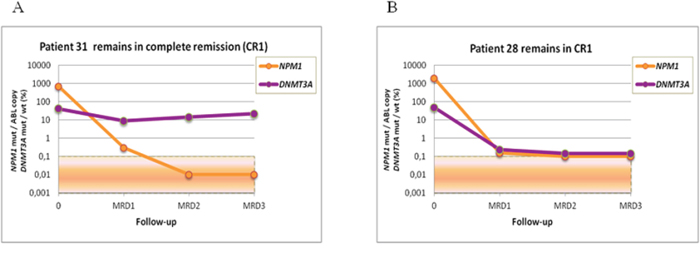
Figure 2: MRD monitoring in AML patients with DNMT3A mutations using NGS and NPM1 mutations using quantitative RTqPCR. A. Discrepancy between DNMT3A and NPM1 mutation rates according to MRD stages in patient 31. B. Correlation between DNMT3A and NPM1 mutations rates according to MRD stages in patient 28.
In the 3 patients for which cell subpopulations were available, DNMT3A mutations were found in the whole peripheral blood and bone marrow collected at which time-point, but not in the DNA extracted from a skin biopsy. These findings suggest that DNMT3A mutations were somatically acquired (Figure 3). The percentage of mutations in which gene in these patients were comparable in all the bone marrow cell subpopulations analyzed. In complete remission, all of the following cell subpopulations collected from peripheral blood (i.e., CD56+ NK cells, CD19+ B lymphocytes, CD14+ monocytes, CD66+ granulocytes, CD34+ CD45 - low blasts), and (i.e., CD34+ CD38- CD123+ and aldehyde dehydrogenase (ALDH) intermediate leukemic stem cells (LSCs) or CD34+ CD38- CD123+ and ALDH high hematopoietic stem cells (HSCs)) isolated from bone marrow harbored DNMT3A mutations but none of the molecular abnormalities (i.e., NPM1, IDH1/2 and FLT3-ITD mutations) identified at AML diagnosis. Interestingly, DNMT3A mutations were not found in CD3+ T lymphocytes of these patients (Table 3).
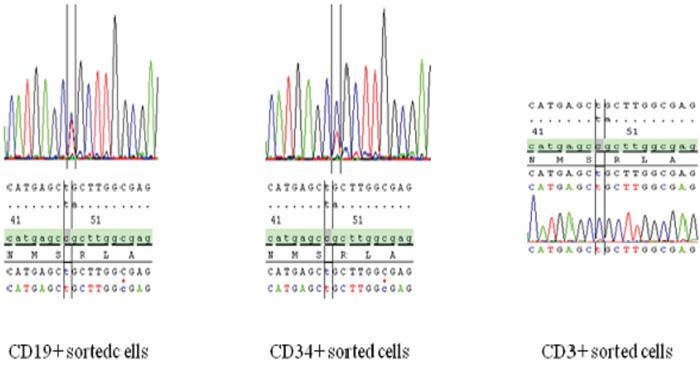
Figure 3: Sequencing results for the different blood fractions showing DNMT3A mutations in all fractions except in the CD3+ T lymphocyte fraction.
Table 3: Molecular abnormalities for the 3 patients who harbored DNMT3A mutations at complete remission
UPN |
Sample type |
Mutation at AML diagnosis |
Cell fraction analyzed |
FLT3 |
NPM1 |
DNMT3A |
|---|---|---|---|---|---|---|
14 |
blood |
DNMT3A R882C |
total |
ND |
ND |
+ |
BM |
total |
+ |
||||
skin |
total |
– |
||||
blood |
CD3 |
– |
||||
blood |
CD19 |
+ |
||||
blood |
CD56 |
+ |
||||
blood |
CD14 |
+ |
||||
blood |
CD66 |
+ |
||||
blood |
CD34 |
+ |
||||
BM |
CD34+CD 38-CD123-ALDH high |
+ |
||||
BM |
CD34+CD 38-CD123-ALDH int |
+ |
||||
29 |
blood |
FLT3-TKD, NPM1A, DNMT3A R882C |
total |
– |
– |
+ |
BM |
total |
+ |
||||
skin |
total |
– |
||||
blood |
CD3 |
– |
||||
blood |
CD19 |
+ |
||||
blood |
CD56 |
+ |
||||
blood |
CD14 |
+ |
||||
blood |
CD66 |
+ |
||||
blood |
CD34 |
+ |
||||
BM |
CD34+CD38-CD123+ ALDH high |
+ |
||||
BM |
CD34+CD 38-CD123+ ALDH int |
+ |
||||
31 |
blood |
FLT3-ITD, NPM1A, DNMT3A R882C |
total |
ND |
ND |
+ |
BM |
total |
+ |
||||
skin |
total |
– |
||||
blood |
CD3 |
– |
||||
blood |
CD19 |
+ |
||||
blood |
CD56 |
+ |
||||
blood |
CD14 |
+ |
||||
blood |
CD66 |
+ |
||||
blood |
CD34 |
+ |
||||
BM |
CD34+CD38-CD123+ ALDH high |
– |
– |
+ |
||
BM |
CD34+CD 38-CD123+ ALDH int |
+ |
For each patient, the mutations that were found at diagnosis (column 3) were analyzed in the following fractions: blood, BM and skin (column 2). Column 4 provides details on the different subpopulations analyzed in the BM, the CD34+ CD38-CD123+ ALDH intermediate and the CD34+ CD38-CD123-ALDH high cells were considered enriched in leukemia-initiating cells and normal hematopoietic stem cells, respectively.
Abbreviation: BM, bone marrow; ND, not determined; UPN, unique patient number.
DISCUSSION
Our data suggest that the use of NGS to monitor MRD based on IDH1/2 mutations is feasible and effective, as this method enabled us to predict relapse in the majority of patients, with an area under the curve of 0,7971 (95% CI: 0,6693 – 0,9250), and a success rate of 100% if we include the patient who developed MDS. In our cohort IDH1/2 mutations-based MRD better predicted relapse than NPM1 mutations-based MRD.
IDH1/2 mutated patients may benefit from new targeted therapies with specific molecules inhibiting of IDH1 or IDH2 mutant proteins [15–18].
Of these targeted inhibitors, 3734 AG-120 is effective at lowering 2-HG levels and restoring cellular differentiation in primary AML cells. This therapy could be more personalized by monitoring IDH1/2 mutation levels by NGS during and after treatment.
In contrast, DNMT3A mutations were not a suitable markers for MRD monitoring because of the persistence of a preleukemic clone carrying DNMT3A mutations in 40% of the patients who were in complete remission after a median follow-up of 1439 days (range: 1154–2637 days) which likely reflects the intraclonal molecular heterogeneity of hematopoiesis. Changes in gene mutation frequency were reported between AML diagnosis and relapse, and the expansion of a subclone initially present at a low frequency at the time of diagnosis has been observed at relapse [19].
Our results of the sorted cell populations confirm the molecular heterogeneity of hematopoietic clones at complete remission Liran et al. [20] also reported the presence of DNMT3A mutations at a high allelic frequency in highly purified HSCs, progenitors and mature blood cell fractions in AML patients in complete remission but did not observe concurrent NPM1 mutations, present in the blast cells at AML diagnosis. DNMT3A mutant HSCs showed a multi-lineage repopulation advantage over the non-mutated HSCs in xenograft experiments, which suggests that these cells were pre-leukemic HSCs [21]. Altogether, these data suggest that DNMT3A mutations could induce “pre-leukemic” abnormal hematopoiesis but remain insufficient for leukemogenesis.
We were not able to perform the mutation screening in samples collected before the diagnosis of AML in our patient population to determine whether DNMT3A mutations may have preexisted in the patients whose samples showed a discrepancy between DNMT3A and NPM1 mutation MRD levels. However, the observed DNMT3A variant allele frequency in the patients who were in complete remission after chemotherapy ranged from 5 to 45% at post-induction and increased during follow-up, unlike other MRD markers.
Although we could not evaluate whether mutated DNMT3A was present in HSCs before AML diagnosis, 3 groups independently reported the emergence of neoplastic blood cell clones with aging [22–24]. Jaiswal et al. reported that DNMT3A mutation were the most frequent mutations observed with aging and that patients with DNMT3A mutations had a 10- to 50-fold higher propensity for developing hematologic cancer [24]. Similarly, Genovese et al. reported that the frequency of mutations among individuals older than 65 years was 10% and that the most frequent mutations affected DNMT3A gene [23]. They also reported that DNMT3A mutation was associated with increased risk for developing hematologic cancer that was related to the earlier clone. Our patients were not over 65 years of age and demonstrated an elevated VAF during the CR stage that was higher than the level observed with aging, which suggests that the mutant HSCs were resistant to chemotherapy. How these abnormal hematopoietic clones will be involved in relapse or in the occurrence of new hematological malignancies should be monitored on a long term period.
PATIENTS AND METHODS
Patients and samples
This retrospective study included 94 samples from 31 NPM1 mutation-positive patients (23–70 years old; median: 60 yrs) who were newly diagnosed with AML from the Acute Leukemia French Association (ALFA) - 0701 trial.
Molecular analysis
NPM1 mutation monitoring by RTqPCR was performed as previously described [25]. IDH1/2 and DNMT3A mutation monitoring was performed by NGS using an Ion Torrent ProtonTM instrument (life technologies). To obtain very high coverage (i.e., approximately 2 million reads per sample), 24 samples were analyzed per run. Bioinformatic analysis was performed as described in our previous work [26].
Samples from three patients in CR who had persistent DNMT3A mutations but no other abnormalities were more extensively investigated. DNMT3A quantification by NGS was performed on the following samples: skin, whole peripheral blood, whole bone marrow and blood subpopulations.
Flow cytometry
The following blood subpopulations were sorted using positive selection immuno-bead kits (Easy Sep Stem Cell): Neutrophils targeted by CD66b antibodies (Ab), monocytes targeted by CD14 Ab, T lymphocytes targeted by CD3 Ab, B lymphocytes targeted by CD19 Ab and NK cells targeted by CD56 Ab. The bone marrow subpopulations that were notably enriched in leukemia-initiating cells included CD34+, CD38-, CD123+, and ALDH intermediate cells. These cells were sorted using a FACS ARIA Sorp based on the membrane expression levels of CD34, CD38, and CD123 and the level of ALDH activity [27].
CONCLUSION
The NGS technique is an effective tool to monitor MRD in AML patients, but choosing the appropriate MRD markers is crucial to avoid results that are not related to the disease status. Altogether, our findings show that DNMT3A mutation does not participate to relapse or leukemia progression during our period of clinical follow up. Screening of leukemia-initiating mutations, such as DNMT3A, NPM1 or IDH1/2 mutations should be performed at diagnosis but only NPM1 and IDH1/2 are robust target for MRD monitoring.
ACKNOWLEDGMENTS AND FUNDING
This work was supported by the Association Laurette Fugain, the Ligue Contre le Cancer (North Center), the SIRIC ONCOLille - the North-West Canceropole and the Institut National du Cancer, France.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Gilliland DG, Jordan CT, Felix CA. The molecular basis of leukemia. Hematol Educ Program Am Soc Hematol Am Soc Hematol Educ Program 2004:80–97.
2. Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, Tallman MS, Sun Z, Wolniak K et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010; 18:553–567.
3. Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 2013; 368:2059–2074.
4. Perea G, Lasa A, Aventín A, Domingo A, Villamor N, Queipo de Llano MP, Llorente A, Juncà J, Palacios C, Fernández C, Gallart M, Font L, Tormo M et al. Prognostic value of minimal residual disease (MRD) in acute myeloid leukemia (AML) with favorable cytogenetics [t(8;21) and inv(16)]. Leukemia 2006; 20:87–94.
5. Grimwade D, Jovanovic JV, Hills RK, Nugent EA, Patel Y, Flora R, Diverio D, Jones K, Aslett H, Batson E, Rennie K, Angell R, Clark RE et al. Prospective minimal residual disease monitoring to predict relapse of acute promyelocytic leukemia and to direct pre-emptive arsenic trioxide therapy. J Clin Oncol Off J Am Soc Clin Oncol 2009; 27:3650–3658.
6. Krönke J, Schlenk RF, Jensen K-O, Tschürtz F, Corbacioglu A, Gaidzik VI, Paschka P, Onken S, Eiwen K, Habdank M, Späth D, Lübbert M, Wattad M et al. Monitoring of minimal residual disease in NPM1-mutated acute myeloid leukemia: a study from the German-Austrian acute myeloid leukemia study group. J Clin Oncol Off J Am Soc Clin Oncol 2011; 29:2709–2716.
7. Buccisano F, Maurillo L, Del Principe MI, Del Poeta G, Sconocchia G, Lo-Coco F, Arcese W, Amadori S, Venditti A. Prognostic and therapeutic implications of minimal residual disease detection in acute myeloid leukemia. Blood 2012; 119:332–341.
8. Hourigan CS, Karp JE. Minimal residual disease in acute myeloid leukaemia. Nat Rev Clin Oncol 2013; 10:460–471.
9. Shayegi N, Kramer M, Bornhäuser M, Schaich M, Schetelig J, Platzbecker U, Röllig C, Heiderich C, Landt O, Ehninger G, Thiede C. The level of residual disease based on mutant NPM1 is an independent prognostic factor for relapse and survival in AML. Blood 2013; 122:83–92.
10. Chou W-C, Peng K-Y, Lei W-C, Ko BS, Tsay W, Kuo CH, Tien HF. Persistence of mutant isocitrate dehydrogenase in patients with acute myeloid leukemia in remission. Leukemia 2012; 26:527–529.
11. Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, Kandoth C, Payton JE, Baty J, Welch J, Harris CC, Lichti CF, Townsend RR et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med 2010; 363:2424–2433.
12. Thol F, Damm F, Lüdeking A, Winschel C, Wagner K, Morgan M, Yun H, Göhring G, Schlegelberger B, Hoelzer D, Lübbert M, Kanz L, Fiedler W et al. Incidence and prognostic influence of DNMT3A mutations in acute myeloid leukemia. J Clin Oncol Off J Am Soc Clin Oncol 2011; 29:2889–2896.
13. Renneville A, Boissel N, Nibourel O, Berthon C, Helevaut N, Gardin C, Cayuela JM, Hayette S, Reman O, Contentin N, Bordessoule D, Pautas C, Botton Sd et al. Prognostic significance of DNA methyltransferase 3A mutations in cytogenetically normal acute myeloid leukemia: a study by the Acute Leukemia French Association. Leukemia 2012; 26:1247–1254.
14. Hou H-A, Kuo Y-Y, Liu C-Y, Chou WC, Lee MC, Chen CY, Lin LI, Tseng MH, Huang CF, Chiang YC, Lee FY, Liu MC, Liu CW et al. DNMT3A mutations in acute myeloid leukemia: stability during disease evolution and clinical implications. Blood 2012; 119:559–568.
15. Wang F, Travins J, DeLaBarre B, Penard-Lacronique V, Schalm S, Hansen E, Straley K, Kernytsky A, Liu W, Gliser C, Yang H, Gross S, Artin E et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science 2013; 340:622–626.
16. Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, Tsoi J, Clark O, Oldrini B, Komisopoulou E, Kunii K, Pedraza A, Schalm S et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 2013; 340:626–630.
17. Pirozzi CJ, Reitman ZJ, Yan H. Releasing the Block: Setting Differentiation Free with Mutant IDH Inhibitors. Cancer Cell 2013; 23:570–572.
18. Chaturvedi A, Araujo Cruz MM, Jyotsana N, Sharma A, Yun H, Görlich K, Wichmann M, Schwarzer A, Preller M, Thol F, Meyer J, Haemmerle R, Struys EA et al. Mutant IDH1 promotes leukemogenesis in vivo and can be specifically targeted in human AML. Blood 2013; 122:2877–2887.
19. Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS, Ritchey JK, Young MA, Lamprecht T, McLellan MD, McMichael JF, Wallis JW, Lu C et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 2012; 481:506–510.
20. Shlush LI, Zandi S, Mitchell A, Chen WC, Brandwein JM, Gupta V, Kennedy JA, Schimmer AD, Schuh AC, Yee KW, McLeod JL, Doedens M, Medeiros JJ et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 2014; 506:328–333.
21. Chan SM, Majeti R. Role of DNMT3A, TET2, and IDH1/2 mutations in pre-leukemic stem cells in acute myeloid leukemia. Int J Hematol 2013; 98:648–657.
22. Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, McMichael JF, Schmidt HK, Yellapantula V, Miller CA, Ozenberger BA, Welch JS, Link DC et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med 2014; 20:1472–1478.
23. Genovese G, Kähler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, Chambert K, Mick E, Neale BM, Fromer M, Purcell SM, Svantesson O, Landén M et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 2014; 371:2477–2487.
24. Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A, Higgins JM, Moltchanov V, Kuo FC et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 2014; 371:2488–2498.
25. Lambert J, Lambert J, Nibourel O, Pautas C, Hayette S, Cayuela JM, Terré C, Rousselot P, Dombret H, Chevret S, Preudhomme C, Castaigne S, Renneville A. MRD assessed by WT1 and NPM1 transcript levels identifies distinct outcomes in AML patients and is influenced by gemtuzumab ozogamicin. Oncotarget 2014; 5:6280–6288.
26. Abdelhamid E, Figeac M, Renneville A, Quief S, Villenet C, Boyer T, Nibourel O, Coiteux V, Cassinat B, Lippert E, Helevaut N, Soua Z, Preudhomme C. Quantification of JAK2V617F mutation by next-generation sequencing technology. Am J Hematol 2013; 88:536–537.
27. Gerber JM, Smith BD, Ngwang B, Zhang H, Vala MS, Morsberger L, Galkin S, Collector MI, Perkins B, Levis MJ, Griffin CA, Sharkis SJ, Borowitz MJ et al. A clinically relevant population of leukemic CD34(+)CD38(-) cells in acute myeloid leukemia. Blood 2012; 119:3571–3577.