
INTRODUCTION
Hepatocellular carcinoma (HCC) is among the top three causes of cancer death in the Asian Pacific region [1]. Hepatitis B virus (HBV) of the hepadnavirus family confers major risk for HCC. In most high-risk HCC regions, HBV is associated with most cases of cirrhosis and at least 80% of HCC cases [2]. However, how HBV contributes to development of HCC is unclear.
MicroRNAs (miRNAs) are small single-stranded noncoding RNAs that regulate gene expression by interacting preferentially with the 3′untranslated regions (3′-UTRs) of target mRNAs, which may result in either inhibition of the target protein translation or degradation of the target mRNA [3, 4]. miRNAs and their target corresponding mRNAs form complex regulatory networks that are involved in cell proliferation, apoptosis, differentiation stress responses and other biological processes [5, 6]. miRNAs aberrant expression also contributes to a range of human pathologies, including cancer [7]. More and more reports have shown deregulation of miRNAs in human HCCs in recently years. Discovery of the critical role of miRNAs in modulating gene expression has not only changed our concept of gene expression regulation, but has also offered a new opportunity for designing anticancer strategies and therapies in HCC [8, 9].
As a major pathogen, did HBV involved in the dysregulation of miRNAs expression in HCC? It has been reported that HBV didn’t encoded their own miRNAs but manipulate cellular miRNA expression [10]. miR-148a expression reduced growth, epithelial-to-mesenchymal transition, invasion, and metastasis of HBx-expressing HCC cells via inhibiting HPIP-mediated mTOR signaling [11]. miR-29c may contribute to tumor suppressive miRNA in the development and progression of HBV-related HCC by targeting TNFAIP3 [12]. miR-181 induces carcinogenesis in HBV-related HCC by targeting E2F5 and Fas [13, 14].
It has been reported that miR-331-3p promotes proliferation and metastasis of HCC by targeting the PH domain and leucine-rich repeat protein phosphatase (PHLPP) [15]. Recently, a miRNA microarray analysis showed that miR-331-3p was upregulated in a chronic HBV infection model HepG2.2.15 [16] cell line compared to the parental control HepG2 cell line [17]. Thus, the pathogenesis of miR-331-3p dysregulation in HBV-related HCC and the carcinogenesis of HBV and miR-331-3p in HCC development and progression warrants study.
To further study the function of miR-331-3p in HCC cell lines, we searched in the database for miR-331-3p candidate target genes, and detected gene expression in miR-331-3p over-expression SMMC7721 cells or HepG2 and HepG2.2.15 cells by using qRT-RCR. ING5 can be downregulated by miR-331-3p and also inhibited by HBV, so we chosen ING5 for our next step research. The inhibitor of growth 5 (ING5) gene encodes a 28-kD protein [18] and its over-expression in colorectal cancer cells diminished colony-forming efficiency, decreased the number of cells in the S phase, and induced apoptosis in a p53-dependent manner [19]. Altered ING5 expressions have been reported in certain cancers including gastric carcinogenesis, human head and neck squamous cell carcinoma, and oral squamous cell carcinoma, functioned as a suppressor by inhibiting cell growth and inducing apoptosis [20–22]. However, as a tumor suppressor gene, ING5 functions have not been reported in HCC. The discovery of HBV, miRNAs, and target gene interactions may offer insight into mechanisms of HCC as well as provide novel therapeutic strategies for treating HCC.
RESULTS
HBV upregulates expression of miR-331-3p by enhancing its promoter activity
Firstly, expression of miR-331-3p in a panel of cell lines including HepG2, HepG2.2.15 was measured using qRT-PCR. miR-331-3p expression was greater in HepG2.2.15 cells compared to HepG2 cells (Figure 1A), that consistent with previous miRNAs microarray data [17]. In a similar approach, another HCC cell line SMMC7721 cells were transfected with pCH9 (vector control) or pCH9/3091 (HBV-expressing) plasmids, and their miR-331-3p expression was measured. Again, miR-331-3p expression was increased in SMMC7721 transiently expressing HBV compared to controls (Figure 1B). miR-331-3p expression was also suppressed in tetracycline (Tet) treated HepAD38 compared to those in untreated cells (Figure 1C). HBsAg and HBeAg were measured by ELISA in three cell lines to confirm HBV expression (Figure 1D). Therefore, HBV promoted miR-331-3p expression in different HCC cell lines.
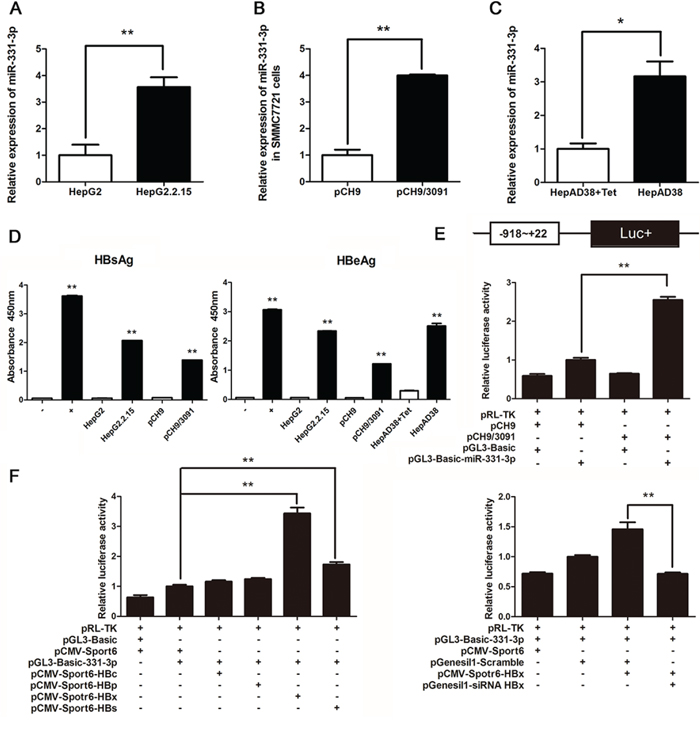
Figure 1: HBV upregulates miR-331-3p expression by enhancing its promoter activity. A–C. Relative miR-331-3p expression in HepG2 and HepG2.2.15 cells (A), SMMC7721 cells transfected with pCH9 or pCH9/3091 plasmids (B) and HepAD38 cells with/without Tet treatment (C) miRNA was normalized to U6 RNA. D. HBsAg and HBeAg expression in HCC cell lines. E. miR-331-3p promoter activity in SMMC7721 cells transfected with pCH9 or pCH9/3091 measured by Dual luciferase reporter analysis. F. miR-331-3p promoter activity in SMMC7721 cells co-transfected with DNAs or RNAs as indicated and measured by Dual luciferase reporter analysis. *P < 0.05, **P < 0.01.
To investigate the mechanism of miR-331-3p upregulation by HBV, we constructed pGL3-Basic-miR-331-3p vector which contains the miR-331-3p promoter for the luciferase reporter according to luciferase reporter assay. miR-331-3p promoter activity was higher in the pCH9/3091 and pGL3-Basic-miR-331-3p co-transfected group compared to pCH9 and pGL3-Basic-miR-331-3p co-transfected group (Figure 1E).
To investigate the possible mechanism by which HBV regulate miR-331-3p promoter activity, viral proteins HBc, HBp, HBx and HBs expression plasmids [23] were transfected respectively in HCC cells expressing miR-331-3p. Dual luciferase reporter assay found that viral protein HBx significantly enhanced miR-331-3p promoter activity while HBx knockdown inhibited its promoter activity (Figure 1F). These data suggested that HBV, especially HBx, could upregulate miR-331-3p expression by increasing its promoter activity.
miR-331-3p promotes proliferation of HCC cells
After we confirmed a correlation between miR-331-3p and HBV expression, we functionally characterized miR-331-3p by studying its effect on HCC cell proliferation. SMMC7721 cells were transiently transfected with pTARGET, pTARGET-miR-331-3p, inhibitor NC or miR-331-3p inhibitor, and miR-331-3p relative expression was measured by qRT-PCR. Cell proliferation was measured using MTS assay. miR-331-3p expression was greater in pTARGET-miR-331-3p transfected SMMC7721 cells compared to the vector pTARGET transfected cells. As shown in Figure 2A, miR-331-3p overexpression significantly promoted cell proliferation. When SMMC7721 cells were transfected miR-331-3p inhibitor, miR-331-3p expression was lower and cell proliferation was repressed compared to those in NC cells (Figure 2B).
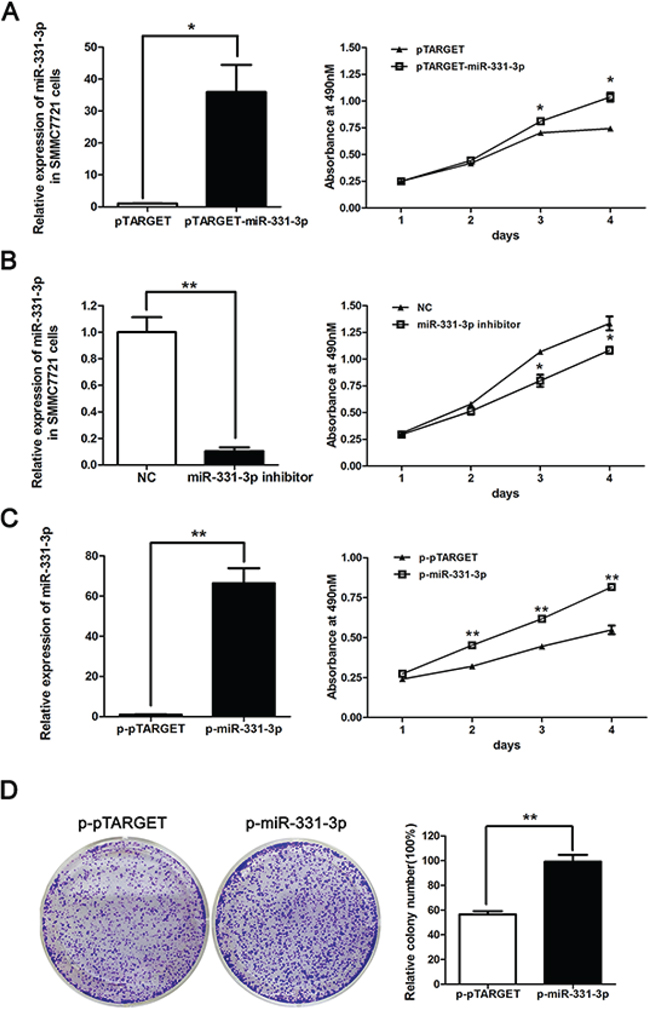
Figure 2: miR-331-3p promotes proliferation of HCC cells. A. Relative miR-331-3p expression in pTARGET-miR-331-3p and pTARGET transfected SMMC7721 cells and effect of miR-331-3p over-expression on cell proliferation measured by MTS assay. B. Relative miR-331-3p expression in miR-331-3p inhibitor or NC transfected SMMC7721 cells and effect of miR-331-3p inhibitor on cell proliferation measured by MTS assay. C. Relative miR-331-3p expression in p-miR-331-3p compares to p-pTARGET stable cell lines, p-miR-331-3p and p-pTARGET cell proliferation were measured by MTS assay. miRNA was normalized to U6 RNA. D. Representative pictures of colony formation assay with p-pTARGET and p-miR-331-3p. Colonies were counted and values are reported as ratios. *P < 0.05, **P < 0.01.
To confirm these data, SMMC7721 cell lines stably expressing miR-331-3p, p-miR-331-3p and its negative control p-pTARGET, were constructed. Overexpression of miR331-3p was confirmed using qRT-PCR. MTS and colony formation assay results indicated that both cell and colony numbers were increased in p-miR-331-3p-expressing cells compared to control cells (Figure 2C and 2D). Therefore, miR-331-3p can promote cell proliferation in HCC cell lines.
ING5 is a target gene of miR-331-3p
To better understand mechanisms of growth regulation by miR-331-3p, a bioinformatic website miRBase (http://mirbase.org/index.shtml) was used to search for downstream genes of miR-331-3p. Using website data, we screened several target genes and compared their expression in pTARGET or pTARGET-miR-331-3p transfected SMMC7721 cells. We found that ING5, VHL, ERBB2 and SPOP expression were inhibited by miR-331-3p overexpression. DOHH, a gene that has been reported to be a target gene of miR-331-3p [25] was included as a positive control. We then examined expression of these genes in HepG2 and HepG2.2.15 cells. Again ING5, VHL and ERBB2 expression were suppressed in the HBV-expressing HepG2.2.15 cells that have elevated expression of miR-331-3p (Figure 3A). We finally chose ING5 for subsequent studies.
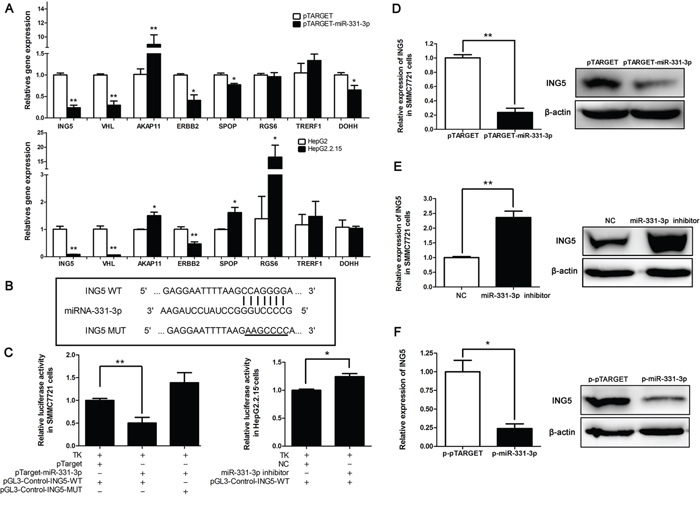
Figure 3: ING5 is a target gene of miR-331-3p. A. Relative gene expression in pCH9 and pCH9/3091 transfected SMMC7721 cells (upper panel) and in HepG2 and HepG2.2.15 cells (lower panel). B. Schematic diagram of predicted miR-331-3p binding site WT or MUT in the ING5 3′-UTR. C. Luciferase reporter assays in SMMC7721 (left panel) or HepG2.2.15 cells (right panel), co-transfected with DNAs or RNAs as indicated. D. ING5 mRNA and protein expression in SMMC7721 cells transfected with pTARGET-miR-331-3p or its control. E. ING5 mRNA and protein expression in SMMC7721 cells transfected with miR-331-3p inhibitor or NC. F. ING5 mRNA and protein expression in p-pTARGET and p-miR-331-3p. β-actin was used as an internal quantitative control. *P < 0.05, **P < 0.01.
MiRNA regulates gene expression by interacting preferentially with the 3′-untranslated regions (3′-UTRs) of its target mRNAs. To confirm that ING5 is a target gene of miR-331-3p, pGL3-Control-ING5-WT containing the 3′-UTR binding site (CCAGGGG) for miR-331-3p, and the pGL3-Control-ING5-MUT containing a mutated site (AAGCCCC) was constructed (Figure 3B). The 3′-UTR binding site was predicted in miRDB. A luciferase assay showed that luciferase activity was lower in SMMC7721 cells co-transfected with pTARGET-miR-331-3p and pGL3-Control-ING5-WT than that in SMMC7721 cells co-transfected with pTARGET and pGL3-Control-ING5-WT, and recovered in SMMC7721 cells co-transfected with pTARGET-miR-331-3p and pGL3-Control-ING5-MUT. When HepG2.2.15 cells were co-transfected with miR-331-3p inhibitor and pGL3-Control-ING5-WT, luciferase activity was increased (Figure 3C). These data suggested that miR-331-3p significantly decreased luciferase activity of the ING5 WT 3′-UTR but not the mutant 3′-UTR.
Next, qRT-PCR and Western blot analyses were used to assess effects of miR-331-3p on ING5 expression in HCC cells. Our data showed that ING5 expression decreased in SMMC7721 cells overexpressing miR-331-3p at both mRNA and protein levels (Figure 3D). On the other hand, when miR-331-3p expression was inhibited we observed an increase of ING5 expression in the SMMC7721 cells (Figure 3E). This also occurred in p-pTARGET and p-miR-331-3p cells (Figure 3F). Taken together, we conclude that ING5 is a target gene of miR-331-3p and ING5 expression is inhibited by miR-331-3p in HCC cell lines.
HBV represses ING5 expression by upregulating miR-331-3p
We next investigated the regulating effect between HBV and ING5. At first, we examined ING5 expression in HBV-expressing HCC cell lines. Both qRT-PCR and Western blot results showed that ING5 expression was inhibited in HepG2.2.15 and HBV transiently transfected SMMC7721 cells (Figure 4A and 4B). Upregulated ING5 expression was also observed in Tet treated HepAD38 cells compared to untreated cells (Figure 4C). We concluded that ING5 expression could be inhibited by HBV.
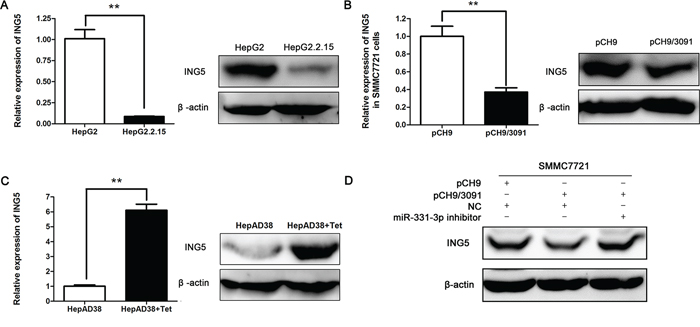
Figure 4: HBV represses ING5 expression by upregulating miR-331-3p. A–C. ING5 mRNA and protein expression in HepG2 and HepG2.2.15 cells (A), pCH9 and pCH9/3091 transfected SMMC7721 cells (B) and HepAD38 cells treated with Tet or not (C) β-actin was used as an internal quantitative control. *P < 0.05, **P < 0.01. D. ING5 protein expression in SMMC7721 cells cotransfected with DNAs or RNAs as indicated.
We next assessed whether HBV inhibited ING5 expression by regulating miR-331-3p. To this end, SMMC7721 cells were co-transfected with pCH9/3091 and miR-331-3p inhibitor. As shown in Figure 4D, ING5 protein expression was reduced in SMMC7721 cells expressing only HBV (co-transfection with NC and pCH9/3091) and reversed when the cells express both HBV and miR-331-3p inhibitor (co-transfection with miR-331-3p inhibitor and pCH9/3091). This result suggested that HBV represses ING5 expression by upregulating miR-331-3p.
ING5 inhibits HCC cell proliferation
To assess how ING5 modulates HCC cell proliferation, we constructed a pcDNA3.1-ING5 vector that could over express ING5 (the 3′-UTR sequence of ING5 was not included) and an ING5 siRNA. As expected, ING5 over-expression inhibited cell proliferation and ING5 silencing promoted cell proliferation (Figure 5A and 5B).
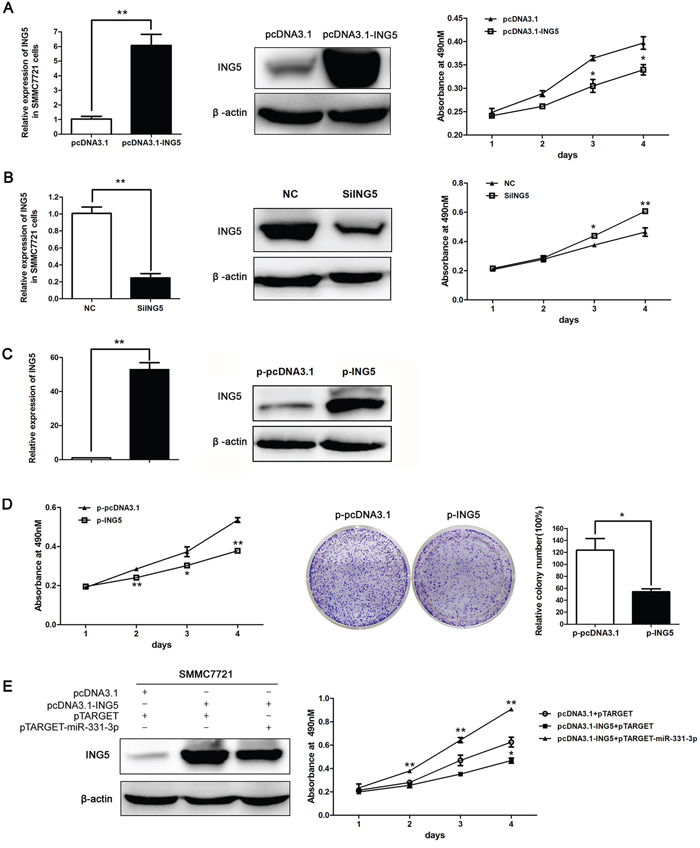
Figure 5: ING5 inhibited HCC cell proliferation. A. ING5 mRNA and protein expression in SMMC7721 cells transfected with pcDNA3.1 or pcDNA3.1-ING5, effect of ING5 overexpression on cell proliferation was measured by MTS assay. B. ING5 mRNA and protein expression in siING5 transfected SMMC7721 cells compared to NC; effect of ING5 silencing on cell proliferation was measured by MTS assay. C. ING5 mRNA and protein expression in p-pcDNA3.1 and p-ING5 stable cell lines. (A-C) β-actin was used as an internal quantitative control. D. p-ING5 and p-pcDNA3.1 cell proliferation measured by MTS and colony formation assays. Colonies were counted and values are reported as ratios. E. ING5 protein expression in SMMC7721 cells co-transfected with DNAs as indicated. MTS assay was used to measure cell proliferation of SMMC7721 cells co-transfected with DNAs as indicated. *P < 0.05, **P < 0.01.
Then, we constructed an ING5 stably expressing cell line p-ING5 and a control group p-pcDNA3.1 based on SMMC7721. Over-expression efficiency was measured with qRT-PCR and Western blot (Figure 5C). MTS and colony formation assay data indicated that cell proliferation and number of colonies decreased in p-ING5 compared to p-pcDNA3.1 (Figure 5D). Thus, ING5 can repress HCC cell proliferation.
Finally, we transfected SMMC7721 with three vector groups as indicated in Figure 5E. ING5 expression was increased in pcDNA3.1-ING5 and pTARGET co-transfected groups and decreased in pcDNA3.1-ING5 and pTARGET-miR-331-3p co-transfected groups as measured by Western blot analysis. MTS assay indicated that cell proliferation was decreased by ING5 and increased by miR-331-3p (Figure 5E). Therefore, miR-331-3p promotes cell proliferation at least partly by inhibiting ING5.
ING5 induces HCC cell apoptosis
As the ING is known as an apoptosis inducer [20–22, 24], whether miR-331-3p/INR5 could inhibit apoptosis in the HCC? Cell apoptosis analysis was performed. We found that HCC cells showed enhanced apoptosis rates when ING5 was overexpressed compared to its control group (Figure 6A). And apoptosis rate was recovered when miR-331-3p was overexpressed in ING5 overexpression cells (Figure 6B). These results indicate that miR-331-3p/INR5 could inhibit apoptosis in the HCC.
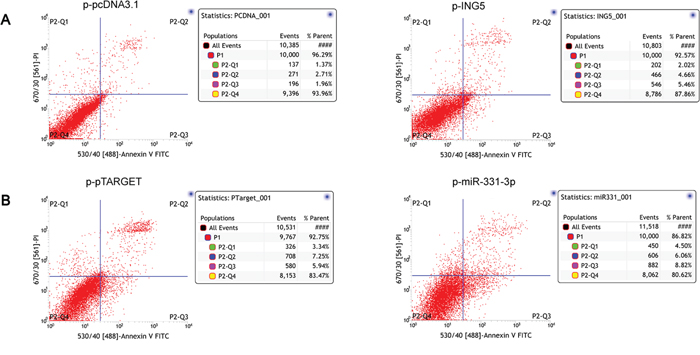
Figure 6: ING5 induces HCC cell apoptosis. A. ING5 stable overexptession HCC cells (p-ING5) apoptosis were analyzed. p-pcDNA3.1 stable HCC cells were used as control. B. p-miR-331-3p, p-pTARGET (control) plasmids were transfected into ING5 stable overexptession HCC cells, apoptosis were analyzed by flow cytometry.
miR-331-3p or ING5 over-expression influence tumor growth in nude mice
p-pTARGET, p-miR-331-3p, p-pcDNA3.1 or p-ING5 cells were injected subcutaneously into the upper left dorsal flank of nude mice. At 5 weeks after inoculation, mice injected with p-miR-331-3p cells had larger tumors compared to p-pTARGET (Figure 7A). Tumor growth curves showed that tumors grew faster in the p-miR-331-3p injected group (Figure 7B). At 4 weeks after inoculation, mice injected with p-ING5 cells had smaller tumors (Figure 7C) and these grew more slowly (Figure 7D).
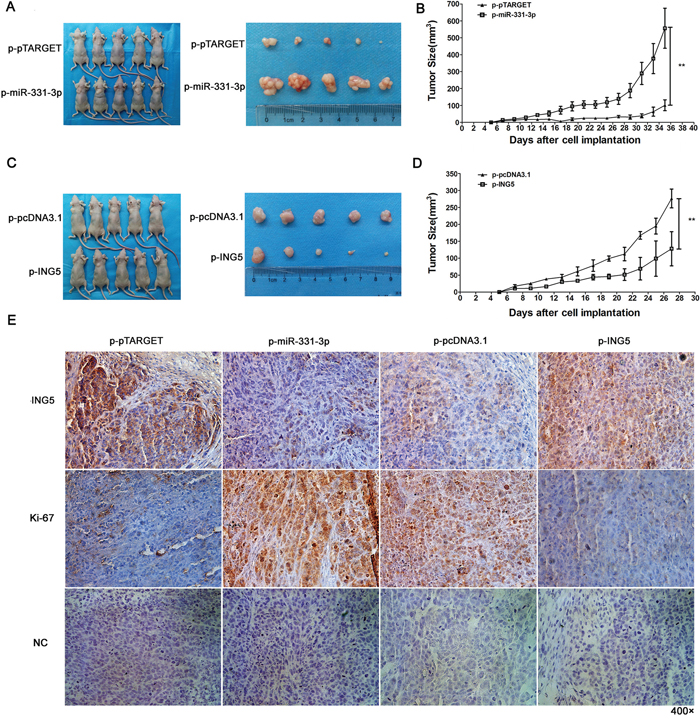
Figure 7: miR-331-3p or ING5 overexpression influences tumor growth in nude mice. A. Representative pictures of tumors formed in p-pTARGET and p-pTARGET-miR-331-3p injected nude mice. B. Tumor growth curve of each group in A. C. Representative pictures of tumors formed in p-pcDNA3.1 and p-ING5 injected nude mice. D. Tumor growth curve of each group in C. *P < 0.05, **P < 0.01. E. Ki-67 and ING5 stained sections of transplanted tumors. Original magnification: 400×.
To better understand the molecular mechanism of miR-331-3p and ING5 on tumorigenesis in vivo, Ki-67 and ING5 expression was measured in tumor tissues via immunohistochemistry. ING5-staining decreased in the p-miR-331-3p group and increased in the p-ING5 group compared with control. Also, staining intensity and the number of Ki-67 positive tumor cells increased in the p-miR-331-3p group and decreased in the p-ING5 group compared with their corresponding controls (Figure 7E). Therefore, miR-331-3p promoted tumorigenesis of hepatoma cells in vivo by regulating ING5 expression.
ING5 was downregulated in human HCC tissue with HBV infection
Because miR-331-3p was reported to be upregulated in human HCC tissue [15], we asked whether ING5 expression was correlated with miR-331-3p in HBV-HCC tissue. Immunohistochemistry revealed that ING5 expression was decreased in HCC tissue compared to corresponding pericarcinous tissue (Figure 8).
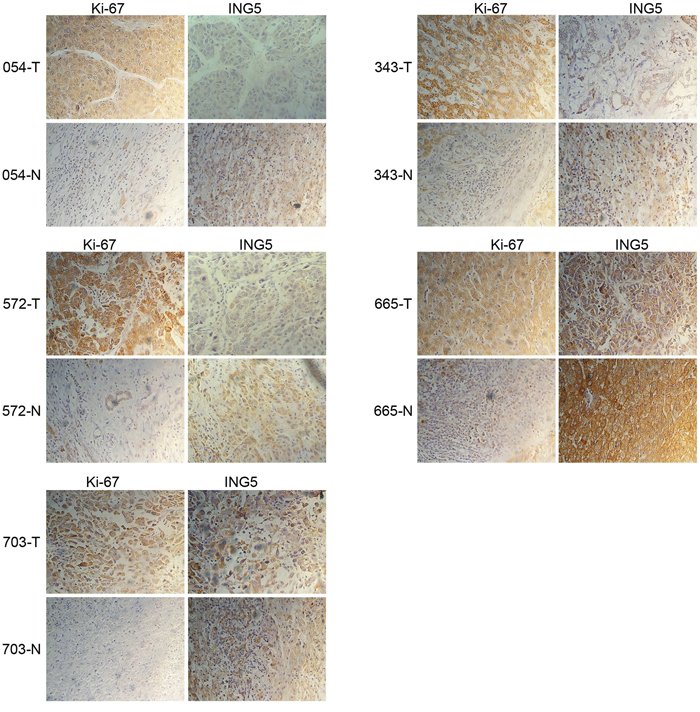
Figure 8: ING5 is downregulated in HBV-infected HCC tissue. Human HBV-infected HCC tumor and pericarcinous tissue sections were immunohistochemistry staining with Ki-67 and ING5 (magnification 400×).
DISCUSSION
It has been reported that 5% of the world’s population (350 million people) is chronically infected with HBV, and locations with high incidence and mortality rates for HCC have high prevalence of chronic HBV infection [2]. miRNA dysregulation is known to be involved in human cancers, including HCC [25–28]; therefore, investigation of the association between abnormal expression of miRNAs and HBV in HCC may lead to new strategies for HCC prevention and therapy.
Here, we present evidence that miR-331-3p is upregulated in HCC cells lines that differentially express HBV. We also studied associations between HBV and miR-331-3p and showed that HBV increases miR-331-3p expression by enhancing its promoter activity. Furthermore, over-expression of miR-331-3p promoted proliferation of HCC cells both in vitro and in vivo.
Various studies showed that the dysregulated of miR-331-3p expression was associated with a variety of human cancers including prostate cancer, glioblastoma multiforme and gastric cancer and played a role as a tumor suppressor [29–31]. However, a recently study demonstrated that miR-331-3p promoted proliferation and metastasis of HCC [15]. In this report, we show that miR-331-3p expression was stimulated by HBV, which in turn inhibited the expression of a tumor suppressor gene ING5 and promoted proliferation of HCC. Our data suggest that miR-331-3p might act as an oncogenic factor in HCC cells. miR-331-3p may play different roles in different tissues and different types of cancers, so do other miRNAs. It has been reported for another miRNA called miR-143. miR-143 was dramatically upregulated in HBV-related HCC and promoted cancer cell invasion, migration and tumor metastasis by repression of FNDC3B expression [26], but miR-143 also repressed glioma cell migration, invasion, tube formation and slowed tumor growth and angiogenesis in a manner associated with N-RAS downregulation [32]. miR-17–5p, another well-known miRNA, has been reported to act as both an oncogene and a tumor suppressor in different cellular contexts by targeting different genes [33–35]. Thus, miRNAs have complicated functions in human cancer and these warrant investigation.
ING5 was reported to be a tumor suppressor gene that inhibited cell growth and induced apoptosis in certain cancers [20–22], but the effects of ING5 in HCC are poorly understood. In our research, we confirmed for the first time that ING5 was a target gene of miR-331-3p, and that miR-331-3p overexpression inhibited ING5. When miR-331-3p was suppressed, ING5 expression increased. ING5 expression was also inhibited by HBV, overexpression of ING5 inhibited cell growth and induced apoptosis of HCC cells. Thus, the proliferation effect of miR-331-3p depends on ING5 inhibition. As shown in Figure 5E, in pTARGET-miR-331-3p and pcDNA3.1-ING5 co-transfected groups, ING5 expression was greater than negative controls, but MTS assay indicated that cell proliferation was the greatest among the three. Thus, miR-331-3p-induced proliferation not only depends on suppression of ING5, but may require inhibition of other target mRNAs or signaling pathways. miRNA and its target mRNAs often form complex regulatory networks. Our data suggest that HBV upregulates miR-331-3p expression, which in turn inhibits multiple target genes that may be involved in HBV-associated HCC.
After confirming the HBV-miR-331-3p-ING5 interactions, we studied clinical diagnostic and therapeutic values of miR-331-3p and ING5. Chang and colleagues documented that [15] miR-331-3p was significantly upregulated (more than 2-fold; i.e., log2 [fold change] > 1) in 79 of 120 HCC cases. We measured ING5 expression in HCC tumor tissue derived from HBV infections by western blot and immunohistochemistry, and found that ING5 expression was decreased in human tumour samples compared to corresponding pericarcinous tissue. So, although we know more about HBV-related HCC, treatment is still problematic. We identified a link among HBV, miR-331-3p, and ING5 that is a novel constituent of HCC and this may offer a novel therapeutic strategy for HCC and may better define mechanisms underlying HCC on how it develops from HBV infection.
MATERIALS AND METHODS
Cell culture and transfection
All the cells lines used for this study were routinely used and preserved in our lab, including human hepatoma cell lines HepG2, HepG2.2.15, HepAD38 [36] and SMMC7721. HepG2, HepG2.2.15, and HepAD38 cell lines were cultured in minimum essential media (MEM) (Hyclone, China) supplemented with 10% fetal bovine serum (FBS) (Life Technologies, Grand Island, NY), 100 units/ml penicillin and 100 μg/ml streptomycin (Hyclone) and 1.2% sodium pyruvate (Hyclone). SMMC7721 cells were cultured in RPMI-1640 medium (Hyclone) supplemented with 10% FBS (Life Technologies) and 100 units/mL penicillin and 100 μg/ml streptomycin. HepG2, HepG2.2.15, HepAD38, and SMMC7721 cells were all maintained in a humidified incubator at 37°C with 5% CO2. Transfections were performed with Lipofectamine 2000 (Life Technologies) according to the manufacturer’s instructions.
RNA extraction and qRT-PCR
Total RNA from cell lines containing miRNA was isolated with Trizol reagent (Life Technologies) and treated with DNase (Promega, Madison, WI). First-strand cDNA was generated with a miRNA cDNA Kit or a BioRT cDNA First Strand Synthesis Kit according to the manufacturer’s instructions (CWBio, Hangzhou Bioer Technology, China). Quantitative real time polymerase chain reaction (qRT-PCR) was performed using miRNA Real-Time PCR Assay Kit or UltraSYBR mixture (CWBio) to confirm expression of miRNAs or mRNAs. A cycle threshold (CT) was assigned at the beginning of the logarithmic phase of PCR amplification, and duplicate CT values were analyzed using the 2−ΔΔCT method [37]. U6 and β-actin mRNA were used for normalization. The forward and reverse primers of qRT-PCR are given in Table 1.
Table 1: Primer sequences used for PCR or constructions of various plasmids
Amplifier primers |
Primer sequence (5′-3′) |
|---|---|
Real-time PCR primer |
|
miR-331-3p-F |
GCCCCTGGGCCTATCCTAGAA |
U6-F |
AGAGCCTGTGGTGTCCG |
U6-R |
CATCTTCAAAGCACTTCCC |
β-actin-F |
GTGGATCAGCAAGCAGGAGT |
β-actin-R |
TGTGTGGACTTGGGAGAGGA |
ING5-F |
ACCAGAGGACGGAAGATAAG |
ING5-R |
TGCACTTGCTGTAGGCGTTC |
VHL-F |
ACATCGTCAGGTCGCTCTAC |
VHL-R |
ATCTCCCATCCGTTGATGTG |
AKAP11-F |
AGCTGGAGCAGTCTTGGTTT |
AKAP11-R |
ACTTCGTCCTCATGCTCTTC |
ERBB2-F |
ACCTGCTGAACTGGTGTATG |
ERBB2-R |
TGACATGGTTGGGACTCTTG |
RGS-F |
TGCCCATCAGAACAGTCAAG |
RGS-R |
ACTGGGTCCTCAATGGAAAG |
DOHH-F |
AGGCCTTCGATGACGATTC |
DOHH-R |
TGTCTTGCAGCACGTCCAC |
SPOP-F |
AGAGTCAACGGGCATATAGG |
SPOP-R |
AGCTTGTCATCAGGGAGAAG |
TRERF1-F |
ATGACCTCCAGCAAAGAGTG |
TRERF1-R |
TTGGCCTCAAACAGAGAGTG |
miR-331-3p over-expression primer |
|
pTARGET-miR-331-3p-F |
CGCCGCTCGAGATAATATCCTAAACAAAGCA |
pTARGET-miR-331-3p-R |
ACGCGTCGACTTTTAGGGCTAAGTTGCTTC |
miR-331-3p promoter primer |
|
pGL3-Basic-miR-331-3p-F |
CGGGGTACCAACCGCCTGACCAACATGGAG |
pGL3-Basic-miR-331-3p-R |
CCGCTCGAGTCCCTGGGACCATACCTAGAAC |
ING5 3 ’UTR primer |
|
pGL3-Control-ING5-WT-F |
AGCTCTAGAAGACCCATCTCTTGGATTG |
pGL3-Control-ING5-WT-R |
AGCTCTAGATCCAACACAATCTCTCCTG |
pGL3-Control-ING5-MUT-F |
TGAGAGGAATTTTAAGAAGCCCCAAGTGTAAG |
pGL3-Control-ING5-MUT-R |
CTTACACTTGGGGCTTCTTAAAATTCCTCTCA |
ING5 over-expression primer |
|
pcDNA3.1-ING5-F |
CGGGGTACCGCCACCATGGCGACCGCCATGTAC TTGG |
pcDNA3.1-ING5-R |
CCGCTCGAGCTACTTCTTCTTCCTCTTTTCC |
HBV assay
HBV surface antigen (HBsAg) or HBe antigen (HBeAg) expression was determined by ELISA using the conditioned medium. Conditioned medium from each cell line was assayed according to the supplier’s recommendations (Diagnostic kit for Hepatitis B Virus surface antigen ELISA, diagnostic kit for Hepatitis B e antigen ELISA, Shanghai KeHua B-engineering, China) 48 h after transfection. Absorbance at 450 nm was measured using a Synergy HT Multi-Detection Microplate Reader (BioTek). Each cell line was tested in three independent experiments.
miRNA target predictions
Predictions of miR-331-3p targets was performed using databases including miRbase (http://mirbase.org/), miRDB (http://mirdb.org/miRDB/, ref), http://microRNA.org-Targets and Expression (http://www.microrna.org/), and TargetScan (http://www.targetscan.org/).
Plasmid construction
All sequences of cloning primers are illustrated in Table 1. The HBV expression plasmid pCH9/3091, constructed by Nassal’s group (Heidelberg University, Germany), was a gift from Dr. Lan Lin (Southwest Hospital affiliated with the Third Military Medical University, China). A fragment of the miR-331-3p promoter region, designed using Eukaryotic Promoter Database (http://epd.vital-it.ch/), was amplified by PCR using the genomic DNA of HepG2.2.15 cells as the template. The amplified fragment was cloned into the Kpn I/Xho I sites of the pGL3-Basic vector (Promega) immediately upstream of the luciferase gene to generate pGL3-Basic-miR-331-3p. The miR-331-3p expression vector (pTARGET-miR-331-3p), a fragment encompassing the mature miR-331-3p sequence and its 5′-and 3′-flanking regions (694 bp in total) was amplified and then cloned into pTARGET™ Vector (Promega). The ING5 3′-UTRs fragment containing miR-331-3p binding site (CCAGGGG) was amplified by PCR from genomic DNA of HepG2.2.15 cells, and cloned into the Xba I site of pGL3-Control dual-luciferase miRNA target expression vector (Promega) immediately downstream of the luciferase gene. pGL3-Control-ING5-MUT, which carried the mutated 3′-UTR sequence (AAGCCCC) was generated based on pGL3-Control-ING5-WT plasmid by site-specific mutagenesis. The ING5 expression vector (pcDNA3.1-ING5) was created by cloning the ING5 coding sequence into the Kpn I/Xho I site of pcDNA3.1 (Life Technologies). All constructed vectors were confirmed by DNA sequencing.
RNA interference
MiR-331-3p inhibitor, siING5 and its negative control (NC) were designed and synthesized by Invitrogen. Sequences were as follows, miR-331-3p inhibitor: 5′-UUCUAGGAUAGGCCCAGGGGC-3′, random microRNA inhibitor NC: 5′-CAGUACU UUUGUGUAGUACAA-3′, siING5: 5′-UUUCUUAUCU UCCGUCCUCUG-3′, siHBx: GUUUAAGGACUGGG AGGAGC, random scramble siRNA NC: 5-CGCG AAUACGGAAAAGGAAUG-3.
Luciferase reporter assay
For the luciferase reporter assay, SMMC7721 cells were seeded in a 24-well plate at a density of 45% confluent and co-transfected with 250 ng pTARGET-miR-331-3p or pTARGET vector, 150 ng of pGL3-Control-ING5-WT or pGL3-Control-ING5-MUT and 25 ng pRL-TK plasmid expressing Renilla luciferase (Promega). HepG2.2.15 cells were seeded in 24 well plates at a density of 60% confluent and co-transfected with 70 pmol miR-331-3p inhibitor or inhibitor NC, 150 ng of pGL3-Control-ING5-WT and 50 ng pRL-TK. Cells were collected 48 h after transfection and analyzed using the Dual-Luciferase Reporter Assay System (Promega, Madison, WI). Relative luciferase activity was normalized to Renilla luciferase activity. Transfections were performed in triplicates and repeated at least 3 times in independent experiments.
Western blot
Cells were lysed with 1% RIPA Lysis Buffer (Beyotime, China) 48 h after transfection. Supernatants were collected, and protein was measured using the BCA Assay Kit (Beyotime). Protein samples were separated by 10% SDS-PAGE and then transferred to a PVDF membrane. The membrane was blocked with 5% milk, incubated overnight at 4°C with a primary rabbit antibody against human ING5 (Proteintech, China, 1:1000 dilution), washed three times in TBST and then incubated with a goat anti-rabbit HRP secondary antibody (Bioword, USA, 1:5000 dilution). Finally, bound antibody was detected using an ECL Detection Reagent (Millipore, Billerica, MA). Data were normalized to β-actin (Bioword, 1:5000 dilution).
Stable cell line generation
SMMC7721 cells were transfected with pTARGET, pTARGET-miR-331-3p, pcDNA3.1 or pcDNA3.1-ING5 and selected with G418 (1,000 g/ml). Two weeks later, few cells survived, and G418 was reduced to 500 μg/ml. Stable cell lines p-miR-331-3p and p-ING5, which could stably express miR-331-3p or ING5 were established and expression was measured via Real time RCR and Western blot, respectively.
MTS assay
SMMC7721 cells were trypsinized and seeded into 96-well culture plates 24 h after transfected specific DNAs or RNAs as indicated in result (4,000 cells/well). Cells were harvested at different time points (24, 48, 72, and 96 h) for cell proliferation assay using an MTS kit (cellTiter96AQ, Promega, Madison, WI) following the manufacturer’s protocol and the absorption was read at 490 nm.
Colony formation assay
Twenty-four hours after transfection, cells were trypsinized and seeded into 6-well plates with a density of 2,000 per well. When cells were visible colonies (about 1 week), colonies were washed with PBS three times. Cell colonies were fixed and stained with a crystal violet cell colony staining kit (GenMed Scientifics, city, state) according to the manufacturer’s instructions. Colonies were counted under a microscope. Clone formation rate was calculated according to the formula: Clone formation rate = clone forming number/Inoculation cell number x 100%.
Tumor transplantation
Female BALB/c nude mice (4–6 weeks-of-age) were purchased from the Laboratory Animal Services Center of Chongqing Medical University. Animal handling and experimental procedures were approved by the Animal Experimental Ethics Committee of Chongqing Medical University. A total of 3 × 106 p-pTARGET or p-miR-331-3p cells, 5 × 106 p-pcDNA3.1 or p-ING5 cells were injected subcutaneously into the upper left dorsal flank of nude mice (N = 5). Tumor volumes were measured every 2 days. Four (p-pcDNA3.1 and p-ING5) or five (p-pTARGET and p-miR-331-3p) weeks later, mice were euthanized and tumors were dissected. Tumor volumes were calculated using the equation V (mm3) = A × B 2/2, where A is the largest diameter, and B is the perpendicular diameter.
Apoptosis assay
Cells in 6-well plates were transfected for 24 h followed by serum deprivation for another 48 h. The cells were then harvested by trypsinization, washed with PBS, and resuspended in 1 ml PBS. Apoptosis in HCC cells was quantified by staining with fluorescein isothiocyanate (FITC)-Annexin V and PI. The stained cells were immediately analyzed by flow cytometry.
Immunohistochemistry
Paraformaldehyde-fixed, paraffin-embedded tissues of transplanted tumors were sectioned at 4.5 μm thickness and Ki-67 and ING5 expression were measured (Bioword, 1:50 dilution; Proteintech, 1:80 dilution, respectively). Visualization was achieved using 3,3′-diaminobenzidine substrate and sections stained with no primary antibodies were negative staining controls. The staining images were collected using a microscope (Carl Zeiss, Germany) and at a original magnification of 400×.
Patient samples
HBV-HCC tissues and paired pericarcinous tissues were obtained from patients who underwent surgery for HCC at the 1st or 2nd Affiliated Hospitals of Chongqing Medical University between 2010 and 2012, with the approval of the Institutional Ethical Review Board of Chongqing Medical University. Patients offered informed consent and had no history of chemotherapy or radiation therapy before surgery. All liver specimens were immediately collected after surgery and stored at −80°C until further use.
Statistical analysis
Data are expressed as means ± standard deviation. Statistical analysis was performed with an independent t-test. p < 0.05 was considered statistically significant.
ACKNOWLEDGMENTS
This work was supported by the Major National S&T Program (2013ZX10002002, ALH), the major project of Chongqing Science & Technology Commission (cstc2013jcyjC10002, ALH), the Natural Science Foundation Project of CQ CSTC (2010BB5359), and the Scientist Culture Plan of Chongqing Medical University (162014)
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Yuen MF, Hou JL, Chutaputti A. Hepatocellular carcinoma in the Asia pacific region. J Gastroenterol Hepatol. 2009; 24:346–353.
2. McGlynn KA, London WT. The global epidemiology of hepatocellular carcinoma: present and future. Clin Liver Dis. 2011; 15:223–243.
3. Bartel D. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell. 2004; 116:281–297.
4. Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006; 6:857–866.
5. Gaál Z, Oláh E. MicroRNAs and their role in malignant hematologic diseases. Orv Hetil. 2012; 153:2051–2059.
6. Lin S, Pan L, Guo S, Wu J, Jin L, Wang JC, et al. Prognostic role of microRNA-181a/b in hematological malignancies: a meta-analysis. PLoS One. 2013; 8:e59532.
7. Lujambio A, Lowe SW. The microcosmos of cancer. Nature. 2012; 482:347–355.
8. Hou J, Lin L, Zhou W, Wang Z, Ding G, Dong Q, et al. Identification of miRNomes in human liver and hepatocellular carcinoma reveals miR-199a/b-3p as therapeutic target for hepatocellular carcinoma. Cancer Cell. 2011; 19:232–243.
9. Giordano S, Columbano A. MicroRNAs: new tools for diagnosis, prognosis, and therapy in hepatocellular carcinoma?. HEPATOLOGY. 2013; 57:840–847.
10. Lin Z, Flemington EK. MiRNAs in the pathogenesis of oncogenic human viruses. Cancer Lett. 2011; 305:186–199.
11. Xu X, Fan Z, Kang L, Han J, Jiang C, Zheng X, et al. Hepatitis B virus X protein represses miRNA-148a to enhance tumorigenesis. J Clin Invest. 2013; 123:630–645.
12. Wang CM, Wang Y, Fan CG, Xu FF, Sun WS, Liu YG, et al. MiR-29c targets TNFAIP3, inhibits cells proliferation and induces apoptosis in Hepatitis B virus-related hepatocellular carcinoma. Biochem Biophys Res Commun. 2011; 411:586–592.
13. Zou CC, Li YG, Cao YY, Zhang JN, Jiang JR, Sheng YR, et al. Up-regulated MicroRNA-181a induces carcinogenesis in Hepatitis B virus-related hepatocellular carcinoma by targeting E2F5. BMC Cancer. 2014; 14:97.
14. Zou C, Chen J, Chen K, Wang S, Cao Y, Zhang J, et al. Functional analysis of miR-181a and Fas involved in Hepatitis B virus-related hepatocellular carcinoma pathogenesis. Exp Cell Res. 2015; 331:352–361.
15. Chang RM, Yang H, Fang F, Xu JF, Yang LY. MicroRNA-331-3p Promotes Proliferation and Metastasis of Hepatocellular Carcinoma by Targeting PH Domain and Leucine-Rich Repeat Protein Phosphatase. Hepatology. 2014; 60:1251–1263.
16. Sells MA, Chen ML, Acs G. Production of Hepatitis B virus particles in Hep G2 cells transfected with cloned Hepatitis B virus DNA. Proc Natl Acad Sci USA. 1987; 84:1005–1009.
17. Zhang ZZ, Liu X, Wang DQ, Teng MK, Niu LW, Huang AL, Liang Z. Hepatitis B virus and hepatocelluar carcinoma at the miRNA level. Word J Gastroenterol. 2011; 17:3353–3358.
18. Gunduz M, Gunduz E, Rivera RS, Nagatsuka H. The inhibitor of growth (ING) gene family: potential role in cancer therapy. Curr Cancer Drug Targets. 2008; 8:275–284.
19. Shiseki M, Nagashima M, Pedeux RM, Kitahama-Shiseki M, Miura K, Okamura S, et al. p29ING4 and p28ING5 bind to p53 and p300, and enhance p53 activity. Cancer Res. 2003; 63:2373–2378.
20. Xing YN, Yang X, Xu XY, Zheng Y, Xu HM, Takano Y, Zheng HC. The altered expression of ING5 protein is involved in gastric carcinogenesis and subsequent progression. Hum Pathol. 2011; 42:25–35.
21. Li X, Nishida T, Noguchi A, Zheng Y, Takahashi H, Yang X, Masuda S, Takano Y. Decreased nuclear expression and increased cytoplasmic expression of ING5 may be linked to tumorigenesis and progression in human head and neck squamous cell carcinoma. J Cancer Res Clin Oncol. 2010; 136:1573–1583.
22. Cengiz B, Gunduz E, Gunduz M, Beder LB, Tamamura R, Bagci C, Yamanaka N, Shimizu K, Nagatsuka H. Tumor-specific mutation and downregulation of ING5 detected in oral squamous cell carcinoma. Int J Cancer. 2010; 127:2088–2094.
23. Tian YY, Hu Y, Wang ZC, Chen K, Zhang L, Wang LY, Ren M, Huang AL, Tang H. Hepatitis B virus regulates Raf1 expression in HepG2.2.15 cells by enhancing its promoter activity. Arch Virol. 2011; 156:869–874.
24. Bose P, Thakur S, Thalappilly S, Ahn BY, Satpathy S, Feng X, Suzuki K, Kim SW, Riabowol K. ING1 induces apoptosis through direct effects at the mitochondria. Cell Death Dis. 2013; 4:e788.
25. Epis MR, Giles KM, Kalinowski FC, Barker A, Cohen RJ, Leedman PJ. Regulation of expression of deoxyhypusine hydroxylase (DOHH), the enzyme that catalyzes the activation of eIF5A, by miR-331-3p and miR-642-5p in prostate cancer cells. J Biol Chem. 2012; 287:35251–35259.
26. Zhang X, Liu S, Hu T, Liu S, He Y, Sun S. Up-regulated microRNA-143 transcribed by nuclear factor kappa B enhances hepatocarcinoma metastasis by repressing fibronectin expression. Hepatology. 2009; 50:490–499.
27. Zhang L, Yang L, Liu X, Chen W, Chang L, Chen L, Loera S, Chu P, Huang WC, Liu YR, Yen Y. MicroRNA-657 promotes tumorigenesis in hepatocellular carcinoma by targeting transducin-like enhancer protein 1 through nuclear factor kappa B pathways. Hepatology. 2013; 57:1919–1930.
28. Tsai WC, Hsu PW, Lai TC, Chau GY, Lin CW, Chen CM, Lin CD, Liao YL, Wang JL, Chau YP, Hsu MT, Hsiao M, Huang HD, Tsou AP. MicroRNA-122, a tumor suppressor microRNA that regulates intrahepatic metastasis of hepatocellular carcinoma. Hepatology. 2009; 49:1571–1582.
29. Epis MR, Giles KM, Candy PA, Webster RJ, Leedman PJ. MiR-331-3p regulates expression of neuropilin-2 in glioblastoma. J Neurooncol. 2014; 116:67–75.
30. Epis MR, Barker A, Giles KM, Beveridge DJ, Leedman PJ. The RNA-binding protein HuR opposes the repression of ERBB-2 gene expression by microRNAmiR-331-3p in prostate cancer cells. J Biol Chem. 2011; 286:41442–41454.
31. Guo X, Guo L, Ji J, Zhang J, Zhang J, Chen X, Cai Q, Li J, Gu Q, Liu B, Zhu Z, Yu Y. MiRNA-331-3p directly targets E2F1 and induces growth arrest in human gastric cancer. Biochem Biophys Res Commun. 2010; 398:1–6.
32. Wang L, Shi ZM, Jiang CF, Liu X, Chen QD, Qian X, Li DM, Ge X, Wang XF, Liu LZ, You YP, Liu N, Jiang BH. MiR-143 acts as a tumor suppressor by targeting N-RAS and enhances temozolomide-induced apoptosis in glioma. Oncotarget. 2014; 5:5416–5427.
33. He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, Powers S, Cordon-Cardo C, Lowe SW, Hannon GJ, Hammond SM. A microRNA polycistron as a potential human oncogene. Nature. 2005; 435:828–833.
34. O’Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc regulated microRNAs modulate E2F1 expression. Nature. 2005; 435:839–843.
35. Fang L, Li H, Wang L, Hu J, Jin T, Wang J, Yang BB. MicroRNA-17-5p promotes chemotherapeutic drug resistance and tumour metastasis of colorectal cancer by repressing PTEN expression. Oncotarget. 2014; 5:2974–2987.
36. Ladner SK, Otto MJ, Barker CS, Zaifert K, Wang GH, Guo JT, Seeger C, King RW. Inducible expression of human Hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: A novel system for screening potential inhibitors of HBV replication. Antimicrob Agents Chemother. 1997; 4:1715–1720.
37. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔC(T)) method. Methods. 2001; 25:402–408.