
INTRODUCTION
The p53 tumor suppressor activates a series of diverse antiproliferative responses to protect cells from different cellular stresses. One of the most important p53 functions is its ability to activate apoptosis in a stage-, tissue- and stress-signal specific manner, and disruption of this process can promote tumor development [1]. As a hallmark of cancer, mutant p53 was involved in resistance to apoptosis, genomic instability, aberrant cell cycle, invasion and metastasis, tumor microenvironment, and inflammation [2].
Two major apoptotic pathways mediate p53-stimulated apoptosis, including the B-cell leukemia lymphoma 2 (Bcl-2) family and the caspase cascade. Members of Bcl-2 family include the ‘multidomain’ Bcl-2 family member Bax, the ‘BH3-only’ members Puma [3], Noxa [4, 5], and Bid [6]. Bcl-2 belongs to the Bcl-2 family proteins, which is one of the most important regulators of the mitochondrial apoptosis pathway [7–9]. It has been proved that the severity and extensiveness of the phenotype of Bcl-2 deficiency could be greatly affected by multiple genetic elements of the mice, resulting in tissue-specific modulations of the cell death program during development and cellular homeostasis [10]. Deficiencies of different Bcl-2 family proteins in mice result in multiple phenotypes, some of which are lethal [11]. Specifically, Bcl-2 deficiency in mice leads to various abnormalities, such as increased embryonic death, lymphocytopenia, hypopigmentation, polycystic kidney disease (PKD), distorted small intestine, abnormal skeletal development, reduced body weight, postnatal growth retardation and shortened life span [12–18]. These results indicate that the Bcl-2 protein has pleiotropic effects on embryonic development and adult homeostasis in various tissues and cells in vivo.
The interaction between p53 and Bcl-2 has been intensely studied in vitro. Specifically, p53 interacts with members of the Bcl-2 family (such as Bcl-2, Bcl-XL and Mcl1) of apoptosis-regulatory proteins in the cytoplasm to trigger apoptosis and to inhibit autophagy [19, 20]. Furthermore, the functions of Bcl-2 on apoptosis are suppressed by the p53 proapoptotic protein [21]. Interestingly, several independent studies have shown that p53 affects transcriptional activity of a partial Bcl-2 promoter [22–24]. In addition, blocking p53-Bcl-2 interaction decreases Granzyme B-induced Bax activation, cytochrome c release, and hence effector caspase inactivation, resulting in a reduced sensitivity of target cells to both Granzyme B and CTL/NK-mediated cell death [25]. However, it remains unknown the interaction between p53 and Bcl-2 in vivo. To further characterize the potential interaction between p53 and Bcl-2 in vivo, we depleted p53 in Bcl-2-null mice. We found that the Bcl-2 knockout phenotype was alleviated by p53 protein deletion in a tissue specific manner. Loss of p53 in Bcl-2−/− mice inhibited apoptosis induction in spleen and hair follicle melanocytes and keratinocytes, but not in kidney.
RESULTS
Loss of p53 in Bcl-2−/− mice prevents spleen atrophy, but does not restore normal growth and ear length of the mice
Loss of Bcl-2 itself induces striking abnormalities in mice. Although these animals appear normal at birth, they fail to thrive, develop short ears, and their thymus and spleen degenerate; later, their coats turn gray and the mice die within 4 to 16 weeks from renal failure due to polycystic kidney disease. Cell death is prominent in the affected tissues, and the lymphocytes are abnormally sensitive to diverse cytotoxic stimuli [12].
To eliminate any variability due to genetic background, we analyzed only Bcl-2 on a C57BL/6 background [26]. The Bcl-2−/− phenotype was proved more uniformly severe than reported in mixed backgrounds [27]. To determine the effect of p53 loss on the Bcl-2−/− phenotypes in vivo, we generated mice double null for Bcl-2 and p53. For mating, the p53 status was maintained in the heterozygous state to avoid potential broad genomic consequences arising from p53 deficiency.
All Bcl-2−/− mice that we produced had short ears, soon became runts (Figure 1A), and succumbed to polycystic kidney disease between the third and eighth week of life. Strikingly, all the double knockout mice (n = 17) exhibited growth retardation with short ears (Figure 1A), polycystic kidneys, thymus degeneration, low body weight (Figure 1C & Figure 2C, E, F and H) and died from 2 to 12 weeks old (Figure 1B). However, spleen degeneration was rescued (Figure 2A). We stained apoptotic cells in the spleen and kidney of adult mice in order to determine whether the interaction between p53 and Bcl-2 is tissue specific. In spleen, cell morphology is normal in the double-null mice (Figure 2A). Pertinently, apoptotic cells were especially abundant in the Bcl-2−/− spleen compared to the double knockout mice (Figure 2B). In the kidney, TUNEL staining revealed similar numbers of positive cells in wild-type and p53 and p53-null Bcl-2−/− deficient mice (Figure 2D). Thus, loss of p53 is strongly anti-apoptotic in spleen, but not in kidney. As a control, we detected the expression of p53 protein in mouse skin, kidney, spleen and thymus in vivo and found that the expression level of p53 protein is lower in kidney than in skin, spleen and thymus, all of which have same expression levels of p53 protein (Figure 2I).
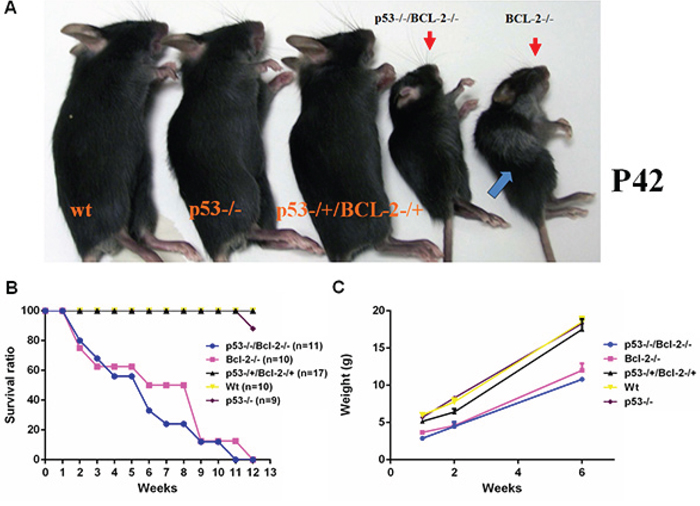
Figure 1: Loss of p53 prevents hair graying caused by Bcl-2 deficiency, but does not restore normal growth. A. wt, p53−/−, p53−/+/Bcl-2−/+, p53−/−/Bcl-2−/− and Bcl-2−/− mice. Both p53−/−/Bcl-2−/− and Bcl-2−/− groups exhibited growth retardation with small body size and short ears. Hair graying is also usually observed in adult Bcl-2-null mice (arrow). B. Mean survival curve of mice of the indicated genotypes. The observed numbers were as shown. C. Mean body weight of mice of the indicated genotypes at different ages. Six mice were observed in each group.
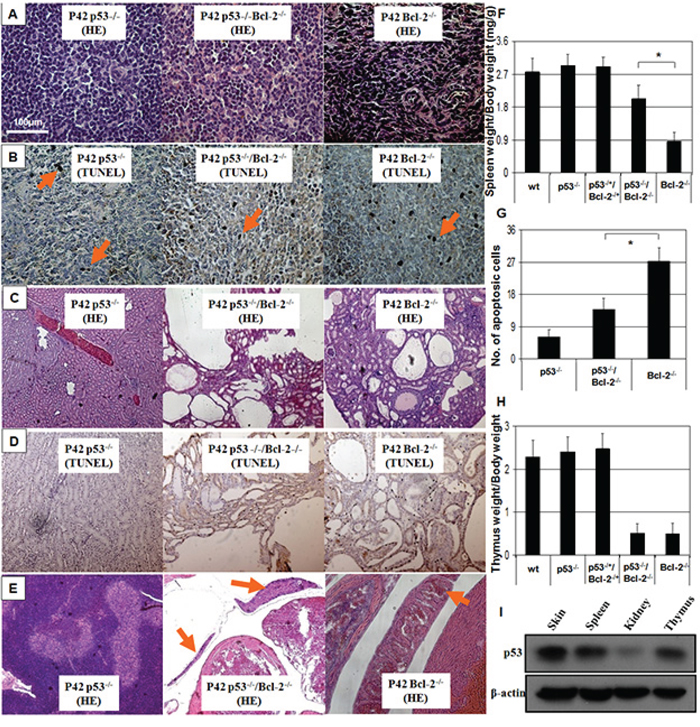
Figure 2: Spleen degeneration in Bcl-2−/− mice is prevented by absence of p53. A. Hematoxylin and eosin staining of spleen sections ( × 100). Splenic degeneration was found in Bcl-2−/−mice and was rescued by p53 deficiency in the double knockout. B. TUNEL staining for spleen sections showed hyper-apoptotic cells in Bcl-2−/−, which was prevented by loss of p53. C. Kidney sections ( × 100) show the abundant large thin-walled cysts in typical Bcl-2−/− and Bcl-2−/−/p53−/− mice. D. TUNEL staining of kidney sections showed hyper-apoptotic cells both in Bcl-2−/− and Bcl-2−/−/p53−/− mice. E. Thymus degeneration was seen in the Bcl-2−/− background with or without p53 ( × 100). F. Spleen weights were evaluated at P42. Six mice were observed in each group. Asterisk indicates statistical significance (P < 0.01). G. Numbers of apoptotic cells in the spleen sections of Bcl-2−/− and Bcl-2−/−/p53−/− mice were calculated. Asterisk indicates statistical significance (P < 0.01). H. Thymus weights were evaluated at P42 (P > 0.05). I. The p53 protein expression in skin, kidney, spleen and thymus in vivo.
Graying is prevented by the loss of p53 in the Bcl-2 deficient mouse
Hair graying is also usually observed in adult Bcl-2-null mice. To determine the potential impact of p53 on the melanocytes in Bcl-2-null background, we observed the hair graying in p53−/−, Bcl-2−/− and double knockout mice at different ages. During the second hair follicle cycle at 5–7 weeks of age, Bcl-2−/− mice (n = 10) developed a gray coat as reported [12, 14, 26, 28], and removal of p53 (n = 9) almost completely prevented the coat color change (Figure 3A). Loss of one allele of p53 was not able to rescue hair graying in Bcl-2 deficient mice (data not shown). The whiskers became gray starting at P20 in Bcl-2−/− mice and were rescued by loss of p53 as well (Figure 3B). We cannot conclude whether hair graying was delayed or prevented by loss of p53 in Bcl-2−/− mice because double knockout mice all died by 12 weeks of age.
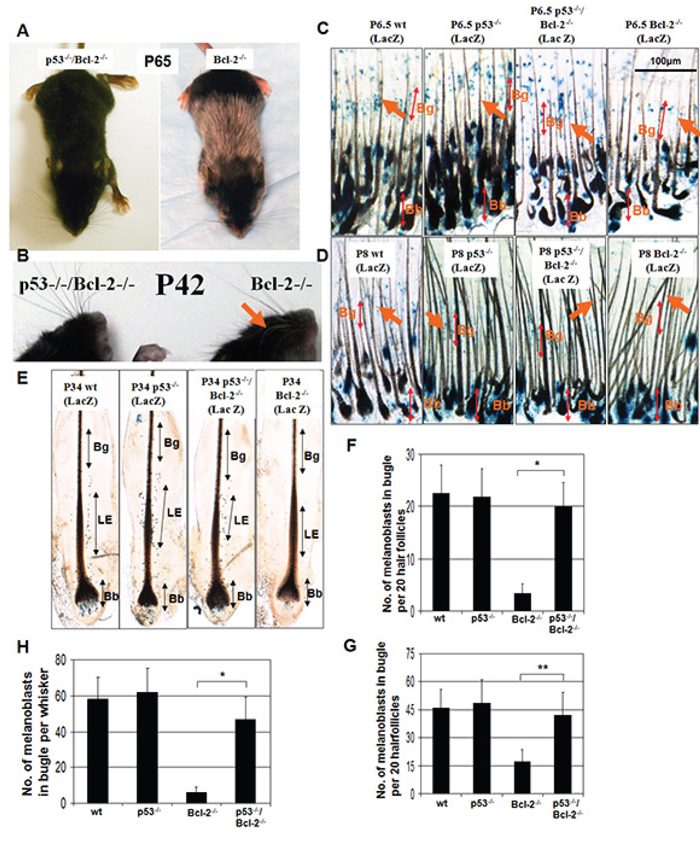
Figure 3: Graying elicited by Bcl-2 deficiency reflects p53-driven melanocyte death. A. Bcl-2−/−, and Bcl-2−/−/p53−/− C57BL/6 mice. Sixty-five day old Bcl-2−/− mice turned gray from the second hair follicle cycle, whereas the coats of Bcl-2−/−/p53−/− mice remained almost completely black although both groups exhibited growth retardation with small body size and short ears. B. Compare with Bcl-2−/−/p53−/− mice, the whiskers of Bcl-2−/− mice also turned gray (arrow). C and D. Distribution of Dct-lacZ+ melanoblasts (arrows) in the bulge region (Bg) (top double arrow) of pelage follicles at P6.5 and P8. Whereas bulb (Bb) melanocytes appear largely unchanged (bottom double arrow), bulge melanoblasts are unchanged in Bcl-2−/−/p53−/− follicles but lower in Bcl2−/− follicles at P6.5 and lost at P8 compared with wt and p53−/− follicles. F and G. The frequency of Dct-lacZ+ melanoblasts per 20 follicles in the bulge region. Asterisks indicate statistical significance (*P < 0.01, **P < 0.05). E. Distribution of Dct-lacZ+ melanoblasts in the niche: lower enlargement (LE) (double arrow) of whisker hair follicles at P34. Compared to wt and p53−/− follicles and whiskers, LE melanoblasts were unchanged in Bcl-2−/−/p53−/− follicles but lower in Bcl2−/− whiskers, and Bb melanocytes appear largely unchanged (bottom double arrow) in Bcl-2−/−/p53−/− follicles, but were lost in Bcl-2−/− whiskers. H. The quantity of Dct-lacZ+ melanoblasts per whisker in the LE. Asterisks indicate statistical significance (P < 0.01).
Death of Bcl2−/− melanocyte stem cells is rescued by the absence of p53
Hair follicles contain a well-demarcated structure for the stem-cell niche (within the lower permanent portion), whereas differentiated melanocytes reside in the hair bulb (at the base of the transient portion of the hair follicle) [26, 29, 30]. Although the graying of Bcl-2−/− mice was originally ascribed to defective melanin synthesis [12], observations support the conclusion that it reflects the loss of melanocytes stem cells in the hair follicle [28]. Taking advantage of the spatial segregation of the stem versus differentiated cell compartments, we used melanocyte-targeted (Dct) lacZ transgenic mice [26, 29, 31, 32] with Bcl-2−/−, p53−/−, or both to examine the impact of aging on these melanocyte compartments.
At P6.5, hair follicle morphogenesis is almost complete. Bcl-2−/− follicles contained melanoblasts in the bugle area (Figure 3C), whereas Bcl-2−/−/p53−/− follicles exhibited significantly more melanoblasts in the similar area (Figure 3C and 3F). In contrast, Bcl-2−/− follicles at P8 showed nearly a complete loss of melanoblasts in the niche (bulge area), whereas melanoblasts still appeared in the niche of Bcl-2−/−/p53−/− follicles (Figure 3D and 3G). However, the number of melanocytes in the hair bulb did not show any differences between Bcl-2−/−/p53−/− and Bcl2−/− mice in both time periods (data not shown). We also examined the numbers of melanocytes in the whiskers of Bcl-2−/− mice at P34. Melanoblasts were lost at this stage in the niche (lower enlargement area) and melanocytes were lost in the bulb as well in Bcl-2−/− follicles, whereas Bcl-2−/−/p53−/− follicles still contained melanoblasts in the niche and melanocytes in bulb (Figure 3E and 3G).
Hair growth is delayed by hyper-apoptosis in Bcl-2−/− follicles, which is partly rescued by the loss of p53
A previous report demonstrated that Bcl-2 protein is highly expressed in p53 null hair follicles. Furthermore, p53 protein is involved in the control of murine hair follicle regression in vivo [33]. To identify the role of Bcl-2 absence in hair growth and the interaction between Bcl-2 and p53 in hair growth, we assayed hair growth in p53−/−, Bcl-2−/− and double knockout mice. Hair follicles are constantly renewing, with alternating phases of growth (anagen), regression (catagen), and rest (telogen). The hair on the adult Bcl-2−/− mice (with or without p53) appeared to be sparser than that of their Bcl-2+/+ counterparts. Hair sparseness could be a function of the ratio of hair follicle cells in the anagen (growth) phase to those in the telogen (resting) phase. To test this, age-matched wild-type (wt), p53−/−, Bcl-2−/−/p53−/− and Bcl-2−/− mice (18 days old) were shaved in a 2-cm2 area on the back near the tail using an electric razor and then depilated. The subjective element in estimating the area is minimized by deficient regrowth as the first appearance of hair, and dividing the shaved area to be scored into eight equal portions with a transparent screen for hair-growth assay as described [34, 35]. A depilation-induced murine hair cycle is twenty-five days. In this period, the anagen phase is from day 1 to day 12, the catagen phase is from day 17 to day 18, and the telogen phase is day 0 or day 25 [36]. We measured hair regrowth one hair cycle (24 days) after depilation. Almost no hair growth was observed in Bcl-2−/− mice (Figure 4C and 4E). Hair growth declined linearly Bcl-2−/−/p53−/− mice (Figure 4D and 4E), whereas wt or p53−/− mice displayed robust hair growth (Figure 4A, 4B, 4E) and exhibited no hair cycle changes as reported [33]. Histology and apoptosis of hair follicles were observed on the age-matched wt, p53−/−, Bcl-2−/−/p53−/− and Bcl-2−/− mice. We couldn’t find any marked defect in the hair follicles of all groups of mice (Figure 4F). As expected, Bcl-2−/− mice, compared to wt and p53−/− mice, were characterized by the significantly increased (P < 0.01) number of TUNEL-positive cells and this increase was partly prevented by the loss of p53 (Figure 4G and 4H). This suggests that deletion of Bcl-2 induces apoptosis in follicles and retards the hair growth. This process can be partially prevented by loss of p53.
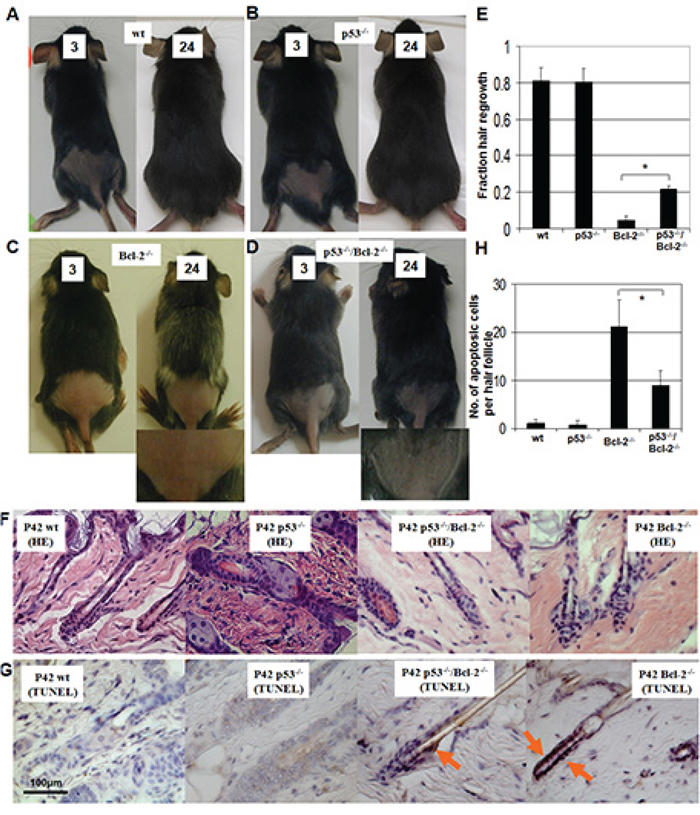
Figure 4: Hair regrowth in wild-type (wt), p53−/−, Bcl-2−/−/p53−/− and Bcl-2−/− mice. Hair regrowth assays were done in wt A. p53−/− B. Bcl-2−/− C. and Bcl-2−/−/p53−/− D. mice. Mice were shaved the fraction of a 2-cm2 back area near the tail at 18 days old and observed every day for 24 days. The hair-growth assay was performed as described [34, 35]. Days after depilation (3 or 24 days) are indicated. An amplified picture is shown for Bcl-2−/− C. and Bcl-2−/−/p53−/− D. Each condition was done in triplicate per group. E. Mean hair growth is shown for wt, p53−/−, Bcl-2−/−, and Bcl-2−/−/p53−/− mice at P42. Asterisks indicate statistical significance (P < 0.01). F. Photomicrographs of histological sections of the hair follicles at P42. HE staining, 400 ×. G. TUNEL staining of hair follicle sections showed hyper-apoptotic cells in Bcl-2−/− mice, which was partly prevented by the loss of p53. H. Numbers of hair follicle apoptotic cells (TUNEL-positive) in Bcl-2−/− and Bcl-2−/−p53−/− mice were calculated compared to controls. Asterisk indicates statistical significance (P < 0.01).
Loss of p53 inhibted the cell death induced by Bcl-2 knockdown
Our in vivo studies demonstrated that loss of p53 inhibited the Bcl-2-null-induced melanocyte death (Figure 3). To identify whether this melanocyte lineage specific interaction between p53 and Bcl-2 is detectable in vitro, we silenced the expression of Bcl-2 in mouse primary melanocytes isolated from wild-type p53 mice or p53-null mice and then stained with toluidine blue to observe cell growth (Figure 5A) and counted cell numbers. Specifically, Bcl-2 siRNA was transfected into wild-type and p53−/− primary melanocytes. Cell number was counted at 48 hours after transfection. As shown in Figure 5B, more than half of the wild-type primary melanocytes died in comparison to the death of 20% of p53−/− primary melanocytes (Figure 5B and 5C). This result indicates that the majority of Bcl-2-induced apoptosis is regulated by the optional p53 in vitro.
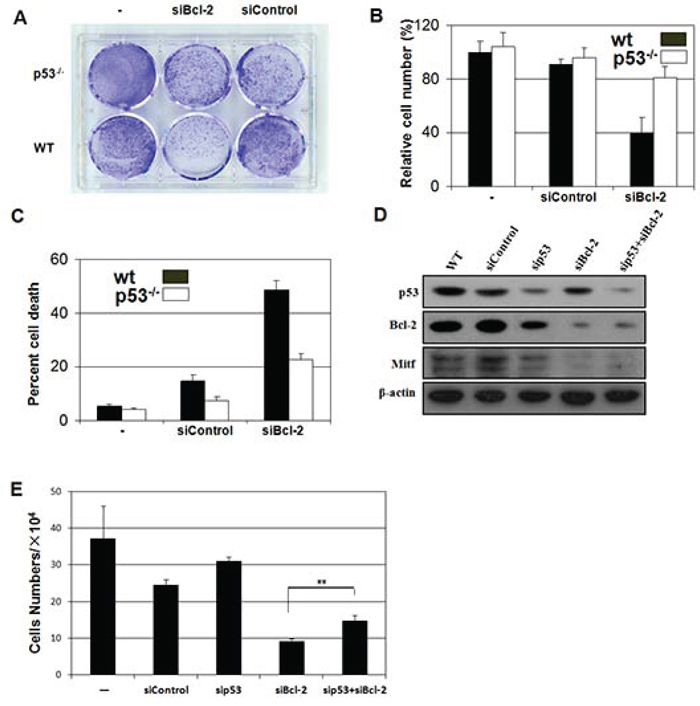
Figure 5: Loss of p53 inhibits the apoptosis induced by absence of Bcl-2 in vivo or Bcl-2 knockdown in vitro. A. p53−/− melanocytes are relatively resistant to cell death driven by knockdown of Bcl-2. Mouse primary melanocytes were plated and transfected with control or siRNA as indicated. Cells were fixed and visualized by dye staining. B. p53−/− mouse primary melanocytes were refractory to proliferation after knockdown of Bcl-2. Equal numbers of mouse primary melanocytes were transfected with vector or siRNA as indicated. C. Wild-type, but not p53−/−, mouse primary melanocytes showed reduced survival in response to the siBcl-2. Viability and apoptosis in mouse primary melanocytes was determined by flow cytometry 48 hr after transfection. D. Immunoblot analysis (IB) of whole cell lysates (WCL) derived from B16 cells transfected with the indicated siRNA constructs. E. Numbers of survival B16 cells by knockdown of Bcl-2 or/and p53. Equal numbers of B16 cells were transfected with vector or siRNA as indicated. The cells per well were determined under the microscope. Asterisks indicate statistical significance (**P < 0.05).
Bcl-2-targeted therapeutics are being widely explored [37]. To identify the dependence of p53 status in Bcl-2 targeted therapeutics in melanoma, we silenced the expression of p53, Bcl-2 or both in B16 melanoma cells using siRNA approaches (Figure 5D) and then counted the cell numbers at 48 hours after silencing. In these experiments, Mitf expression was monitored to evaluate the silencing effects of Bcl-2 [38]. We found that silencing Bcl-2 inhibited B16 melanoma cell growth, especially in p53-absent cells (Figure 5E).
DISCUSSION
In this study, we deleted the p53 gene in Bcl-2−/− mice to investigate the genetic interaction between p53 and Bcl-2 in vivo. We found the Bcl-2 knockout phenotype was partially alleviated by the deficiency of p53. Previous reports demonstrated p53 can either induce the expression of pro-apoptotic Bcl-2 proteins, or directly regulate a variety of Bcl-2 proteins in the cytoplasm [39]. In the current study, we found that loss of p53 rescued melanocyte stem cells from cell death and reduced hyper-apoptosis of spleenocytes in the Bcl-2−/− mouse. These results are consistent with previous reports that the absence of p53 resulted in a broader resistance to death stimuli in vivo [39]. However, the absence of p53 could not prevent Bcl-2 deficient mice from polycystic kidney disease and thymus degeneration, indicating that Bcl-2 deficiency-induced phenotype is p53-independent in kidney and thymus. Interestingly, previous reports showed that the p53 response to γ-irradiation in vivo was also in a tissue-specific manner. Thus, tissue specific role of p53 in apoptosis may be a generable phenotype [40]. Previous reports have demonstrated that loss of Bim rescue all defect in Bcl-2-null mice and indicate that Bim levels set the threshold for initiation of apoptosis [27]. Thus, our results also demonstrate the different role of p53 protein and Bim protein in apoptosis. Our results further confirmed the gene-gene interaction is tissue specific. Multiple molecular mechanisms might involve in these tissue specific interactions. First of all, the expression levels of p53 are different in different tissues. For example, we found that the expression of p53 protein in mouse kidney is lower than in spleen, skin and thymus (Figure 2I). We further found that the interactions between p53 protein and Bcl-2 protein are different in kidney, spleen, skin and thymus (Figure 1, 2, 3 and 4). Second, co-factors in a protein complex are usually tissue specific. Other mechanisms, such as different transcriptional regulations in different tissues and different microRNAs expressions in different tissues, may also involve in the Bcl-2/p53 interactions, all of which are deserved to be further studied. Further experiments are required to provide the detailed genetic and biochemical mechanisms for the tissue specific interaction between p53 and Bcl-2 in the regulation of cellular apoptosis.
Our in vitro data suggest that the majority of Bcl-2-induced apoptosis is regulated by the optional p53 (Figure 5). This finding may encourage us to evaluate the therapeutic potential by the induction of apoptosis with p53- and Bcl-2-targeting in a tissue specific manner. Furthermore, our results preliminary suggest that Bcl-2-targeted therapeutics is more effective in melanomas with wild-type p53 than in melanomas with mutant p53 (Figure 5E). Due to loss of p53 function, through p53 mutation itself or perturbations in p53 signaling pathways, is a common feature in the majority of human cancers. Our results indicate that p53 status should be detected before Bcl-2-targeted therapeutics, which is suitable to be used in tumors with wild-type p53.
MATERIALS AND METHODS
Animals
p53-deficient (−/−) mice were C57BL/6 TSG-p53® N12 and purchased from Taconic Farms (Hudson, NY, USA). These p53-deficient mice were originally generated by Donehower et al. [41]. Bcl-2−/+ (C57BL/6J) mice were purchased from the Jackson laboratory. DCT-lacZ transgenic mice were a gift from I. Jackson. The DCT-lacZ transgenic colony (CBA/C57BL6) was backcrossed to C57BL/6J [26].
Skin and hair growth assays
Dorsal skin sections were fixed, embedded in paraffin and stained with hematoxylin and eosin. For the hair growth assay, age-matched p53+/+, p53−/−, Bcl-2−/+, Bcl-2−/−/p53−/− and Bcl-2−/− mice (18 days) were shaved in a 2cm square area on their back near the tail using an electric razor and then depilated by Nair®. The subjective element in estimating the area is minimized by defining regrowth as the first appearance of hair, and dividing the shaved area to be scored into eight equal portions with a transparent screen [34, 35]. We measured hair regrowth 24 days after depilation.
Whole mount β–galactosidase staining
Skin samples from DCT-lacZ, bcl-2−/−/DCT-lacZ, p53−/−/DCT-lacZ, and p53−/−/bcl-2−/−/DCT-lacZ mice were stained as previously described [26]. Briefly, skin was fixed in X-Gal buffer (5 mM K3Fe (CN)8 postassium ferricyanide, 2 mM MgCl2, 0.01% DOC, 0.02% NP40 and 0.05% X-Gal in PBS), incubated at 37° for 6 hours and cut by hand under a dissecting microscope.
Histology
Animals were sacrificed by CO2 inhalation. Sections were immediately placed in 10% buffered formalin until paraffin embedding and sectioning (done by the Rodent Histopathology core service at Harvard Medical School). Hematoxylin/Eosin staining was performed by the histopathology core. Immunohistochemical studies were performed on skin specimens using formalin-fixed, paraffin-embedded tissue. Tissues known to express the antigen of interest were used as positive controls, whereas removal of the primary antibodies in the test tissues was used as negative controls. TUNEL kit was purchased from R & D and applied according to the manufacturer's instructions.
siRNA
siRNA was from Dharmacon (Lafayette, CO). siControl contains four mismatches with all known human, mouse, and rat genes. Primary murine melanocytes were plated at <50% confluence (300,000 cells per 60-mm plate) 16–20 h before transfection in penicillin/streptomycin-free medium at a ratio of 6:1 (vol:vol) Oligofectamine (Invitrogen, Carlsbad, CA) to siRNA and incubated for 5–6 h per the manufacturer's instructions.
Flow cytometry
Cells were fixed in ice-cold 70% ethanol before DNA staining with 50 μg/ml propidium iodide (Sigma Aidrich) in phosphate buffer saline containing 0.5 mg/ml RNase A (Amersha, inc. DNA content was analyzed by flow cytometry (Becton Dickinson FACSCalibur) [42].
Cell growth assay
Cells were plated in 6-well plates and then transfected. 48 h after transfection cells were fixed in cold 100% methanol and cell growth was visualized by staining cells with 0.1% toluidine blue solution followed by destaining with 1% acetic acid. Images were documented with a FluorChem Imaging System (Bio-Rad).
Abbreviations
MSC, melanocyte stem cell; PKD, polycystic kidney disease; CTL, cytotoxic lymphocyte; NK, natural killer; Palb2, Partner And Localizer of BRCA2; DCT, dopachrome tautomerase; WT, wildtype; UV, ultraviolet; DNA, deoxyribonucleic acid; Mitf, microphthalmia-associated transcription factor; Bg, bulge region; Bb, bulb; LE, lower enlargement; HE, Hematoxylin and eosin; siRNA, small interfering RNA.
CONFLICTS OF INTEREST
The authors declare that there is no conflict of interests regarding the publication of this paper.
GRANT SUPPORT
This work was supported a grant (nos. 81302971, 81273764 and 81473682) from the National Science Foundation (NSFC) of China, Shanghai Municipal Health Bureau Fund (nos. 2010Y133 and 20134203) and Shanghai Science and Technology Committee Fund (nos. 12401903500, 14401970200 and 14401972703).
REFERENCES
1. Levine AJ. The first 30 years of p53: growing ever more complex. Nature reviews Cancer. 2009; 9:749–758.
2. Solomon H, Madar S, Rotter V. Mutant p53 gain of function is interwoven into the hallmarks of cancer. The Journal of pathology. 2011; 225:475–478.
3. Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Molecular cell. 2001; 7:683–694.
4. Oda K, Arakawa H, Tanaka T, Matsuda K, Tanikawa C, Mori T, Nishimori H, Tamai K, Tokino T, Nakamura Y, Taya Y. p53AIP1, a potential mediator of p53-dependent apoptosis, and its regulation by Ser-46-phosphorylated p53. Cell. 2000; 102:849–862.
5. Ploner C, Kofler R, Villunger A. Noxa: at the tip of the balance between life and death. Oncogene. 2008; 27:S84–92.
6. Sax JK, Fei P, Murphy ME, Bernhard E, Korsmeyer SJ, El-Deiry WS. BID regulation by p53 contributes to chemosensitivity. Nature cell biology. 2002; 4:842–849.
7. Cory S, Huang DC, Adams JM. The Bcl-2 family: roles in cell survival and oncogenesis. Oncogene. 2003; 22:8590–8607.
8. Green DR. The pathophysiology of mitochondrial cell death. Science. 2004; 305:626–629.
9. Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014; 15:49–63.
10. Ni HM, Chen X, Chen L, DiFrancesca D, Harada H, Yin XM. The impact of genetic background and Bid on the phenotype of Bcl-2-deficiency in mice. Apoptosis: an international journal on programmed cell death. 2008; 13:53–62.
11. Ranger AM, Malynn BA, Korsmeyer SJ. Mouse models of cell death. Nature genetics. 2001; 28:113–118.
12. Veis DJ, Sorenson CM, Shutter JR, Korsmeyer SJ. Bcl-2-deficient mice demonstrate fulminant lymphoid apoptosis, polycystic kidneys, and hypopigmented hair. Cell. 1993; 75:229–240.
13. Nakayama K, Nakayama K, Negishi I, Kuida K, Sawa H, Loh DY. Targeted disruption of Bcl-2 alpha beta in mice: occurrence of gray hair, polycystic kidney disease, and lymphocytopenia. Proceedings of the National Academy of Sciences of the United States of America. 1994; 91:3700–3704.
14. Kamada S, Shimono A, Shinto Y, Tsujimura T, Takahashi T, Noda T, Kitamura Y, Kondoh H, Tsujimoto Y. bcl-2 deficiency in mice leads to pleiotropic abnormalities: accelerated lymphoid cell death in thymus and spleen, polycystic kidney, hair hypopigmentation, and distorted small intestine. Cancer research. 1995; 55:354–359.
15. Amling M, Neff L, Tanaka S, Inoue D, Kuida K, Weir E, Philbrick WM, Broadus AE, Baron R. Bcl-2 lies downstream of parathyroid hormone-related peptide in a signaling pathway that regulates chondrocyte maturation during skeletal development. The Journal of cell biology. 1997; 136:205–213.
16. Fedorov LM, Schmittwolf C, Amann K, Thomas WH, Muller AM, Schubert H, Domen J, Kneitz B. Renal failure causes early death of bcl-2 deficient mice. Mechanisms of ageing and development. 2006; 127:600–609.
17. Yang E. Molecular thanatopsis: a discourse on the BCL2 family and cell death. Blood. 1996; 88:386–401.
18. Dicou E, Perez-Polo JR. Bax-an emerging role in ectopic cell death. Int J Dev Neurosci. 2009; 27:299–304.
19. Green DR. Cytoplasmic functions of the tumour suppressor p53. Nature. 2009; 458:1127–1130.
20. Vaseva AV. The mitochondrial p53 pathway. Biochimica et biophysica acta. 2009; 1787:414–420.
21. Chernigovskaya E, Atochin D, Yamova L, Huang P, Glazova M. Immunohistochemical expression of Bcl-2, p53 and caspase-9 in hypothalamus magnocellular centers of nNOS knockout mice following water deprivation. Biotechnic & histochemistry: official publication of the Biological Stain Commission. 2011; 86:333–339.
22. Miyashita T, Harigai M, Hanada M, Reed JC. Identification of a p53-dependent negative response element in the bcl-2 gene. Cancer research. 1994; 54:3131–3135.
23. Wu Y, Mehew JW, Heckman CA, Arcinas M, Boxer LM. Negative regulation of bcl-2 expression by p53 in hematopoietic cells. Oncogene. 2001; 20:240–251.
24. Bredow S, Juri DE, Cardon K, Tesfaigzi Y. Identification of a novel Bcl-2 promoter region that counteracts in a p53-dependent manner the inhibitory P2 region. Gene. 2007; 404:110–116.
25. Ben Safta T, Ziani L, Favre L, Lamendour L, Gros G, Mami-Chouaib F, Martinvalet D, Chouaib S, Thiery J. Granzyme B-Activated p53 Interacts with Bcl-2 To Promote Cytotoxic Lymphocyte-Mediated Apoptosis. Journal of immunology. 2015; 194:418–428.
26. Nishimura EK, Granter SR, Fisher DE. Mechanisms of hair graying: incomplete melanocyte stem cell maintenance in the niche. Science. 2005; 307:720–724.
27. Bouillet P, Cory S, Zhang LC, Strasser A, Adams JM. Degenerative disorders caused by Bcl-2 deficiency prevented by loss of its BH3-only antagonist Bim. Dev Cell. 2001; 1:645–653.
28. Yamamura K, Kamada S, Ito S, Nakagawa K, Ichihashi M, Tsujimoto Y. Accelerated disappearance of melanocytes in bcl-2-deficient mice. Cancer research. 1996; 56:3546–3550.
29. Nishimura EK, Jordan SA, Oshima H, Yoshida H, Osawa M, Moriyama M, Jackson IJ, Barrandon Y, Miyachi Y, Nishikawa S. Dominant role of the niche in melanocyte stem-cell fate determination. Nature. 2002; 416:854–860.
30. Fuchs E, Merrill BJ, Jamora C, DasGupta R. At the roots of a never-ending cycle. Dev Cell. 2001; 1:13–25.
31. Mackenzie MA, Jordan SA, Budd PS, Jackson IJ. Activation of the receptor tyrosine kinase Kit is required for the proliferation of melanoblasts in the mouse embryo. Dev Biol. 1997; 192:99–107.
32. Fitch KR, McGowan KA, van Raamsdonk CD, Fuchs H, Lee D, Puech A, Herault Y, Threadgill DW, Hrabe de Angelis M, Barsh GS. Genetics of dark skin in mice. Genes Dev. 2003; 17:214–228.
33. Botchkarev VA, Komarova EA, Siebenhaar F, Botchkareva NV, Sharov AA, Komarov PG, Maurer M, Gudkov AV, Gilchrest BA. p53 Involvement in the control of murine hair follicle regression. Am J Pathol. 2001; 158:1913–1919.
34. Harrison DE. Genetic differences in effects of food restriction on aging in mice. J Nutr. 1987; 117:376–382.
35. Harrison DE. Biomarkers of aging: tissue markers. Future research needs, strategies, directions and priorities. Exp Gerontol. 1988; 23:309–325.
36. Handjiski BK, Eichmuller S, Hofmann U, Czarnetzki BM, Paus R. Alkaline phosphatase activity and localization during the murine hair cycle. Br J Dermatol. 1994; 131:303–310.
37. Thomas S, Quinn BA, Das SK, Dash R, Emdad L, Dasgupta S, Wang XY, Dent P, Reed JC, Pellecchia M, Sarkar D, Fisher PB. Targeting the Bcl-2 family for cancer therapy. Expert Opin Ther Targets. 2013; 17:61–75.
38. McGill GG, Horstmann M, Widlund HR, Du J, Motyckova G, Nishimura EK, Lin YL, Ramaswamy S, Avery W, Ding HF, Jordan SA, Jackson IJ, Korsmeyer SJ, Golub TR, Fisher DE. Bcl2 regulation by the melanocyte master regulator Mitf modulates lineage survival and melanoma cell viability. Cell. 2002; 109:707–718.
39. Chipuk JE. Dissecting p53-dependent apoptosis. Cell Death Differ. 2006; 13:994–1002.
40. Coates PJ, Lorimore SA, Lindsay KJ, Wright EG. Tissue-specific p53 responses to ionizing radiation and their genetic modification: the key to tissue-specific tumour susceptibility? J Pathol. 2003; 201:377–388.
41. Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA Jr, Butel JS, Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992; 356:215–221.
42. Cui R, Widlund HR, Feige E, Lin JY, Wilensky DL, Igras VE, D’Orazio J, Fung CY, Schanbacher CF, Granter SR, Fisher DE. Central role of p53 in the suntan response and pathologic hyperpigmentation. Cell. 2007; 128:853–864.