
INTRODUCTION
Hepatocellular carcinoma (HCC), which accounts for approximately 90% of primary liver cancers, is the third most common cause of cancer-related death, with approximately 782,000 newly diagnosed cases and 746,000 deaths in 2012 worldwide [1, 2]. Although surgical therapies are the first option for HCC patients, HCC-associated mortality remains high [1]. In addition, chemotherapy is minimally effective and accompanied by severe toxicity and side effects. Sorafenib extends the survival of advanced HCC patients by only approximately three months compared with placebo [3]. These data underscore the urgent need to develop more effective therapies against liver cancer.
Anticancer agents from natural sources have attracted significant attention worldwide. For example, the biodiversity of chemical components in toad glandular secretion and skin extract possess antineoplastic activity, and these compounds may be used to develop new therapeutics [4, 5]. The dried skin gland secretion of the toad Bufo gargarizans Cantor or Bufo melanostictus Suhneider is called toad venom (also termed Chan’su), and its preparations have been widely used to treat several cancers in China and East/Southeast Asian countries [5]. The main active ingredients derived from Chan’su, bufadienolides, are classical Na+/K+-ATPase inhibitors [6–8] that also exert antineoplastic effects. Specifically, they induce apoptosis [9–11], disrupt the cell cycle [10, 12, 13], induce differentiation [14, 15], and inhibit cancer angiogenesis [16, 17]. The mechanisms of bufadienolides-induced apoptosis are implicated in several pathways, including the mitochondria-mediated pathway [9, 10, 18], the PI3K/Akt signaling pathway [19], the ClC-3 chloride channel [20], the IKKβ/NF-κB signaling pathway [11] and DNA topoisomerase II [21, 22]. While bufadienolides have been reported to disrupt the cell cycle, the underlying mechanisms of this disruption have, to the best of our knowledge, not yet been defined.
In an effort to isolate and identify active compounds in Chan’su, we found arenobufagin, a representative bufadienolide compound, substantially contributes to the anti-cancer effects of Chan’su [19]. Arenobufagin blocked the Na+/K+ pump current in cardiac myocytes [23, 24]. Recently, our group showed that arenobufagin inhibits the growth of a variety of human tumor cells [19] and VEGF-mediated angiogenesis [17]. Arenobufagin has also been shown to induce apoptosis and autophagy via the inhibition of the PI3K/Akt/mTOR pathway [19].
In this study, arenobufagin directly binded with DNA via intercalative binding. This interaction led to double-strand DNA breaks (DSBs) and triggered the DNA damage response (DDR) via the ATM/ATR signal pathway, which subsequently resulted in G2 phase arrest in HCC cells. This study has shed new light on the mechanism by which arenobufagin interacts with DNA to induce cell cycle arrest, and it is also the first to note that bufadienolides may be DNA-targeting agents, which will help elucidate the mechanisms of their anticancer activities.
RESULTS
Arenobufagin inhibits cell cycle transition from G2 to M phase in HCC cells
Arenobufagin significantly inhibited the growth of HCC cell lines, the p53 wild-type cell lines HepG2 and HepG2/ADM and the p53-null cell line Hep3B (Supplementary Figure S1A). The effect of arenobufagin on the cell cycle was assessed by staining these three HCC cell lines, with propidium iodide (PI). As shown in Figure 1A, exposing cells to arenobufagin significantly increased the cell population in the 4N-DNA content phase in a time-dependent manner (Figure 1A, left panel). Quantitatively, arenobufagin treatment for 48 h resulted in 4N-DNA contents of 47.95 ± 1.34% in HepG2 cells, 41.65 ± 0.49% in HepG2/ADM cells, and 40.3 ± 0.99% in Hep3B cells (Figure 1A, right panel). The G2 and mitotic cells were not distinguishable by PI staining, because both populations contain 4N-DNA. Thus, the cells were immunostained with p-Histone H3 (Ser10), an M-phase-specific marker [25], to assess the mitotic index. Arenobufagin significantly decreased the number of mitotic HepG2 and HepG2/ADM cells (Figure 1B) and slightly increased the mitotic index of Hep3B cells to 15.34 ± 0.28%. Paclitaxel, a mitotic inhibitor [26], was used as a positive control. The statistical analysis of the DNA content and mitotic index data indicated that arenobufagin inhibited the G2/M transition in HCC cells, and the majority of cells were arrested in G2 phase rather than in the M phase.
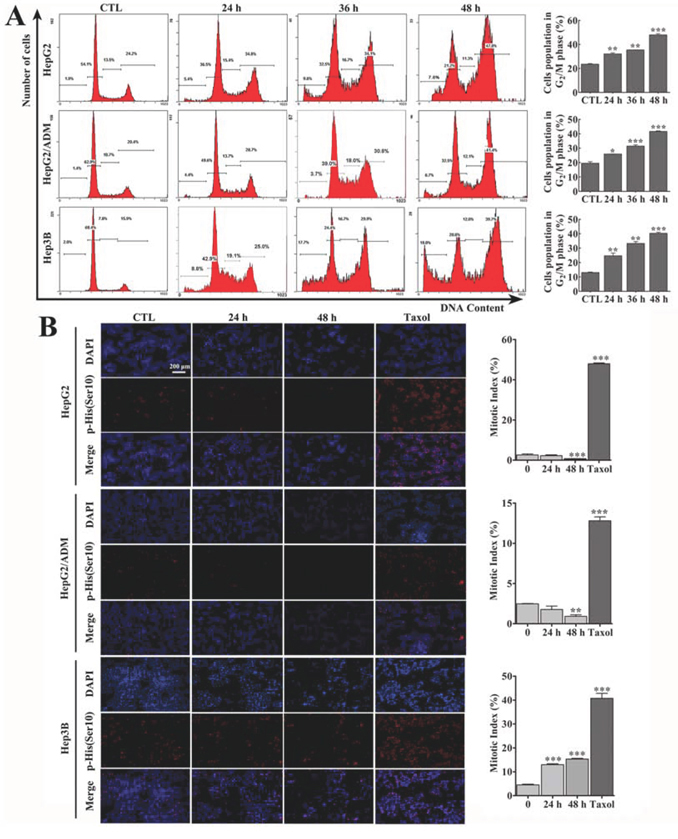
Figure 1: Arenobufagin induces G2 cell cycle arrest in HCC cells. A. After treatment with 10 nmol/L (Hep3B cells) or 20 nmol/L (HepG2 and HepG2/ADM cells) of arenobufagin for 0, 24, 36, and 48 h, the cell cycle distributions were measured using flow cytometry. Representative pictures (left panel) and a quantification of the cell population in the G2/M phase (right panel) are shown. Each column represents the mean ± SD of at least three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 versus the DMSO control. B. Effect of arenobufagin on the mitotic index in HCC cells. Cells were treated with arenobufagin for 0, 24 and 48 h and Taxel for 12 h (25 nmol/L for HepG2 and Hep3B cells, 5 μmol/L for HepG2/ADM cells) as a positive control. Representative pictures are shown (left panel). Original magnification: 100×; Scale bar: 200 μm. The mitotic indexes were calculated using the number of p-Histone H3-positive cells per total number of cells (DAPI-positive cells). Each column represents the mean ± SD of triplicates. **P < 0.01, ***P < 0. 001 versus the DMSO control (right panel).
The role of p53 in the arenobufagin-induced G2 response
As shown in Figure 1, the p53 wild-type cell lines HepG2 and HepG2/ADM remained arrested in the G2 phase following arenobufagin exposure, with only a fraction of cells becoming hypoploid by 48 h (7.8% for HepG2 and 6.7% for HepG2/ADM). However, the p53-null cell line Hep3B responded to arenobufagin with G2 cell cycle arrest accompanied by a substantial increase in the percentage of subG1 phase cells (approximately 20%), indicating that arenobufagin induced apoptosis. To further verify that Hep3B cells underwent apoptosis, Annexin V-FITC staining assay was performed. As shown in Figure 2A, 48 h of arenobufagin treatment increased the percentage of apoptotic cells from 4.5 ± 0.34% to 18.69 ± 0.70% in Hep3B cells, while the percentage of apoptotic cells increased slightly in HepG2 cells (from 2.97 ± 0.21% to 7.36 ± 1.13%) and HepG2/ADM cells (from 3.08 ± 0.34% to 4.99 ± 0.29%).
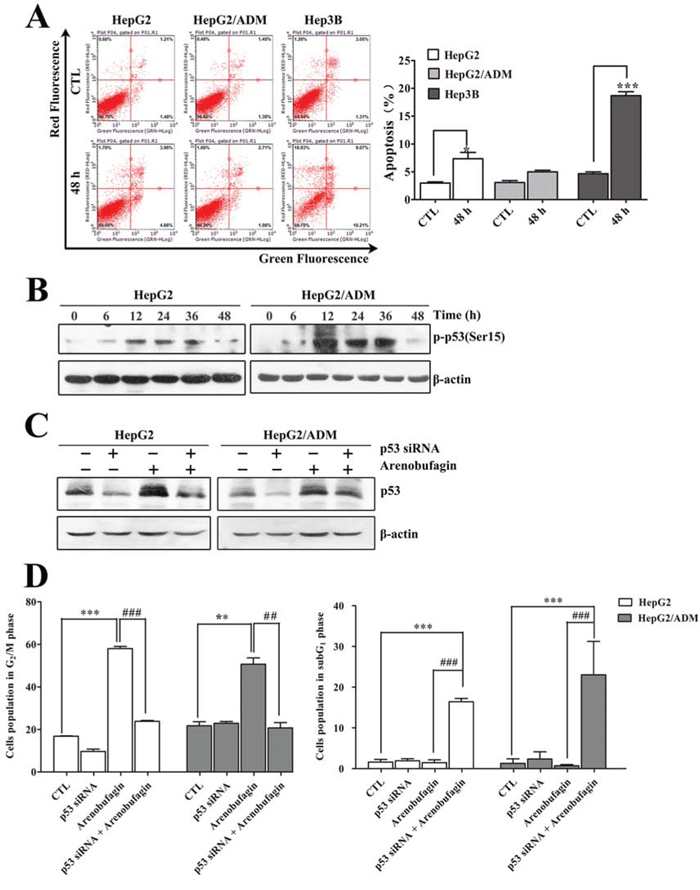
Figure 2: The role of p53 in arenobufagin-induced G2 arrest. A. After treatment with arenobufagin for 48 h, the apoptotic cells were measured using flow cytometry. At least 10,000 cells were analyzed per sample. Representative pictures (left panel) and a quantification of the apoptotic cells (right panel) are shown. Each column represents the mean ± SD of triplicates. *P < 0.05, ***P < 0.001 versus the DMSO control. B. HepG2 and HepG2/ADM cells were incubated with arenobufagin for 0, 6, 12, 24, 36 and 48 h. The total protein cell lysates were harvested and evaluated by Western blotting with the indicated antibodies. C. The knockdown efficiency of p53 by siRNA in HepG2 and HepG2/ADM cells was evaluated by Western blotting. D. The effect of combined p53 siRNA and arenobufagin on the DNA content of HepG2 and HepG2/ADM cells. Cell cycle distributions of cells were assessed by flow cytometry. Each column represents the mean ± SD of at least three independent experiments. **P < 0.01, ***P < 0.001 versus the DMSO control; ##P < 0.01, ###P < 0.001 versus the arenobufagin alone.
Interestingly, we also observed a transient increase in transcriptionally active p53 in HepG2 and HepG2/ADM cells following arenobufagin treatment (Figure 2B). The differences in the p53 wild-type cell lines (HepG2 and HepG2/ADM cells) and the p53-null cell line (Hep3B cells) indicated that p53 may play a role in arenobufagin-induced G2 arrest. To further investigate the function of p53, HepG2 and HepG2/ADM cells were transiently transfected with p53 siRNA. The transfection of p53 siRNA efficiently abrogated both p53 expression and p53 induction upon treatment with arenobufagin (Figure 2C). As shown in Figure 2D and Supplementary Figure S2A, transient transfection with p53 siRNA and arenobufagin treatment reduced the number of cells accumulated in the G2 phase by approximately 35%, whereas the hypodiploid peaks increased by approximately 16% compared with arenobufagin treatment alone. Besides, the Annexin V-FITC staining assay also showed that transient transfection with p53 siRNA and arenobufagin treatment increased the percentage of apoptotic cells compared with arenobufagin treatment alone (Supplementary Figure S2B). Thus, these results indicated that p53 contributed to sustaining arrest at the G2 phase of the cell cycle and blocked the apoptosis in HepG2 and HepG2/ADM cells following arenobufagin treatment.
Arenobufagin inhibits the activation of CDK1-Cyclin B1 complex
To delineate the molecular mechanisms underlying the inhibition of the G2/M transition induced by arenobufagin, we measured the key regulators that promote mitosis: CDK1 and Cyclin B1 [27]. As shown in Figures 3A and 3B, arenobufagin treatment resulted in a marked accumulation of Cyclin B1 protein and the CDK1-Cyclin B1 complex. The CDK1-Cyclin B1 complex is inactive at the G2 phase due to the phosphorylation of CDK1 (Tyr15 and Thr14) [28]. Here, CDK1 phosphorylation at Tyr15 and Thr14 were up-regulated, while the total CDK1 protein level did not change (Figure 3A; Supplementary Figure S3A).
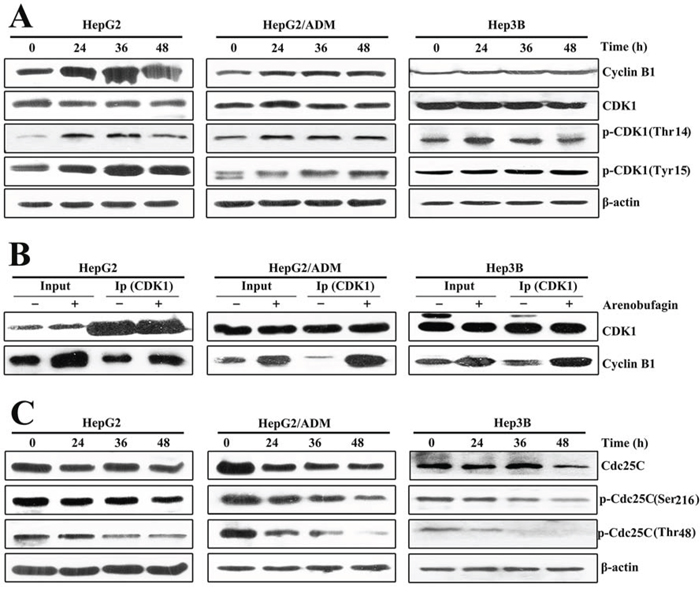
Figure 3: Arenobufagin inhibits the activation of CDK1-Cyclin B1 complex. A. Total cell lysates from HepG2, HepG2/ADM and Hep3B cells treated with arenobufagin for 0, 24, 36 and 48 h. The lysates were evaluated by Western blotting with the indicated antibodies. B. Co-immunoprecipitation of the CDK1-Cyclin B1 complex. Protein extracts (1 mg) were incubated with CDK1 primary antibody. Immunoprecipitated complex were subjected to SDS electrophoresis. Total cell lysates were used as an input control. C. Arenobufagin degraded the expression of Cdc25C protein. Total cell lysates were evaluated by Western blotting with the indicated antibodies.
The protein kinase Cdc25C, which belongs to the dual-specificity Cdc25 phosphatase family, dephosphorylates the two inhibitory residues of CDK1 (Tyr15 and Thr14) to activate the CDK1-Cyclin B1 complex at the G2 phase [29]. Thus, the levels of total Cdc25C and its phosphorylation at Ser216 (active residue) and Thr48 (inhibitory residue) were analyzed [29]. As shown in Figure 3C, both the total and phosphorylated Cdc25C levels time-dependently decreased in arenobufagin-treated HCC cells. Importantly, arenobufagin reduced the level of p-Cdc25C (Thr48) more markedly than those of p-Cdc25C (Ser216) in HCC cells (Figure 3C). We further quantified the ratio of p-Cdc25C (Ser216) to total Cdc25C and the ratio of p-Cdc25C (Thr48) to total Cdc25C (Supplementary Figure S3B). Arenobufagin suppressed both the expression and the phosphatase activity of Cdc25C. Collectively, these results showed that arenobufagin down-regulated Cdc25C, inhibited the dephosphorylation of CDK1-Cyclin B1 complex, and ultimately blocked the G2/M transition in HCC cells.
Arenobufagin activates the ATM/ATR-Chk1/Chk2 signaling pathway
As the key regulators of cell cycle arrest, the checkpoint kinases Chk1 and Chk2 inhibit the G2/M transition [30]. Thus, we monitored the expression levels and phosphorylation status of both Chk1 and Chk2 in arenobufagin-treated HCC cells. As illustrated in Figure 4, the phosphorylation status of the checkpoint kinases p-Chk1 (Ser345) and p-Chk2 (Thr68) significantly increased, whereas the total protein level of Chk1 and Chk2 remained unchanged. ATR-dependent phosphorylation activates Chk1 at residue Ser345 [31], and ATM phosphorylates Chk2 at residue Thr68 [32]. Indeed, ATM and ATR were activated in the treated cells, as indicated by the up-regulation of p-ATM (Ser1981) and p-ATR (Ser428). These kinases were similarly activated in 293 cells treated with UV light for 4 h (Figure 4). These data indicated that arenobufagin treatment activated the ATM/ATR-Chk1/Chk2 signaling pathway.
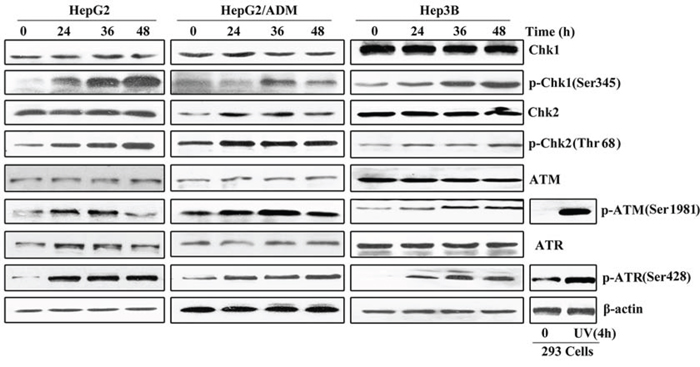
Figure 4: Arenobufagin activates the ATM/ATR-Chk1/Chk2 signaling pathway. HepG2, HepG2/ADM and Hep3B cells treated with arenobufagin for 0, 24, 36 and 48 h. 293 cell extracts were exposed to UV for 4 h. Cells were collected and lysed. The lysates were assayed by Western blotting with the indicated antibodies. The representative pictures from 3 independent experiments are shown.
Arenobufagin induces DSBs
ATM and ATR are activated by phosphorylation after DNA damage [33]. The ability of arenobufagin to cause DNA damage was directly assessed with a comet assay. As shown in Figure 5A, the appearance of DNA in cells treated with arenobufagin for 24 h was similar to that of a comet’s tail and differed from the intact DNA of untreated cells. Quantitatively, arenobufagin increased the Tail Length, Tail DNA%, and Olive Tail Moment in a time-dependent manner (Figure 5A). Furthermore, we analyzed the level of γ-H2AX, a marker of DSBs [34]. We observed a high γ-H2AX signal in cells after 24 h arenobufagin treatment, as well as significantly increased the number of punctate γ-H2AX foci (Figure 5B), indicating that arenobufagin caused DSBs.
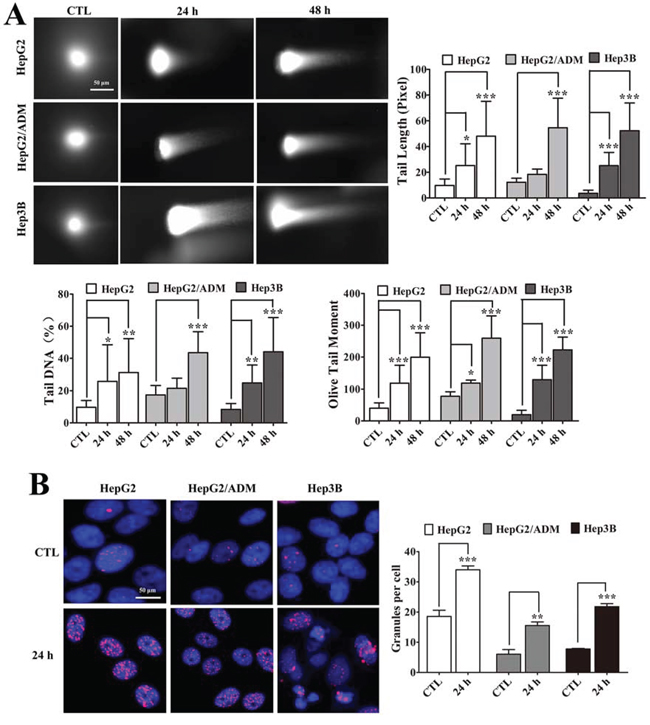
Figure 5: Arenobufagin induces DSBs in cells. A. Cells treated with arenobufagin for 0, 24 and 48 h and then harvested and evaluated with a comet assay. The DNA was stained with Vista Green DNA dye. Representative images of arenobufagin-induced DNA damage are shown. Original magnification: 200×; Scale bar: 50 μm. The percentage of Tail DNA, Tail Length, and Olive Tail Moment were evaluated using MetaXpress software. Each sample includes at least 20 cells. Each column represents the mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001 versus the DMSO control. B. Cells were incubated with arenobufagin for the indicated times and then stained with γ-H2AX and DAPI. Representative images are shown (left panel). Original magnification: 400×; Scale bar: 50 μm. The granules were calculated using the number of γ-H2AX-positive cells per the total number of cells (DAPI-positive cells). Each column represents the mean ± SD of triplicates. **P < 0.01, ***P < 0.001 versus the DMSO control (right panel).
Arenobufagin-induced G2 arrest resulted from DNA damage
Cell cycle arrest can be induced via various signaling pathways, including the DDR. We used caffeine, an inhibitor of ATM/ATR [35], which can block DNA damage response, to assess whether arenobufagin-triggered DNA damage caused the cell cycle arrest at the G2 phase in HCC cells. As shown in Figure 6A, pretreating HepG2 cells with 2 mmol/L caffeine remarkably attenuated the arenobufagin-induced phosphorylation of ATM and ATR. Consistently, pretreatment with caffeine also significantly blocked the arenobufagin-induced increase in the γ-H2AX signal (Figure 6B). Furthermore, combined arenobufagin and caffeine treatment significantly decreased the proportion of cells in G2 phase arrest compared with arenobufagin treatment alone (Figure 6C). Taken together, these data demonstrated that G2 cell cycle arrest was a downstream effect of arenobufagin-induced DNA damage.
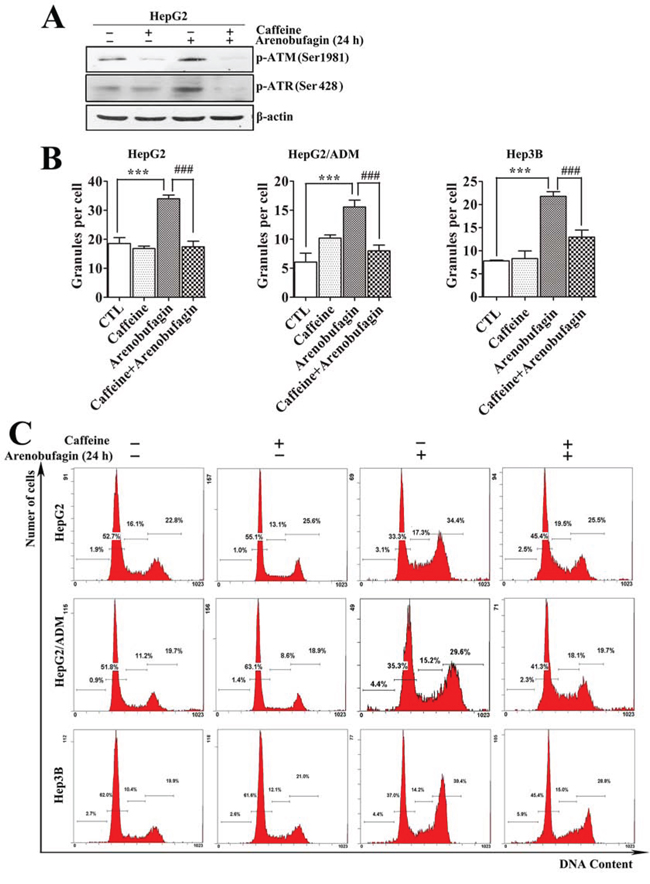
Figure 6: Arenobufagin-induced G2 arrest results from DNA damage. A. Caffeine antagonized the arenobufagin-induced phosphorylation of ATM and ATR. HepG2 cells were pretreated with 2 mmol/L of caffeine for 2 h and then incubated with arenobufagin (20 nmol/L) for 24 h. Cell lysates were harvested and evaluated by Western blotting with the indicated antibodies. B. HCC cells were treated with caffeine combined with arenobufagin (20 nmol/L) and then stained with an antibody to γ-H2AX and the DNA dye DAPI. The granules were calculated using the number of γ-H2AX-positive cells per the total number of cells (DAPI-positive cells). Each column represents the mean ± SD of triplicates. ***P < 0.001 versus the DMSO control; ### P < 0.001 versus the arenobufagin alone. C. Effect of caffeine on arenobufagin-induced G2 arrest. The cell cycle distributions were measured by flow cytometry. Representative images from 3 independent experiments are shown.
Arenobufagin binds with DNA
Because arenobufagin triggered DSBs, we hypothesized that arenobufagin directly binds with DNA. To verify this hypothesis, we assessed the cellular distribution of arenobufagin. Because arenobufagin does not fluoresce, it was conjugated to a D-biotin tag to synthesize the chemical probe biotinylated-arenobufagin, which strongly binds to streptavidin-phycoerythrin (SP). SP can be directly visualized by immunofluorescence [36]. The structure of biotinylated arenobufagin (DB7) is shown in Figure 7A. The cytotoxicities of biotinylated arenobufagin and arenobufagin did not significantly differ, as indicated by an MTT assay (Supplementary Figure S4A). Besides arenobufagin-conjugated chemical probe also induced DNA damage and cell cycle arrest, like arenobufagin (Supplementary Figures S4B and S4C). As shown in Figure 7B, fluorescent biotinylated arenobufagin signals were detected in the cytoplasm after 0.5 h and 1 h of drug exposure. After 6 h, they appeared as punctuate spots that progressively concentrated around the nuclear envelope. Eventually, the biotinylated arenobufagin fluorescence mainly accumulated in the nucleus at 12 h (Figure 7B, arrows).
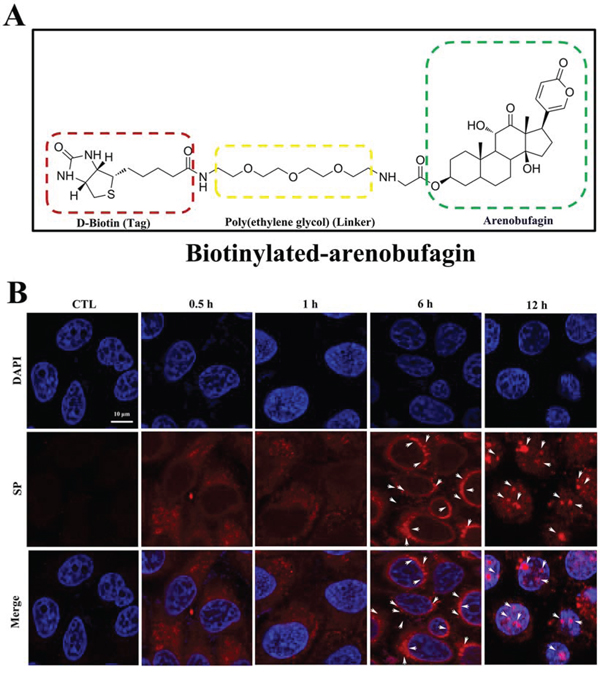
Figure 7: The cellular localization of arenobufagin in live cells. A. The chemical structure of biotinylated arenobufagin. B. HepG2 cells were incubated with biotinylated arenobufagin for various times and then probed with SP. Nuclear DNA was stained with DAPI. Arrows indicated the granules of accumulation of biotinylated-arenobufagin. Original magnification: 630×; Scale bar: 10 μm.
To further test whether arenobufagin directly binds to DNA in vitro, isothermal titration calorimetry (ITC) was employed to evaluate the microscopic thermodynamic parameters driven by the formation of arenobufagin-DNA complex. The results are shown in Figure 8A: Ka = 2.43E5 ± 9.80E4 M−1, ΔH = −4664 ± 728.6 cal/mol, ΔS = 9.00 cal/mol/deg. The enthalpy was less than zero, which indicated that the binding process was driven by enthalpy specifically via the hydrogen bonds. The positive entropy indicated a hydrophobic interaction. Thus, both hydrogen bonds and hydrophobic interactions might contribute to the binding of arenobufagin to DNA and increase the stability of the arenobufagin-DNA complex. According to the equation ΔG = ΔH-TΔS (ΔG is the free energy change, T is the absolute temperature), the value of ΔG was negative, indicating that the binding process was spontaneous.
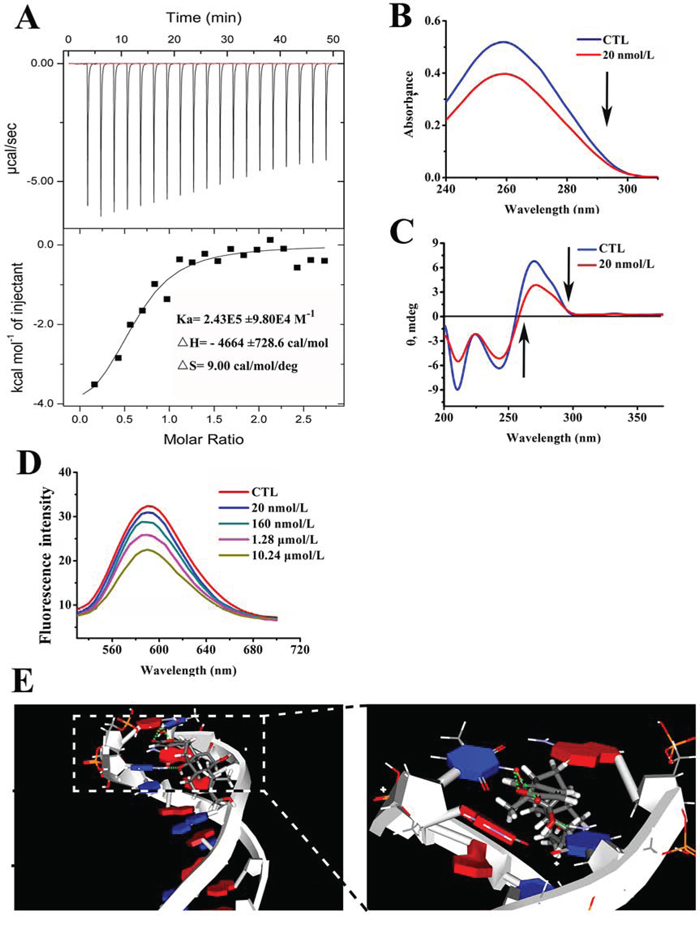
Figure 8: Arenobufagin directly binds with DNA via intercalation. A. Arenobufagin binding to DNA was measured by ITC. A total of 30 μmol/L of DNA was titrated with 0.4 mmol/L of arenobufagin. The resulting thermograms were analyzed based on the one set of binding sites model using Microcal Origin 7.0 (Microcal. Inc.). B. The effect of arenobufagin on the UV absorption spectrum of DNA. 1 mmol/L DNA solution was mixed with 20 nmol/L arenobufagin. After the solution was mixed and equilibrated for approximately 5 min, the absorption spectra were measured at wavelengths ranging from 200 nm to 400 nm. C. The effect of arenobufagin on the CD spectra of DNA. The CD spectra of DNA (1 mmol/L) in 50 mmol/L Tris-HCl (pH = 8.0) with 20 nmol/L of arenobufagin. Each spectrum was analyzed from 200 nm to 370 nm at 25°C with a 10 mm path length cell. D. Fluorescence titration of EB-DNA complex with arenobufagin. EB-DNA complex was excited at 524 nm, and emission spectra was recorded from 530 to 700 nm at 25°C. E. The docked conformations suggested the intercalation between arenobufagin and d(CCGGCGGT)2. The green dotted lines represent the hydrogen bonds formed between arenobufagin and the DNA duplex.
Arenobufagin binds with DNA via intercalative binding
Next, we explored the mode by with arenobufagin binds to DNA. Figure 8B showed the UV-visible absorption spectra of DNA, with the peak position at 259 nm (line blue). Upon the addition of arenobufagin, the absorbance decreased from 0.51892 to 0.39687 without an apparent shift in the position of the absorption peak (Figure 8B). Moreover, the addition of arenobufagin to the DNA solution shifted the CD spectrum intensities from −6.94282 to −6.11314 at the negative band and from 6.39962 to 3.93849 at the positive band (Figure 8C). Hypochromicity in the UV-visible absorption spectrum and significant changes in the CD spectrum are associated with many DNA intercalators [37]. The classical intercalator ethidium bromide (EB) [38] was utilized to test whether arenobufagin intercalated with DNA. As shown in Figure 8D, the distinct quenching of the fluorescence intensity of the EB-DNA system after the continuous addition of arenobufagin suggested that arenobufagin binded with DNA in the same manner as the bound dye EB.
To clarify the mechanism by which arenobufagin intercalated with DNA, a docking study was conducted using the GOLD program. The GC-rich B form of the DNA double helix model (5′-d (CCGGCGGT)-3′) was constructed [39]. As shown in Figure 8E, the pyran moiety of arenobufagin intercalated between GT base pairs via the hydrogen bonds. The specific interactions were the hydrogen bonds between NH2 in the pyridine moiety of G7 and O=C-O in the pyran moiety of arenobufagin in a six-membered ring (Figure 8E). Moreover, a hydrogen bond also formed between the NH (N1) of T8 and OH on C14 of arenobufagin (Figure 8E). These findings agreed with the ITC analysis.
DISCUSSION
Bufadienolides, including cinobufagin, bufalin, resibufogenin, hellebrigenin and bufotalin, are the major pharmacologic constituents of Chan’su [40]. Bufadienolide has been shown to disrupt the cell cycle. Cinobufacini arrested MDA-M-231 cells at the S phase [41], and bufalin arrested endometrial and ovarian cancer cells at the G0/G1 transition [42]. Our previous studies demonstrated that hellebrigenin induced G2/M arrest in HepG2 cells [10], and bufotalin caused G2/M arrest in HepG2/ADM cells [12]. However, these studies simply broached the subject of the effect of bufadienolides on cell cycle disruption and did not define the underlying mechanisms of this effect. Our current study focused on these unaddressed mechanisms and found that the bufadienolide arenobufagin directly binds to DNA via the intercalative binding mode to activate the DDR and ultimately induce G2 arrest in HCC cells. We also demonstrated that the ATM/ATR-Chk1/Chk2-CDC25C signaling pathway may contribute to the G2 cell cycle arrest caused by arenobufagin.
To visualize the localization of arenobufagin, we designed and synthesized a chemical biotinylated-arenobufagin probe using D-biotin as the tag. With the aim of reducing the steric hindrance effect between arenobufagin and D-biotin, poly(ethylene glycol) was employed as a linker group. Based on previous reports, the C-3 position of arenobufagin could be modified without significantly influencing its antitumor activity [20, 43]. Thus, poly(ethylene glycol) was used as a linker between the 3-OH of arenobufagin and D-biotin to form biotinylated-arenobufagin. The live cell images revealed that biotinylated-arenobufagin accumulated mainly in the nucleus. The data from ITC also demonstrated that arenobufagin directly and strongly binds to DNA (the Kd value was approximately 4.12 μmol/L).
Drug-DNA interactions can be classified into intercalation and groove binding [37]. Based on the characteristic parameters, small molecules bind to DNA by intercalative binding as follows: approximately 4 kcal/mol of free-energy cost, association constants (Ka) of 105 to 1011 M−1, and hypochromism in the UV-visible spectrum of DNA [37], which are consistent with our data. Therefore, we predicted that arenobufagin binds with DNA via intercalation. The CD spectrum of DNA exhibits a negative band at 245 nm induced by right-handed helicity and a positive band at 275 nm induced by base stacking, and these bands are sensitive to the small molecules that bind with DNA [44]. The changes in DNA morphology defined by CD signals revealed strong intercalation between arenobufagin and DNA. Consistent with this observation, arenobufagin displaced EB from the DNA solution, supporting the intercalation model. In addition, molecular modeling also revealed that the pyran moiety of arenobufagin intercalated between GT base pairs via the hydrogen bonds, as did the NH (N1) of T8 and OH on C14 of arenobufagin. The negative value of ΔH further demonstrated that the binding process was associated with the formation of hydrogen bonds. Importantly, the thermodynamic parameters obtained from the ITC analysis (ΔH < 0, -TΔS < 0, and ΔG < 0) revealed that the binding progress was energetically favorable and that arenobufagin either specifically binds or maintains the membrane permeability. However, our current data only demonstrated that arenobufagin directly binded to DNA from HepG2 cells. Before getting to the conclusion that arenobufagin is a DNA-targeting agent, we still need to investigate whether arenobufagin also binds to DNA of other cancer cells or non-tumor cells.
It has been demonstrated that small molecules that bind to DNA can block DNA replication or cause DNA lesions. In response to DNA binding agents, cells can arrest cell cycle at G1/S or S phase to prevent incorrect DNA replication, or at G2/M phase to prevent entry into mitosis with damaged DNA [45]. We found that arenobufagin impeded cell cycle progression at the G2 phase, suggesting that arenobufagin intercalated with DNA might not block DNA replication, but instead induced DNA damage. The comet assay confirmed that arenobufagin induced DNA damage. The DNA damage factors phosphorylated ATM, phosphorylated ATR and phosphorylated γH2AX accumulate upon the activation of DNA damage checkpoints [46], as observed in this study. The DDR sensors ATM and ATR block the cell cycle partly via the activation of a signaling cascade that activates the checkpoint kinases Chk2 and Chk1. Chk2 is phosphorylated by ATM at residue Thr68 [32], and Chk1 is activated by the ATR-dependent phosphorylation of residue Ser345 [31]. Active Chk2 and Chk1 phosphorylate Cdc25C on Ser216 and decrease Cdc25C activity, and Cdc25C then sequesters itself in the cytoplasm by binding to 14-3-3 proteins [29]. The inactivated Cdc25C prevents the dephosphorylation of the two inhibitory residues of CDK1 (Tyr15 and Thr14) to maintain the CDK1-Cyclin B1 complex in an inactivated state at G2, thereby inhibiting G2/M transition [29]. Here, Chk1 and Chk2 kinases were activated, the levels of Cdc25C phosphatase were down-regulated, and the two inhibitory residues of CDK1 (Tyr15 and Thr14) were phosphorylated. Overall, these data demonstrated that arenobufagin induced DNA damag and ultimately led to G2 cell cycle arrest via the ATM/ATR-Chk1/Chk2-Cdc25C pathway in HCC cells. Furthermore, consistent with previous reports [47, 48], it was also found that p53 may be an important determinant of sustaining G2 arrest in response to arenobufagin treatment.
Increasing evidence indicates that bufadienolides inhibit Na+/K+-ATPase to exert cardiotonic effect [6, 7]. Arenobufagin blocked the Na+/K+ pump current in single guinea-pig cardiac myocytes with a half-maximal concentration of 0.29 μmol/L [23], which is much higher than the concentration (20 nmol/L) of arenobufagin-induced cell cycle arrest. Furthermore, arenobufagin at the concentration of 30 μmol/L is nontoxic to H9C2 cardiomyocyte cells (Supplementary Figure S1B). Based on these data, arenobufagin may not cause cardiac side effects at the concentration used in this study. Increasing evidence has shown that some Na+/K+ ATPase inhibitors, such as ouabain, bufalin, induce cell cycle arrest in cancer cells, although there is no report on that Na+/K+ ATPase directly regulates the G2/M cell cycle progression [49, 50]. Indeed, it is an interesting issue whether Na+/K+ ATPase-inhibited activity contributes to arenobufagin’s cell cycle effect on HCC cells, which still needs to be studied intensively.
Several documents report that mTORC1 or mTORC2 is involved in G2/M cell cycle progression [51–54]. Our previous data demonstrated that inhibition of mTOR promoted the development of both autophagy and apoptosis in arenobufagin-treated HepG2 cells [20]. However, transient transfection with mTOR siRNA and arenobufagin treatment negligibly changed the cell numbers at G2/M phase compared to arenobufagin treatment alone (Supplementary Figure S5), indicating that mTOR may not intermediate in the arenobufagin-induced G2 arrest.
Our previous pharmacokinetic study showed that arenobufagin was detected in plasma with a peak concentration of 1980 ng/mL (4.75 μmol/L) within 5 min following intraperitoneal administration of 4.0 mg/kg arenobufagin [55], implying that arenobufagin can be absorbed quickly and the in vitro effective concentration (20 nmol/L) for inducing cell cycle arrest and DNA damage is able to be achieved in plasma. Further in vivo mechanistic study is expected whether arenobufagin inhibits solid tumor growth in vivo through inducing DNA damage and cell cycle arrest.
In summary, we found that arenobufagin directly binds to DNA via the intercalative binding mode in vitro. We observed that arenobufagin accumulated mainly in nucleus in live cells, as evidenced by a synthetic tagged biotinylated arenobufagin conjugate. Moreover, arenobufagin also caused DSBs and ultimately lead to G2 cell cycle arrest via the ATM/ATR-Chk1/Chk2-Cdc25C pathway in HCC cells (Figure 9). These results shed new light on the mechanism of arenobufagin-induced cell cycle arrest, which is valuable for the further study of arenobufagin use in clinical anticancer chemotherapy.
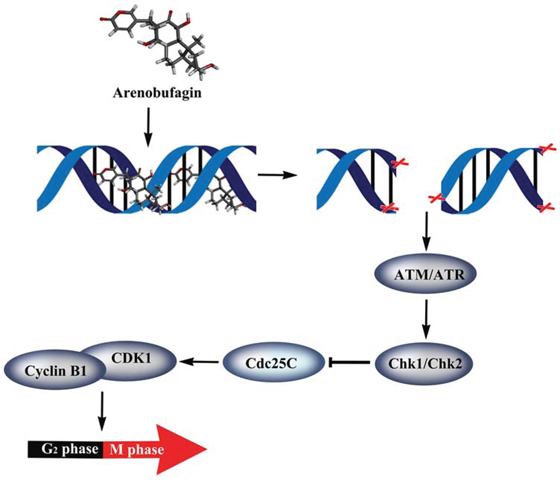
Figure 9: Proposed model for the mechanisms of arenobufagin-induced G2 arrest in HCC cells. Arenobufagin directly binds with DNA via intercalation, leading to DSBs and triggering DDR via the ATM/ATR signaling pathway, which subsequently results in G2 phase arrest in HCC cells.
MATERIALS AND METHODS
Cell lines and cell culture
The human HCC cell lines HepG2 and Hep3B were obtained from the American Type Culture Collection (ATCC, Rockville, MD, USA). The doxorubicin (DOX)-resistant cancer cell line HepG2/ADM was kindly provided by Kwok-Pui Fung (School of Biomedical Sciences, The Chinese University of Hong Kong). All cells were incubated in RPMI-1640 medium, supplemented with 10% (v/v) fetal bovine serum and 1% (v/v) penicillin-streptomycin antibiotic at 37°C in a humidified atmosphere of 5% CO2. The multidrug-resistant characteristics of HepG2/ADM cells were maintained by 1.2 μM DOX.
Reagents
Arenobufagin (purity ≥ 98%) was extracted and isolated from toad venom as previously reported [19]. The DOX and SP conjugates were obtained from Merck Calbiochem (Darmstadt, Germany). PI, ribonuclease A (RNase A), DAPI and caffeine were purchased from Sigma-Aldrich (St. Louis, MO, USA). The OxiSelect™ comet assay kit was purchased from Cell Biolabs (San Diego, CA, USA). The p53 siRNA kit and mTOR siRNA kit were purchased from Genepharma (Shanghai, China). Lipofectamine 2000 reagent and the PureLink® Genomic DNA Kit were purchased from Life Technologies (New York, USA). The antibodies used in the mitotic index assay, Western blotting, and confocal microscopy analysis are listed in Supplementary Table 1. All other chemicals were obtained from Sigma-Aldrich (St. Louis, MO, USA).
Preparation of chemical probe biotinylated arenobufagin (DB7)
The principle and route of the DB7 synthesis is in Supplementary Figure S6. The synthesis of the arenobufagin derivative ClB4 and the linker moiety polyethylene glycol-D-biotin DB6 are shown in the Supplementary Methods. TEA (0.01 mmol) and DB6 (25 mg, 0.06 mmol) in anhydrous THF were added to a stirred solution of ClB4 (20 mg, 0.04 mmol) and NaI (5 mg, 0.03 mmol) in 2 mL anhydrous THF under nitrogen gas. The reaction was heated to reflux for 7 h and monitored by TLC (DCM:MeOH = 20:1). The solution was concentrated under reduced pressure. The residue was purified by flash column chromatography (SiO2 4 g, DCM:MeOH = 8:1 with 0.1% TEA) to yield the light yellow solid DB7 (10 mg, 28%). The structures of ClB4, DB6 and DB7 were identified by 1H NMR, 13C NMR, MS and HRMS (Supplementary Figures S7–S9). All final compounds were purified to > 95% purity.
1H NMR (300 MHz, CDCl3) δ 7.75 (dd, J = 9.9, 2.0 Hz, 1H), 7.38 (s, 1H), 7.16 (t, J = 4.9 Hz, 1H), 6.34 (s, 1H), 6.27 (d, J = 9.7 Hz, 1H), 5.47 (s, 1H), 5.12 (s, 1H), 4.54 – 4.46 (m, 1H), 4.33 (d, J = 3.7 Hz, 1H), 4.30 (s, 1H), 4.08 (t, J = 6.8 Hz, 1H), 3.69 – 3.52 (m, 14H), 3.43 (s, 4H), 3.13 (dd, J = 10.7, 5.9 Hz, 1H), 2.92 (d, J = 4.6 Hz, 1H), 2.88 (d, J = 4.5 Hz, 1H), 2.85 – 2.78 (m, 3H), 2.74 (s, 1H), 2.70 (s, 1H), 2.45 (s, 2H), 2.40 (s, 2H), 2.22 (t, J = 7.2 Hz, 3H), 2.06 (s, 1H), 2.03 (s, 1H), 1.17 (s, 3H), 0.91 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 214.06 (s), 172.06 (s), 164.05 (s), 162.14 (s), 150.36 (s), 147.28 (s), 120.88 (s), 115.87 (s), 85.39 (s), 73.50 (s), 71.38 (s), 70.55 (s), 70.26 (s), 70.17 (s), 62.13 (s), 61.92 (s), 60.29 (s), 55.67 (s), 51.13 (s), 48.84 (s), 40.92 (s), 40.85 (s), 40.67 (s), 39.54 (s), 39.27 (s), 38.67 (s), 36.89 (s), 35.88 (s), 32.90 (s), 32.83 (s), 30.88 (s), 28.30 (s), 28.22 (s), 28.15 (s), 26.42 (s), 25.79 (s), 23.56 (s), 21.72 (s), 17.68 (s); ESI-LRMS (m/z): [M+H]+: 875.4, [M+Na]+: 897.3; HRMS(m/z): calcd for C44H66N4O12S, [M+H]+: 875.4471, found: 875.4461.
Cell cycle analysis
The cells were harvested and fixed in ice-cold 75% ethanol at 4°C overnight. The cells were incubated with 0.02 mg/mL PI and 0.1 mg/mL RNase A at 37°C in the dark for 30 min. The cells were then analyzed by EPICS-XL flow cytometry (Beckman Coulter, Pasadena, California, USA), and the phase distribution of the cell cycle was analyzed using ModFit LT 2.8 software (Becton Dickinson, CA, USA).
Mitotic indexes
The cells were fixed with 4% paraformaldehyde for 10 min and permeabilized with 0.4% Triton X-100 for 30 min. After blocking in 5% bovine serum albumin (BSA, 0.1% Triton X-100) for 1 h, the plates were incubated with an antibody against p-Histone3 (Ser10) diluted in 2% BSA overnight. After washing with ice-cold PBS, the plates were incubated with Alexa Fluor 594 Goat anti-Mouse IgG (H+L) antibody (1:1,000 dilution) for 2 h, and the DNA was stained with DAPI for 5 min. The plates were imaged using an ImageXpress Micro XL (Molecular Devices, Silicon Valley, USA) with a 10× lens. The mitotic indexes were determined by counting the number of p-Histone H3 (Ser10)-positive cells in the number of DAPI-positive cells (which served as the total number of cells). At least 200 cells were analyzed using MetaXpress software (Molecular Devices, Silicon Valley, USA).
Detection of apoptosis
The cells were exposed to arenobufagin for 48 h, and apoptosis was detected using Annexin V-FITC/PI apoptosis detection kit (Biouniquer Tech, Nanjing, Jiangsu, China) according to the manufacturer’s protocol.
Western blotting
The cells were lysed in ice-cold RIPA buffer (1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate, 2 mmol/L EDTA, 25 mmol/L Tris-HCl, pH = 7.5) containing 0.5 mol/L DTT, 0.1 mol/L PMSF, protease and phosphatase inhibitors (Roche Applied Science, Mannheim, Germany) to obtain the total cellular protein. The cell lysates were collected, and the concentrations were determined with a BCA assay (Thermo Fisher Scientific, Waltham, MA, USA). A total of 30–50 μg of the lysates, 293 cell extracts and 293 cell + UV (4 h) extracts (CST, Beverly, MA, USA) were separated by SDS-PAGE and then transferred to PVDF membranes. The membranes were blocked and probed with antibodies against the target proteins and subsequently incubated with either an anti-mouse or anti-rabbit secondary antibody conjugated to HRP. The protein bands were visualized with an ECL kit (Thermo Fisher Scientific, Waltham, MA, USA), and their images were captured on an X-ray film (Kodak, Rochester, New York, USA). The protein levels were quantified using ImageJ software (National Insitutes of Health, Betheda, Maryland, USA).
Small interfering RNA transfection
The cells were transfected with small interfering RNA (siRNA) targeting p53 (100 nmol/L) or negative control siRNA using Lipofectamine® 2000 according to the manufacturer’s protocol. The transfected cells were exposed to arenobufagin for 48 h, followed by Western blotting and cell cycle analyses.
Co-immunoprecipitation
The cells were re-suspended in lysis buffer (50 mmol/L Tris, 150 mmol/L NaCl, 50 mmol/L NaF, 2 mmol/L EGTA, 10% glycerol, 0.25% NP-40, protease and phosphatase inhibitors, pH = 7.5). The cell lysates were collected, and the concentrations were determined with a BCA assay (Thermo Fisher Scientific, Waltham, MA, USA). One milligram of protein extract was incubated with an antibody against CDK1 at 4°C for 2 h before being incubated with G-Sepharose beads overnight. The immunoprecipitated complex were washed, centrifuged and dissolved in 2× loading buffer. The samples were analyzed by SDS polyacrylamide gel electrophoresis and immunoblotting as described above.
Comet assay
The cellular DNA damage in single cell was evaluated as described previously [10]. In brief, the resuspended cells were mixed with melted agarose and then pipetted onto slides. The samples were lysed, denatured, electrophoresed, and stained with Vista Green DNA dye. Images were captured with a Zeiss Axio Imager A2 microscope (Carl Zeiss AG, Oberkochen, Germany). The tail length was defined as the length of the comet tail (Pixel). The tail DNA% was defined the percentage of the intensity of tail DNA to the intensity of cell DNA. The tail moment length was defined as the length from the center of the head to the center of the tail. The Olive tail moment was calculated by multiplying the tail moment length by the tail DNA%. All parameters were evaluated based on at least 20 cells per sample using MetaXpress software (Molecular Devices, Silicon Valley, USA).
γH2AX staining assay
The cells were fixed, permeabilized and incubated overnight with an antibody against p-Histone H2A.X (Ser139). After washing with ice-cold PBS, the cells were incubated with Alexa Fluor 647 donkey anti-rabbit IgG (H+L) (1:1,000 dilution) for 2 h. The DNA was stained with DAPI for 5 min. The plates were then washed and mounted in ice-cold PBS. The cells were photographed with an ImageXpress Micro XL (Molecular Devices, Silicon Valley, USA) with a 40× lens. The granules (red) in individual cells were counted using MetaXpress software (Molecular Devices, Silicon Valley, USA). The quantifiable data were obtained from at least 200 cells per sample.
Cellular distribution of biotinylated arenobufagin
The cells were exposed to 1 μmol/L biotinylated arenobufagin for various time points, fixed and incubated with SP (1:50 diluted with PBS). After washing three times with PBS, the cellular distribution of biotinylated-arenobufagin was imaged using a confocal microscope (Zeiss LSM700, Germany) with a 63× lens at an excitation wavelength of 488 nm.
Preparation of DNA from HepG2 cells
The DNA from HepG2 cells was purified using the PureLink® Genomic DNA Kit according to the manufacturer’s instructions. In brief, cells were harvested, re-suspended in PBS, and digested with Proteinase K and RNase A at 55°C. Binding buffer containing ethanol was added to the mixed lysate to allow the DNA to bind to the column. The proteins and impurities were removed by wash buffers. The DNA bound to the silica-based membrane in the column and then was eluted in low-salt buffer (50 mmol/L Tris-HCl, pH = 8.0). The purified DNA concentrations were spectrophotometrically determined using the molar extinction coefficient ε260 = 6600 M−1 cm−1. All DNA used in subsequent experiments was purified from HepG2 cells.
Isothermal titration calorimetry
The DNA from HepG2 cells was titrated against arenobufagin in 50 mmol/L Tris-HCl (pH = 8.0) by ITC using a MicroCal™iTC200 instrument (GE Health Care/Microcal, Northampton, MA, USA). A total of 30 μmol/L of DNA was injected into a 200 μL calorimetric cell and titrated against 0.4 mmol/L of arenobufagin in a 40 μL syringe at 25°C under constant stirring at 1,000 rpm. The blank titration of DNA was conducted in buffer containing DMSO. The resulting thermograms were analyzed with one set of binding site models using Microcal Origin 7.0.
UV spectroscopy
The spectrophotometric measurements were recorded using a JASCO J-810 spectropolarimeter (Jasco Corporation, Tokyo, Japan) at 25°C. We mixed 1 mmol/L of DNA and 20 nmol/L of arenobufagin as described above in 50 mmol/L of Tris-HCl (pH = 8.0). After the solution was mixed and equilibrated for approximately 5 min, the absorption spectra were measured at wavelengths ranging from 200 nm to 400 nm. A DNA solution of the same concentration without arenobufagin was used as the blank.
Circular dichroic spectroscopy
Circular dichroism (CD) measurements were performed using a JASCO J-810 spectropolarimeter (Jasco Corporation, Tokyo, Japan) at 25°C. The CD scans were recorded within a wavelength range of 200 to 400 nm at sensitivity of 5 mdeg. All measurements were performed in a cuvette with a volume of 400 μL in 50 mmol/L Tris-HCl (pH = 8.0). Individual titrations were performed with 1 mmol/L DNA in a reaction mixture containing 20 nmol/L of arenobufagin. A DNA solution of the same concentration without arenobufagin was used as the blank. The spectra were measured based on an average of three runs.
Fluorescence spectroscopy
The fluorescence emission spectra of the EB displacement assay were recorded on a RF-5301PC spectrofluorophotometer (Shimadzu, Japan) equipped with a xenon flash lamp. The EB-DNA complex was excited at 524 nm, and the emission spectra were recorded between 530 and 700 nm. A solution containing 0.006 μmol/L of EB and 50 μmol/L of DNA was titrated with increasing concentrations of arenobufagin, and the final reaction mixture volume was 3 mL and contained 50 mmol/L Tris-HCl (pH = 8.0). Appropriate blanks corresponding to the buffer were subtracted to correct for the background fluorescence.
Molecular modeling
The 3D structures of arenobufagin were sketched using SYBYL 7.0 (Tripos Associates; St. Louis, MO, USA) and optimized with the Tripos force field. The partial atomic charges were calculated using the Gasteiger-Marsili method. The intercalation between DNA and arenobufagin was modeled by building a B-form duplex DNA model with the sequence 5′-d(CCGGCGGT)-3′ using the BIOPOLYMER module in SYBYL. Hydrogen atoms were then added, and Kollman All-Atom charges were assigned to the DNA molecule. To generate an intercalation cavity, arenobufagin was manually docked into DNA: the pyran ring was positioned between two base pairs (CCGGCGGT), while the other moieties were located in the major groove. This initial complex structure was then optimized via energy minimization with the Tripos force field by employing the Powell method with an energy-gradient-convergence criterion of 0.05 kcal/(mol A°). Arenobufagin and nonpolar hydrogens were removed from the energy-minimized complex, and the residual DNA was assigned Kollman United-Atom charges. The resulting DNA structure and intercalation cavity were to study the docking of arenobufagin with the GOLD program. To simulate the interaction between arenobufagin and DNA, arenobufagin was treated as a flexible ligand and docked into the intercalation cavity of DNA based on default parameters. The docking results were quantified by GOLDSCORE. The complex of the docking result with the best score was then further analyzed to explore the potential key interactions between arenobufagin and DNA.
Statistical analysis
All experiments were performed at least three times. The quantifiable data were derived from three independent experiments. The statistical analysis was conducted with a one-way ANOVA with post hoc comparisons and Tukey’s test using GraphPad Prism 5 software, and values are presented as the mean ± SD. P value ≤ 0.05 was considered to indicate significant differences.
ACKNOWLEDGMENTS
We thank Dr. Kwok-Pui Fung (School of Biomedical Sciences, The Chinese University of Hong Kong) for providing the DOX-resistant cancer cell line HepG2/ADM. We also thank Dr. Yan Li, Dr. Hang Ning and Mr. Wen-pen Li (Molecular Devices, Shanghai, China) for their assistance with the analysis of the comet assay as well as the γH2AX and p-Histone (Ser10) measurements. This study was supported by the Science and Technology Program of China (2012ZX09103101-053), the National Science Foundation of China (30901847 and 81573455), Guangdong Province (S2013050014183 and 2013CXZDA006), Guangzhou City (2011Y1-00017-11 and 2011J2200045), the Specialized Research Fund for the Doctoral Program of Higher Education of China (20124401110008), and the Program for New Century Excellent Talents in University (D.M. Zhang).
CONFLICTS OF INTEREST
All authors declare no conflicts of interest.
REFERENCES
1. Park JW, Chen M, Colombo M, Roberts LR, Schwartz M, Chen PJ, Kudo M, Johnson P, Wagner S, Orsini LS, Sherman M. Global patterns of hepatocellular carcinoma management from diagnosis to death: the BRIDGE Study. Liver Int. 2015; DOI: 10.1111/liv.12818.
2. Anestopoulos I, Voulgaridou GP, Georgakilas AG, Franco R, Pappa A, Panayiotidis MI. Epigenetic therapy as a novel approach in hepatocellular carcinoma. Pharmacology & Therapeutics. 2015; 145:103–119.
3. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, Schwartz M Porta C, Zeuzem S, Bolondi L, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008; 359:378–390.
4. Clarke BT. The natural history of amphibian skin secretions, their normal functioning and potential medical applications. Biol Rev Camb Philos Soc. 1997; 72:365–379.
5. Qi J, Tan CK, Hashimi SM, Zulfiker AM, Good D, Wei MQ. Toad glandular secretions and skin extractions as anti-inflammatory and anticancer agents. Evidence-Based Complementary and Alternative Medicine. 2014; DOI: 10.1155/2014/312684.
6. Mijatovic T, Dufrasne F, Kiss R. Cardiotonic steroids-mediated targeting of the Na+/K+-ATPase to combat chemoresistant cancers. Curr Med Chem. 2012; 19:627–646.
7. Katz A, Lifshitz Y, Bab-Dinitz E, Kapri-Pardes E, Goldshleger R, Tal DM, Karlish SJ. Selectivity of digitalis glycosides for isoforms of human Na+/K+-ATPase. J Biol Chem. 2010; 285:19582–19592.
8. Moreno YBL, Katz A, Miklos W, Cimmino A, Tal DM, Ainbinder E, Zehl M, Urban E, Evidente A, Kopp B, Berger W, Feron O, Karlish S, et al. Hellebrin and its aglycone form hellebrigenin display similar in vitro growth inhibitory effects in cancer cells and binding profiles to the alpha subunits of the Na+/K+-ATPase. Molecular cancer. 2013; 12:33. doi: 10.1186/1476-4598-12-33.
9. Qi F, Li A, Zhao L, Xu H, Inagaki Y, Wang D, Cui X, Gao B, Kokudo N, Nakata M, Tang W. Cinobufacini, an aqueous extract from Bufo bufo gargarizans Cantor, induces apoptosis through a mitochondria-mediated pathway in human hepatocellular carcinoma cells. J Ethnopharmacol. 2010; 128:654–661.
10. Deng LJ, Hu LP, Peng QL, Yang XL, Bai LL, Yiu A, Li Y, Tian HY, Ye WC, Zhang DM. Hellebrigenin induces cell cycle arrest and apoptosis in human hepatocellular carcinoma HepG2 cells through inhibition of Akt. Chem Biol Interact. 2014; 219:184–194.
11. Yu ZL, Guo W, Ma XC, Zhang BJ, Dong PP, Huang L, Wang XL, Wang C, Huo XK, Yu WD, Yi CH, Xiao Y, Yang WJ, et al. Gamabufotalin, a bufadienolide compound from toad venom, suppresses COX-2 expression through targeting IKKβ/NF-κB signaling pathway in lung cancer cells. Molecular cancer. 2014; 13:203.
12. Zhang DM, Liu JS, Tang MK, Yiu A, Cao HH, Jiang L, Chan JYW, Tian HY, Fung KP, Ye WC. Bufotalin from Venenum Bufonis inhibits growth of multidrug resistant HepG2 cells through G2/M cell cycle arrest and apoptosis. European journal of pharmacology. 2012; 692:19–28.
13. Liu JS, Zhang DM, Chen MF, Li MM, Luo QD, Kurihara H, Ye WC. Anti-angiogenetic effect of arenobufagin in vitro and in vivo. Yao Xue Xue Bao. 2011; 46:527–533.
14. Zhang L, Nakaya K, Yoshida T, Kuroiwa Y. Induction by bufalin of differentiation of human leukemia cells HL60, U937, and ML1 toward macrophage/monocyte-like cells and its potent synergistic effect on the differentiation of human leukemia cells in combination with other inducers. Cancer Res. 1992; 52:4634–4641.
15. Kurosawa M, Tani Y, Nishimura S, Numazawa S, Yoshida T. Distinct PKC isozymes regulate bufalin-induced differentiation and apoptosis in human monocytic cells. Am J Physiol Cell Physiol. 2001; 280:C459–64.
16. Lee DY, Yasuda M, Yamamoto T, Yoshida T, Kuroiwa Y. Bufalin inhibits endothelial cell proliferation and angiogenesis in vitro. Life Sci. 1997; 60:127–134.
17. Li MM, Wu S, Liu Z, Zhang W, Xu J, Wang Y, Liu JS, Zhang DM, Tian HY, Li YL, Ye WC. Arenobufagin, a bufadienolide compound from toad venom, inhibits VEGF-mediated angiogenesis through suppression of VEGFR-2 signaling pathway. Biochem Pharmacol. 2012; 83:1251–1260.
18. Qi FH, Li AY, Zhao L, Xu HL, Inagaki Y, Wang DL, Cui XY, Gao B, Kokudo N, Nakata M, Tang W. Cinobufacini, an aqueous extract from Bufo bufo gargarizans Cantor, induces apoptosis through a mitochondria-mediated pathway in human hepatocellular carcinoma cells. Journal of ethnopharmacology. 2010; 128:654–661.
19. Zhang DM, Liu JS, Deng LJ, Chen MF, Yiu A, Cao HH, Tian HY, Fung KP, Kurihara H, Pan JX, Ye WC. Arenobufagin, a natural bufadienolide from toad venom, induces apoptosis and autophagy in human hepatocellular carcinoma cells through inhibition of PI3K/Akt/mTOR pathway. Carcinogenesis. 2013; 34:1331–1342.
20. Liu JS, Zhang DM, Li Y, Chen WM, Ruan ZX, Deng LJ, Wang LW, Tian HY, Yiu A, Fan CL, Luo H, et al. Discovery of Bufadienolides as a Novel Class of ClC-3 Chloride Channel Activators with Antitumor Activities. Journal of medicinal chemistry. 2013; 56:5734–5743.
21. Hashimoto S, Jing Y, Kawazoe N, Masuda Y, Nakajo S, Yoshida T, Kuroiwa Y, Nakaya K. Bufalin reduces the level of topoisomerase II in human leukemia cells and affects the cytotoxicity of anticancer drugs. Leuk Res. 1997; 21:875–883.
22. Watabe M, Nakajo S, Yoshida T, Kuroiwa Y, Nakaya K. Treatment of U937 cells with bufalin induces the translocation of casein kinase 2 and modulates the activity of topoisomerase II prior to the induction of apoptosis. Cell Growth Differ. 1997; 8:871–879.
23. Cruz Jdos S, Matsuda H. Arenobufagin, a compound in toad venom, blocks Na+-K+ pump current in cardiac myocytes. European journal of pharmacology. 1993; 239:223–226.
24. Cruz Jdos S, Matsuda H. Depressive effects of arenobufagin on the delayed rectifier K+ current of guinea-pig cardiac myocytes. European journal of pharmacology. 1994; 266:317–325.
25. Goto H, Tomono Y, Ajiro K, Kosako H, Fujita M, Sakurai M, Okawa K, Iwamatsu A, Okigaki T, Takahashi T, Inagaki M. Identification of a novel phosphorylation site on histone H3 coupled with mitotic chromosome condensation. J Biol Chem. 1999; 274:25543–25549.
26. Horwitz SB. Taxol (paclitaxel): mechanisms of action. Ann Oncol. 1994; 5:S3–6.
27. Gavet O, Pines J. Progressive activation of Cyclin B1-CDK1 coordinates entry to mitosis. Developmental Cell. 2010; 18:533–543.
28. Nurse P. A long twentieth century of the cell cycle and beyond. Cell. 2000; 100:71–78.
29. Perdiguero E, Nebreda AR. Regulation of Cdc25C activity during the meiotic G2/M transition. Cell Cycle. 2004; 3:733–737.
30. Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell. 2003; 3:421–429.
31. Zhao H, Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol. 2001; 21:4129–4139.
32. Matsuoka S, Rotman G, Ogawa A, Shiloh Y, Tamai K, Elledge SJ. Ataxia telangiectasia-mutated phosphorylates Chk2 in vivo and in vitro. Proc Natl Acad Sci U S A. 2000; 97:10389–10394.
33. Durocher D, Jackson SP. DNA-PK, ATM and ATR as sensors of DNA damage: variations on a theme? Current Opinion in Cell Biology. 2001; 13:225–231.
34. Lobrich M, Shibata A, Beucher A, Fisher A, Ensminger M, Goodarzi AA, Barton O, Jeggo PA. GammaH2AX foci analysis for monitoring DNA double-strand break repair: strengths, limitations and optimization. Cell Cycle. 2010; 9:662–669.
35. Sarkaria JN, Busby EC, Tibbetts RS, Roos P, Taya Y, Karnitz LM, Abraham RT. Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res. 1999; 59:4375–4382.
36. Jeffery DA, Bogyo M. Chemical proteomics and its application to drug discovery. Curr Opin Biotechnol. 2003; 14:87–95.
37. Palchaudhuri R, Hergenrother PJ. DNA as a target for anticancer compounds: methods to determine the mode of binding and the mechanism of action. Current Opinion in Biotechnology. 2007; 18:497–503.
38. Olmsted J, Kearns DR. Mechanism of ethidium bromide fluorescence enhancement on binding to nucleic acids. Biochemistry. 1977; 16:3647–3654.
39. Tu LC, Chen CS, Hsiao IC, Chern JW, Lin CH, Shen YC, Yeh SF. The beta-carboline analog Mana-Hox causes mitotic aberration by interacting with DNA. Chem Biol. 2005; 12:1317–24.
40. Qi FH, Li AY, Inagaki Y, Kokudo N, Tamura S, Nakata M, Tang W. Antitumor activity of extracts and compounds from the skin of the toad Bufo bufo gargarizans Cantor. International Immunopharmacology. 2011; 11:342–349.
41. Ma LN, Song B, Jin H, Pi J, Liu L, Jiang JH, Cai JY. Cinobufacini induced MDA-MB-231 cell apoptosis-associated cell cycle arrest and cytoskeleton function. Bioorganic & Medicinal Chemistry Letters. 2012; 22:1459–1463.
42. Takai N, Ueda T, Nishida M, Nasu K, Narahara H. Bufalin induces growth inhibition, cell cycle arrest and apoptosis in human endometrial and ovarian cancer cells. International Journal of Molecular Medicine. 2008; 21:637–643.
43. Kamano Y, Yamashita A, Nogawa T, Morita H, Takeya K, Itokawa H, Segawa T, Yukita A, Saito K, Katsuyama M, Pettit GR. QSAR evaluation of the Ch’an Su and related bufadienolides against the colchicine-resistant primary liver carcinoma cell line PLC/PRF/5(1). J Med Chem. 2002; 45:5440–5447.
44. Ivanov VI, Minchenkova LE, Schyolkina AK, Poletayev AI. Different conformations of double-stranded nucleic acid in solution as revealed by circular dichroism. Biopolymers. 1973; 12:89–110.
45. Laiho M, Latonen L. Cell cycle control, DNA damage checkpoints and cancer. Ann Med. 2003; 35:391–397.
46. Harper JW, Elledge SJ. The DNA damage response: Ten years after. Molecular Cell. 2007; 28:739–745.
47. Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998; 282:1497–1501.
48. Lukin DJ, Carvajal LA, Liu WJ, Resnick-Silverman L, Manfredi JJ. p53 promotes cell survival due to the reversibility of its cell-cycle checkpoints. Molecular Cancer Research. 2015; 13:16–28.
49. Xu ZW, Wang FM, Gao MJ, Chen XY, Hu WL, Xu RC. Targeting the Na+/K+-ATPase α1 subunit of hepatoma HepG2 cell line to induce apoptosis and cell cyclearresting. Biol. Pharm. Bull. 2010; 33:743–751.
50. Babula P, Masarik M, Adam V, Provaznik I, Kizek R. From Na+/K+-ATPase and cardiac glycosides to cytotoxicity and cancer treatment. Anti-Cancer Agents in Medicinal Chemistry. 2013; 13:1069–1087.
51. Ramirez-Valle F, Badura ML, Braunstein S, Narasimhan M, Schneider RJ. Mitotic raptor promotes mTORC1 activity, G2/M cell cycle progression, and internal ribosome entry site-mediated mRNA translation. Mol Cell Biol. 2010; 30:3151–3164.
52. Selvarajah J, Elia A, Carroll VA, Moumen A. DNA damage-induced S and G2/M cell cycle arrest requires mTORC2-dependent regulation of Chk1. Oncotarget. 2015; 6:427–440.
53. Chang L, Graham PH, Hao J, Ni J, Bucci J, Cozzi PJ, Kearsley JH, Li Y. PI3K/Akt/mTOR pathway inhibitors enhance radiosensitivity in radioresistant prostate cancer cells through inducing apoptosis, reducing autophagy, suppressing NHEJ and HR repair pathways. Cell Death Dis. 2014; 5:e1437.
54. Pan ST, Qin Y, Zhou ZW, He ZX, Zhang X, Yang T, Yang YX, Wang D, Qiu JX, Zhou SF. Plumbagin induces G2/M arrest, apoptosis, and autophagy via p38 MAPK- and PI3K/Akt/mTOR-mediated pathways in human tongue squamous cell carcinoma cells. Drug Des Devel Ther. 2015; 9:1601–1626.
55. Li G, Han W, Jiang W, Zhang D, Ye W, Chen X, Ma A. Quantitative determination of arenobufagin in rat plasma by ultra fast liquid chromatography-tandem mass spectrometry and its application in a pharmacokinetic study. J Chromatogr B Analyt Technol Biomed Life Sci. 2013; 939:86–91.