
INTRODUCTION
The signal transducer and activator of transcription 3 (STAT3) has been implicated in intracellular signaling transduction by different cytokines and growth factors. In normal cells, STAT3 activation is tightly controlled to prevent dysregulated gene transcription, whereas hyperactivation of STAT3 plays an important role in tumor cell proliferation, survival, invasion and immunosuppression, in diverse types of human cancers including hepatocellular carcinoma (HCC) [1, 2]. More and more evidences reveal that blocking STAT3 activation is beneficial for cancer therapy [3–6]. Suppressor of cytokine signaling 3 (SOCS3), which is a physiologic negative regulator of STAT3, has received increasing attention. Some pharmacologically safe and effective compounds including MicroRNA let7, nuclear receptor peroxisome proliferators-activated receptor gamma (PPAR γ) ligand, and angiostatic chemokine platelet factor-4 have already been reported to block STAT3 activation through enhancing expression of SOCS3 [7–9].
SOCS3 can be induced strongly by a variety of cytokines and other stimulators, then act to negatively regulate Janus tyrosine kinase 2 (JAK2) -STAT3 activity by inactivating JAK2 or blocking recruitment sites for STAT3 and also may target signaling complexes for ubiquitination and degradation [10, 11]. Loss of SOCS3 in HCC is associated with STAT3 over phosphorylation and poor prognosis [12–14]. Accumulating studies support that inhibition of SOCS3 expression promoted STAT3 activation, enhanced hepatic fibrosis, increased proliferation and tumor aggressiveness [12, 15, 16]. Moreover, the importance of STAT3 to hepatocarcinogenesis was illustrated by the fact that hepatocyte-specific deletion of SOCS3 in a mouse HCC model results in larger and more numerous tumors [17]. So restoration of SOCS3 should be a potential therapeutic strategy for HCC prevention and treatment. To further understand the contribution of SOCS3 to HCC progression, it is essential to better define the detailed regulatory mechanism of SOCS3 expression.
Farnesoid X receptor (FXR; NR1H4), a member of the nuclear receptor superfamily, is highly expressed in the gut-liver axist. FXR can be activated by a wide variety of compounds such as chenodeoxycholic acid (CDCA) and specific synthetic agonist GW4064 [18]. The ligand-activated FXR binds to its responsive elements as a classical heterodimer with retinoid X receptor alpha (RXR α) or as a monomer to coordinating the expression of target genes [19, 20]. In recent years, the understanding of the role of FXR in the liver has developed from that as a metabolic regulator to the novel function as a cell protector required for participating in carcinogenesis including promoting liver regeneration, suppressing hepatic inflammation, and regulating cell growth and differentiation, and thus may be a promising target for the prevention/treatment of liver cancer [21]. However, it is not well known whether the anti-tumor effect of FXR involves the regulation of SOCS3 or STAT3.
In our previous study, we have demonstrated that FXR activation led to an increased expression of SOCS3 in hepatocytes and mice liver, then to protect against the development of LPS-induced liver inflammation [22]. Furthermore, in the present study we continued to investigate the ability of FXR to influence HCC cell growth through the up-regulation of SOCS3 and the inhibition of STAT3 activation. This study will advance our understanding of the molecular mechanism of liver carcinogenesis targeting FXR and SOCS3.
RESULTS
FXR agonist GW4064 inhibits HCC cells growth and induces cell cycle arrested
The elucidation of the mechanism how FXR controls the proliferation of HCC cells is useful to establish the therapy for liver cancer. MTS assay revealed that FXR specific agonist GW4064 drastically decreased the rate of cell proliferation of HepG2 cells and Huh7 cells when compared with the corresponding control on the 48 or 72 h (Fig. 1A). Meanwhile as shown in Fig. 1B, the GW4064-mediated growth inhibition of HCC cells was associated with suppressing entry into the S phase. We also examined the effect of GW4064 on the alterations of p21 expression which controls cell proliferatrion and cell cycle progression. There was an obvious up-regulation in p21 at both transcriptional and translational levels. Besides, upon FXR activation, STAT3 activation, as assessed by the amount of Tyr705-phosphorylated protein, was inhibited, whereas total STAT3 protein remained unchanged (Fig. 1C and 1D). And we also observed that these events accompanied by an increased expression of SOCS3.
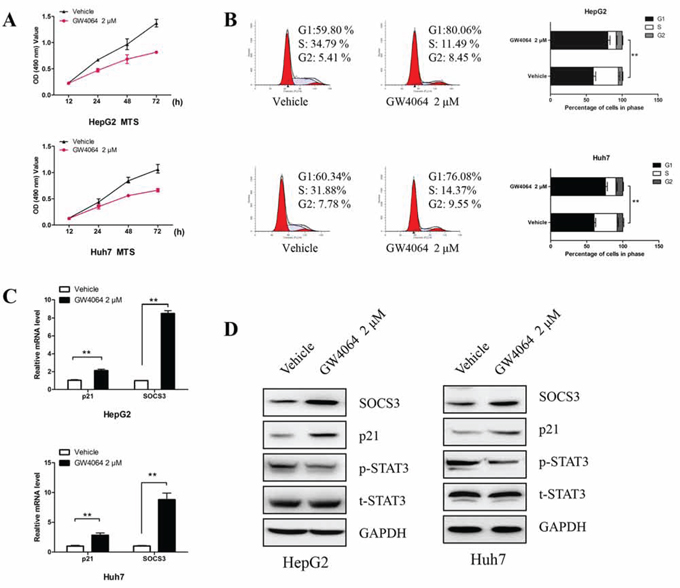
Figure 1: Activation of FXR in HCC cells inhibits cell growth, induces cell cycle arrest, up-regulates SOCS3 and p21, and represses STAT3 activation. A. HepG2 cells and Huh7 cells were treated with vehicle DMSO (0.1%) or GW4064 (2 μM) for different times, and then the cell proliferation was determined with MTS assay. Data are means ± SEM from three independent experiments in triplicate. B–D. HepG2 cells and Huh7 cells were treated with GW4064 (2 μM) or vehicle DMSO for 24 h. Then the cell cycle was analysed using PI staining and flow cytometry. The percentage of cells in each cell cycle phases was quantified (B). The mRNA levels of SOCS3 and p21 were examined by RT-qPCR, taking GAPDH as a control. Data are means of three separated experiments ± SEM, **P < 0.01 (C). The protein levels of SOCS3, p21, total STAT3 (t-SATA3) and phosphoraylated STAT3 (p-STAT3) were assayed by Western blot, taking GAPDH as a loading control (D).
Up-regulation of SOCS3 is involved in the FXR-mediated cell growth repression in HCC cells
A recently study has demonstrated that SOCS3 regulated p21 gene expression and induced cell cycle arrest primarily through its negative regulation of STAT3 signaling [23]. To investigate whether SOCS3 is involved in the anti-proliferative effect of FXR, we decreased SOCS3 expression through siRNA approach (Fig. 2A). As expected, Fig. 2B and 2C showed that the effect of GW4064-induced cell proliferation inhibition and cell cycle arrest at G1 phase were markedly relieved by disruption of the SOCS3 gene. Knockdown of SOCS3 also attenuated phosphorylation of STAT3 and the expression levels of p21 (Fig. 2D). These observations convincingly suggest that SOCS3 plays an important role in the FXR-mediated anti-HCC effects. Namely, FXR exerts the inhibitory ability on HCC, at least partially, through induction of SOCS3.
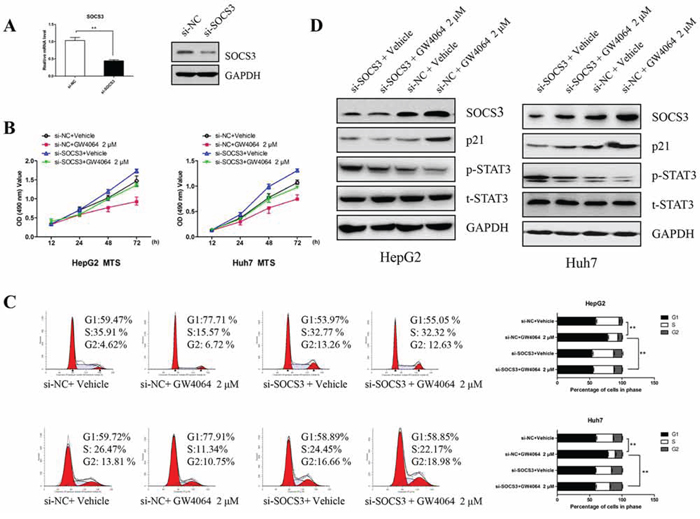
Figure 2: GW4064 influences cell proliferation in HCC cells partly through SOCS3. A. Huh7 cells were transiently transfected with the siRNA targeting SOCS3 for 48 h, the expression of SOCS3 was examined by RT-qPCR and Western blot. B. After silencing SOCS3 expression for 48 h, HepG2 cells and Huh7 cells were treated with GW4064 (2 μM) or vehicle DMSO for different times. Then the cell proliferation was detected by MTS assay. Data are means ± SEM from three experiments in triplicate. C. After interfering SOCS3 expression for 48 h, the cells were treated by GW4064 (2 μM) or vehicle DMSO for another 24 h. Then the percentage of cells in each cell cycle phases was determined. Data are means of three separated experiments ± SEM, **P < 0.01. D. Western blot were assayed for the expression of SOCS3 and p21 and the phosphorylation of STAT3.
IR9 is a likely FXRE within the human SOCS3 promoter region
The effect of GW4064-mediated SOCS3 induction was decreased in the presence of the specific FXR siRNA, further supporting the direct involvement of this nuclear receptor (Fig. 3A and 3B). As a classical transcriptional factor, FXR usually regulates the transcription of target genes via directly binding to FXR-responsive element (FXRE). As shown in Fig. 3C, treatment with GW4064 had an apparent stimulatory effect on the SOCS3 promoter (pSOCS3/2510) transcriptional activity. However, the transcriptional activity was dramatically diminished when the sequence (−2173 to −610) in the SOCS3 promoter region was deleted (pSOCS3/947) and resulted in failure in response to GW4064, which suggested that this DNA fragment in the region might harbor a key positive regulatory element which is sufficient for SOCS3 transcriptional activation. Sequence analysis of the SOCS3 promoter region with a Web-based algorithm (NUBIScan) predicted a potential FXRE/IR9 (an inverted repeat spaced by nine nucleotides, −1878 to −1858) in the human SOCS3 promoter region, and its sequence and location were shown in Fig. 3D.
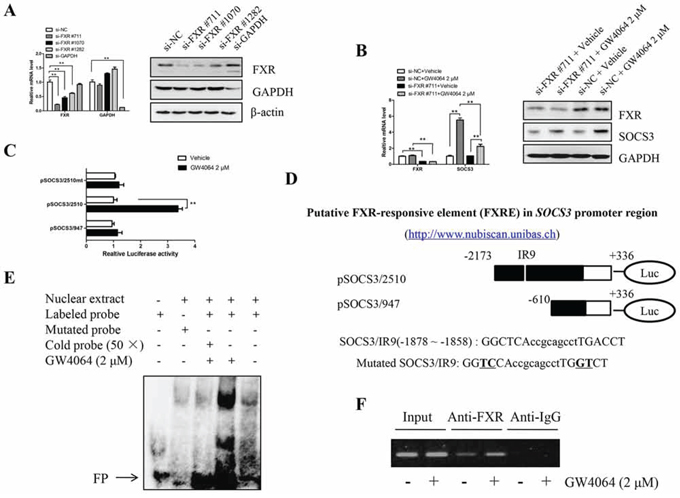
Figure 3: FXR binds to FXRE in human SOCS3 promoter region. A. Huh7 cells were transiently transfected with the siRNAs for FXR (targeting 711, 1070, 1282 sites respectively), the expression of FXR was examined by RT-qPCR and Western blot 48 h after transfection, and the one (numbered as si-FXR#711) with the best silence effect was selected to be used in the subsequent experiments. The siRNA targeting GAPDH is a positive control. B. After interfering FXR expression (si-FXR#711) for 48 h, Huh7 cells were treated with GW4064 (2 μM) or vehicle DMSO for another 24 h, RT-qPCR and Western blot were assayed for FXR and SOCS3 expression. Data are means of three separated experiments ± SEM, **P < 0.01. C. HepG2 cells were transiently transfected with the luciferase reporter plasmid pSOCS3/2510, pSOCS3/947 or pSOCS3/2510mt, followed by treatment with GW4064 (2 μM) for 24 h. Then the luciferase assay was performed. Results represent three independent experiments performed in triplicate. **P < 0.01. D. Prediction of a putative FXRE in human SOCS3 promoter region via an in silico analysis with a Web-based algorithm (NUBIScan). E. EMSA for analyzing the binding between the FXRE/IR9 probe and the nuclear proteins derived from HepG2 cells treated with vehicle DMSO or GW4064 (2 μM) for 24 h. FP: free probe. F. ChIP assay with chromatin isolated from HepG2 cells treated with vehicle DMSO or GW4064 (2 μM) for 24 h. Antibody directed against FXR was used for immune precipitation, taking IgG as a negative control. Final DNA extractions were amplified by PCR with the primer pairs covered the IR9 sequence in SOCS3 promoter region. Total extract (input) was used as positive PCR control.
Moreover, the mutation of FXRE/IR9 in SOCS3 promoter region abolished the GW4064-induced luciferase activity, indicating that FXRE/IR9 is very vital for FXR-enhanced transcriptional activation of SOCS3 gene promoter (Fig. 3C, pSOCS3/2510mt). To determine whether FXR directly binds to this element, EMSA and ChIP were performed. The sequences of the probes used in EMSA were showed in Fig. 3D. Interaction of SOCS3/IR9 probe with the nuclear extract of HepG2 cells yielded a DNA/protein band of expected mobility. The binding was greatly increased when the nuclear extract derived from GW4064-treated (Fig. 3E). Furthermore, this binding was sequence specific because (1) the binding was significantly weakened by a 50-fold excess of the unlabeled IR9 probe (cold probe); and (2) there was no obvious interaction between the nuclear extract and the mutated IR9 probe. Subsequently, ChIP assays showed that FXR could directly bind to the FXRE/IR9 in SOCS3 promoter region in HepG2 cells, and the binding was markedly enhanced after treatment with GW4064 (Fig. 3F). The above results suggest that FXR induces SOCS3 expression via directly and specifically binding to the FXRE/IR9.
FXR agonist suppresses HCC xenograft and represses STAT3 activation in vivo
The above studies clearly clarified that FXR induced SOCS3 expression and this induction played an important role in FXR-mediated growth suppression of HCC cells in vitro. Then we examined the influence of FXR agonist on tumor growth and SOCS3 expression in HCC tumor xenograft model. We injected HepG2 cells into the intrascapular region of nude mice and followed by administration of GW4064. In consistent with the above in vitro findings, we observed that GW4064 induced a regression in tumor growth during this period (Fig. 4A). As shown in Fig. 4B–4D, tumor size and mass were noticeably decreased in mice treated with GW4064 compared with the vehicle-treated group, at the time of killing day. Remarkably, this was accompanied by the up-regulation of SOCS3 and p21 expression in the xenograft tumors, the phosphorylation of STAT3 was lessened as well (Fig. 4E). These data suggest that FXR agonist can exert anti-HCC effects via up-regulating SOCS3 expression in vivo.
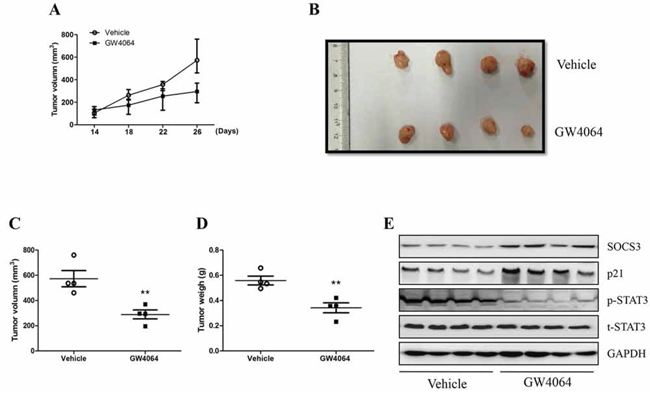
Figure 4: FXR ligand GW4064 represses the growth of HCC xengrafts and inhibits STAT3 activation in vivo. HepG2 cells (1.0 × 106/per mouse) were injected subcutaneously into the flank of nude mice. When palpable tumor were formed, GW4064 (25 mg/kg/day) or vehicle DMSO alone were administrated by intraperitoneally injected once every two day for 2 weeks. A. Tumor gowth was monitored over time. B–D. Tumors were photographed and harvested for analysis of the differences of tumor size and mass. Data are means ± SEM, n = 4, **P < 0.01. E. Tumor samples were subjected to quantitative western blot analysis for SOCS3 and p21expression and STAT3 activation.
FXR and SOCS3 expression levels are positively correlated in human HCC specimens
We examined the expression profile of FXR and SOCS3 and the activation of STAT3 in 66 HCC samples and the corresponding peritumoral tissues using IHC assay (Fig. 5). Statistical analysis with chi-square test showed that there was a positive correlation between FXR and SOCS3 expression (Fig. 5I, P < 0.01). Additionally, tumoral FXR and SOCS3 expression were significantly lower than that of the peritumoral tissue, whereas STAT3 was over-activated in HCC lesions, indicating that the dysexpression of FXR and SOCS3 might be involved in the development and/or progression of HCC.
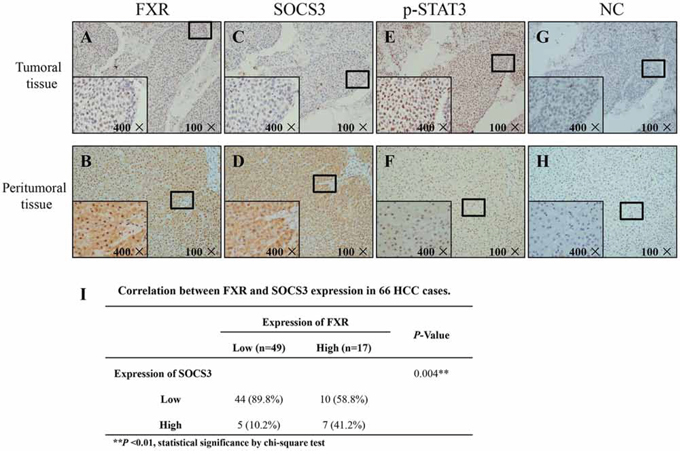
Figure 5: FXR and SOCS3 expression levels are positively correlated in human HCC specimens. Expression profile of FXR and SOCS3 and activation of STAT3 on formalin-fixed paraffin-embedded clinical samples were investigated by Immunohistochemistry. A–H. Representative images are shown. Negative control (NC): the primary antibody was replaced by rabbit IgG (isotype control). I. Correlation between FXR and SOCS3 expression in 66 HCC cases. **P < 0.01, Statistical significance by chi-square test.
DISCUSSION
Several studies support a role for SOCS3 as a tumor suppressor in different types of cancers [24, 25]. Increasing evidences have displayed over-expression of SOCS3 exhibited preclinical anti-tumor activity against HCC, malignant pleural mesotheliom and inflammation-associated colon cancer [26–28]. Up-regulation of SOCS3 will no doubt aid to the development of novel therapeutic strategies. In this study, we illustrate that FXR activation represses STAT3 activation and increases p21 expression via induction of SOCS3, then influences the cell cycle to suppress HCC cells growth. In addition, activation of FXR reduces the in vivo tumor growth rate in a mouse xenograft model. Taken together, these results suggest that FXR may serve as a key transcriptional regulator of cell proliferation in HCC by stimulating SOCS3 expression, and FXR-SOCS3 pathway may be a novel target for the treatment/prevention of HCC.
In the previous study, we for the first time discovered that both SOCS3 mRNA and protein were highly induced in HepG2 cells and mice livers in response to FXR ligand(s) [22]. Reporter assay revealed that SOCS3 promoter activity was significantly increased with FXR agonist treatment. As a classical transcriptional factor, FXR usually regulates the transcription of target genes via directly binding to FXRE such as IR1 (inverted repeat separated by 1 bp) [20], DR8 (directed repeat separated by 8 bp) [29], and ER8 (everted repeat separated by 8 bp) [30]. In this study, as summarized in Fig. 3, IR9 is identified as a novel FXRE that is involved in up-regulation of SOCS3 in HCC cells, which means SOCS3 is a new direct target gene of FXR. Besides, previous studies have demonstrated that another nuclear receptor PPAR γ could promote SOCS3 expression [8, 31], and FXR could up-regulate PPAR γ [32, 33]. So FXR may also indirectly enhance the expression of SOCS3 via inducing PPAR γ.
Given the great physiological importance in metabolism homeostasis, as well as in the regulation of inflammation and fibrosis, FXR plays a suppressive role in the liver carcinogenesis. Numerous studies showed that mice displayed spontaneous development of liver tumors in the absence of FXR [22, 34–38], we have reported that the inhibition of FXR promoted the proliferation, migration, and invasion of HCC cells [39]. FXR can protect against HCC by several mechanisms, including antagonizing nuclear factor-κB (NF-κB) activation to less hepatic inflammatory response [40], inhibiting a small subunit of proteasome gankyrin to subsequent protection of tumor suppressor proteins (Rb, p53, HNF4α) from degradation [41] or interacting with Wnt/β-catenin activation and E-cadherin expression [42, 43]. FXR directly induces the expression of HCC suppressors such as SHP (small heterodimer partner) [35] and NDRG2 (N-myc downstream regulated gene 2) [44] as well. Although multiple factors participate in the execution of FXR activation, its downstream genes that it regulates in HCC progression remains largely unexplored. Here we reported that SOCS3, the feedback inhibitor of the STAT3 pathway, was a direct FXR target gene. Knockdown of SOCS3 by siRNA substantially attenuated the GW4064-mediated repression of STAT3 activation and HCC cell growth. Our data displayed FXR-SOCS3 signaling as a novel mechanism in FXR-mediated anti-HCC effects. Although further research is needed, these studies strongly indicate that FXR is a negative modulator of hepatic cell hyperproliferation, ergo therapeutic modulation of FXR and SOCS3 could be profitable in patient with liver carcinoma, namely, which provides a new view about hepatic cancer treatment when targeting this FXR-SOCS3 signaling.
MATERIALS AND METHODS
Cell lines and cell culture
HCC cell lines HepG2 and Huh7 obtained from American Type Culture Collection were cultured in Dulbeco’s Modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% Penicillin/Streptomycin (complete medium) in a humidified atmosphere of 5% CO2 at 37°C. The cells grown to 70%–80% confluence were cultured in DMEM supplemented with 0.5% FBS and without antibiotics (conditioned medium), then treated with 2 μM FXR agonist GW4064 (Sigma–Aldrich, St. Louis, MO, USA) or vehicle dimethylsulphoxide (DMSO, 0.1%).
Western blot analysis
HCC cells or mice xgnograft tumors were lysed and the protein concentrations were normalizated by the BCA protein assay (Beyotime Inst Biotech, Beijing, China). Western blot analysis was performed as described previously [21]. Rabbit anti-FXR antibody (ab28676), rabbit anti-SOCS3 antibody (ab16030), rabbit anti-GAPDH antibody (ab181602) and rabbit IgG (isotype control, ab172730) were purchased from Abcam (San Francisco, CA, USA). The rabbit antibodies for total STAT3 (t-STAT3, #9132) and phosphorylated STAT3 (p-STAT3, Tyr705, #9145) were from Cell Signaling Technology (Beverly, MA, USA). Rabbit anti-P21 antibody (10355–1-AP) was bought from Proteintech (Chicago, IL, USA). The enhanced chemiluminescence detection reagents (Pierce, Rockford, IL, USA) were used to visualize the signals.
RNA extraction and Real-time quantitative polymerase chain reaction (RT-qPCR) assay
Total RNA was isolated with TRIzol reagent and the first-strand cDNA was synthesized using reverse transcriptase (Invitrogen, Carlsbad, CA, USA). The evaluation of gene expression was performed by real-time quantitative Polymerase Chain Reaction analysis. The mRNA levels of these genes were normalized to GAPDH mRNA levels. The primer sequences used for RT-qPCR are listed in Table 1.
Table 1: The primer sets for RT-qPCR
Gene |
Product size (bp) |
Forward primer |
Reverse primer |
|---|---|---|---|
SOCS3 |
136 |
5′-ATCCTGGTGACATGCTCCTC-3′ |
5′-GGCACCAGGTAGACTTTGGA-3′ |
p21 |
98 |
5′-CTGGAGACTCTCAGGGTGAA-3′ |
5′-GGATTAGGGCTTCCTCTTGGA-3′ |
GAPDH |
159 |
5′-CAATGACCCCTTCATTGACC-3′ |
5′-GACAAGCTTCCCGTTCTCAG-3′ |
Cell proliferation MTS assay
HepG2 cells and Huh7 cells were seeded in 96-well plates and the following day cells were treated with GW4064 (2 μM) or vehicle (0.1% DMSO). After 12, 24, 48, or 72 h, cell proliferation assay was performed using a MTS assay (Promega, Madison, WI, USA) according to the manufacturer’s instructions. Briefly, 2 × 103 cells per well were seeded in 96-well plates and cultured for overnight. Then, the cells were treated with GW4064 or DMSO. After 12, 24, 48, or 72 h, MTS reagent (20 μL) was added to the cells in each well followed by incubation for 2 h, and the absorbance was determined at 490 nm using a microplate reader.
Cell cycle assay
After treated with GW4064 (2 μM) for 24 h, the harvested cells were fixed with 70% ethanol in PBS at −20°C overnight then stained with propidium iodide (PI) using Cell Cycle Phase Determination Kit (Cayman, Ann Arbor, MI, USA), and the samples were then analyzed for cell cycle phase distribution using a FACScan flow cytometer. The data were analyzed by using the Cell Quest computer program (BD).
SOCS3 gene silencing by small interfering RNA (siRNA)
The siRNA sequences used for targeting human SOCS3 (sense 5′-CCAAGAACCUGCGCA UCCAdTdT-3′; antisense, 5′-UGGAUGCGCAGGUUCUUGGdTdT-3′) were synthesized by Genepharma (Shanghai, China). A non-targeting siRNA pool was used as a negative control (NC). The experiment details were performed as previous description [22].
Plasmid vector construction and luciferase assay
Human SOCS3 promoter region containing fragments (−2173 to +336 and −610 to +336) were amplified by PCR using genomic DNA of Huh7 cell as template. The primer sequences used for plasmid construction are listed in Table 2. The fragments were digested with Kpn I and Nhe I then cloned into pGL3-basic vector and the resulting plasmid were named as pSOCS3/2510 and pSOCS3/947. Site-directed mutation in pSOCS3/2510 at the IR9/FXRE (−1878 to −1858 from GGCTCAccgcagcctTGACCT to GGTCCAccgcagcctTGGTCT, the mutated bases were underlined) was constructed by employing TaKaRa mutant BEST Kit (TaKaRa, Dalian, China), and the resulting plasmid was named as pSOCS3/2510mt. Cell extracts were prepared after transfection, the luciferase and β-galactosidase (β-gal) activity assays were performed as described [45].
Table 2: The primer sets for plasmids construction
Plasmid |
Forward primer |
Reverse primer |
|---|---|---|
pSOCS3/2510 |
5′-ggGGTACCTCTGACTCCCTGGTTCAAGC-3′ |
5′-cgGCTAGCCTTCCTACCTGGTCCCGAAT-3′ |
pSOCS3/947 |
5′-ggGGTACCACTGTCGCACGTCTCCAAC-3′ |
5′-cgGCTAGCCTTCCTACCTGGTCCCGAAT-3′ |
Electrophoretic mobility shift assay (EMSA)
The EMSA was performed using the LightShift chemiluminescent EMSA kit (Pierce, Rockford, IL, USA), details were performed as previous description [45]. The DNA probe (SOCS3/IR9, 5′-GATCAT GGCTCAccgcagcctTGACCTCCCT-3′), containing a putative FXR response element (the underlined nucleotides), was derived from human SOCS3 promoter region and was end-labeled with biotin. The mutated probe (5′-GATCATGGTCCAccgcagcct TGGTCTCCCT-3′, the mutant bases were underlined) was also included. For competition experiments, the corresponding unlabeled oligonucleotide (cold probe) was used at 50 × excess concentrations over the labeled probe in the binding reaction.
Chromatin immunoprecipitation assay (ChIP)
ChIP assay was performed using a ChIP assay kit (Upstate Biotechnology, Lake Placid, NY, USA) as described previously [45]. Final DNA extractions were PCR amplified using the primer pairs that covered the putative FXRE/IR9 sequence in the SOCS3 promoter region (forward primer, 5′-TCTCACTCTGTTGCCCAGAC-3′; reverse primer, 5′-GTGGCCTGTGCCTGTAGTC-3′).
Xenotransplantation of HCC cells in nude mice
HepG2 cells (1.0 × 106/per mouse) in 100 μL phosphate buffered saline (PBS) were injected subcutaneously into the flank of 6-week-old male nude mice (4 mice/group). When palpable tumor were formed (about 2 weeks), GW4064 (25 mg/kg/day) or Vehicle DMSO treatment were started by intraperitoneally injected once every two day for 2 weeks. Tumor growth was monitored by caliper measurements along two orthogonal axes as described [46]. Tumor volume was calculated as V (mm3) = (length × width2)/2. Tumors were carefully excised, measured, imaged and collected for Western blot.
Immunohistochemistry (IHC) test
A total of 66 patients underwent surgery at the Department of Hepatobiliary Surgery Institute, Southwest Hospital, Third Military Medical University, China, for HCC from 2013 to 2014. Expression profile of FXR and SOCS3 and activation of STAT3 on formalin-fixed paraffin-embedded clinical samples were investigated by IHC. For immunohistochemical staining and scoring, previously described protocols were followed [47]. The immunohistochemical grade was quantified according to the proportion of stained cells into 4 bins as 0–3+ (0: no expression; 1+: weak expression; 2+: moderate expression and 3+: strong expression) (seen in Supplementary Fig. S1). For statistical analysis, as well as to reduce intraobserver variability, the immunohistochemical scores were further grouped into two categories: low (grade 0 or 1+) or high (grade 2+ or 3+). The primary antibody was replaced by rabbit IgG (isotype control) in negative control (NC) sections.
Statistical analysis
Data were analysed using SPSS 17.0 or GraphPad Prism. When two groups were compared, Student’s t test was used. When more than two groups wre compared, one-way ANOVA followed by Tukey’s Test was carried out. P value <0.05 was taken to be statistically significant.
ACKNOWLEDGMENTS AND FUNDING
We thank Dr. Yuanyin Zhao and Changjiang Hu for kindly assistant on experiment performance and data analysis.
GRANT SUPPORT
This work was supported by the National Natural Science Foundation of China (No. 81402394 and 81273226), the Scientific Funds of Third Military Medical University (2012XJQ02) and Natural Science Foundation of Chongqing, China (cstc2014jcyjA10093).
CONFLICTS OF INTEREST
The authors declare that they have no conflict of interest.
REFERENCES
1. Subramaniam A, Shanmugam MK, Perumal E, Li F, Nachiyappan A, Dai X, Swamy SN, Ahn KS, Kumar AP, Tan BK, Hui KM, Sethi G. Potential role of signal transducer and activator of transcription (STAT)3 signaling pathway in inflammation, survival, proliferation and invasion of hepatocellular carcinoma. Biochimica et biophysica acta. 2013; 1835:46–60.
2. Yu H, Lee H, Herrmann A, Buettner R, Jove R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nature Reviews Cancer. 2014; 14:736–746.
3. Xiang M, Su H, Hong Z, Yang T, Shu G. Chemical composition of total flavonoids from Polygonum amplexicaule and their pro-apoptotic effect on hepatocellular carcinoma cells: Potential roles of suppressing STAT3 signaling. Food and chemical toxicology: an international journal published for the British Industrial Biological Research Association. 2015; 80:62–71.
4. Dai X, Ahn KS, Kim C, Siveen KS, Ong TH, Shanmugam MK, Li F, Shi J, Kumar AP, Wang LZ, Goh BC, Magae J, Hui KM, Sethi G. Ascochlorin, an isoprenoid antibiotic inhibits growth and invasion of hepatocellular carcinoma by targeting STAT3 signaling cascade through the induction of PIAS3. Molecular oncology. 2015; 9:818–833.
5. Siveen KS, Sikka S, Surana R, Dai X, Zhang J, Kumar AP, Tan BK, Sethi G, Bishayee A. Targeting the STAT3 signaling pathway in cancer: role of synthetic and natural inhibitors. Biochimica et biophysica acta. 2014; 1845:136–154.
6. Zuo M, Li C, Lin J, Javle M. LLL12, a novel small inhibitor targeting STAT3 for hepatocellular carcinoma therapy. Oncotarget. 2015; 6:10940–10949.
7. Liang P, Cheng SH, Cheng CK, Lau KM, Lin SY, Chow EY, Chan NP, Ip RK, Wong RS, Ng MH. Platelet factor 4 induces cell apoptosis by inhibition of STAT3 via up-regulation of SOCS3 expression in multiple myeloma. Haematologica. 2013; 98:288–295.
8. Yu JH, Kim KH, Kim H. SOCS 3 and PPAR-gamma ligands inhibit the expression of IL-6 and TGF-beta1 by regulating JAK2/STAT3 signaling in pancreas. The international journal of biochemistry & cell biology. 2008; 40:677–688.
9. Patel K, Kollory A, Takashima A, Sarkar S, Faller DV, Ghosh SK. MicroRNA let-7 downregulates STAT3 phosphorylation in pancreatic cancer cells by increasing SOCS3 expression. Cancer letters. 2014; 347:54–64.
10. Zhang JG, Farley A, Nicholson SE, Willson TA, Zugaro LM, Simpson RJ, Moritz RL, Cary D, Richardson R, Hausmann G, Kile BJ, Kent SB, Alexander WS, Metcalf D, Hilton DJ, Nicola NA, et al. The conserved SOCS box motif in suppressors of cytokine signaling binds to elongins B and C and may couple bound proteins to proteasomal degradation. Proceedings of the National Academy of Sciences of the United States of America. 1999; 96:2071–2076.
11. Larsen L, Ropke C. Suppressors of cytokine signalling: SOCS. APMIS: acta pathologica, microbiologica, et immunologica Scandinavica. 2002; 110:833–844.
12. Niwa Y, Kanda H, Shikauchi Y, Saiura A, Matsubara K, Kitagawa T, Yamamoto J, Kubo T, Yoshikawa H. Methylation silencing of SOCS-3 promotes cell growth and migration by enhancing JAK/STAT and FAK signalings in human hepatocellular carcinoma. Oncogene. 2005; 24:6406–6417.
13. Wu WY, Li J, Wu ZS, Zhang CL, Meng XL, Lobie PE. Prognostic significance of phosphorylated signal transducer and activator of transcription 3 and suppressor of cytokine signaling 3 expression in hepatocellular carcinoma. Experimental and therapeutic medicine. 2011; 2:647–653.
14. Yang SF, Yeh YT, Wang SN, Hung SC, Chen WT, Huang CH, Chai CY. SOCS-3 is associated with vascular invasion and overall survival in hepatocellular carcinoma. Pathology. 2008; 40:558–563.
15. Wu WY, Kim H, Zhang CL, Meng XL, Wu ZS. Loss of suppressors of cytokine signaling 3 promotes aggressiveness in hepatocellular carcinoma. Journal of investigative surgery: the official journal of the Academy of Surgical Research. 2014; 27:197–204.
16. Ogata H, Chinen T, Yoshida T, Kinjyo I, Takaesu G, Shiraishi H, Iida M, Kobayashi T, Yoshimura A. Loss of SOCS3 in the liver promotes fibrosis by enhancing STAT3-mediated TGF-beta1 production. Oncogene. 2006; 25:2520–2530.
17. Ogata H, Kobayashi T, Chinen T, Takaki H, Sanada T, Minoda Y, Koga K, Takaesu G, Maehara Y, Iida M, Yoshimura A. Deletion of the SOCS3 gene in liver parenchymal cells promotes hepatitis-induced hepatocarcinogenesis. Gastroenterology. 2006; 131:179–193.
18. Maloney PR, Parks DJ, Haffner CD, Fivush AM, Chandra G, Plunket KD, Creech KL, Moore LB, Wilson JG, Lewis MC, Jones SA, Willson TM. Identification of a chemical tool for the orphan nuclear receptor FXR. Journal of medicinal chemistry. 2000; 43:2971–2974.
19. Shen H, Zhang Y, Ding H, Wang X, Chen L, Jiang H, Shen X. Farnesoid X receptor induces GLUT4 expression through FXR response element in the GLUT4 promoter. Cellular physiology and biochemistry: international journal of experimental cellular physiology, biochemistry, and pharmacology. 2008; 22:1–14.
20. Laffitte BA, Kast HR, Nguyen CM, Zavacki AM, Moore DD, Edwards PA. Identification of the DNA binding specificity and potential target genes for the farnesoid X-activated receptor. The Journal of biological chemistry. 2000; 275:10638–10647.
21. Gadaleta RM, Cariello M, Sabba C, Moschetta A. Tissue-specific actions of FXR in metabolism and cancer. Biochimica et biophysica acta. 2015; 1851:30–39.
22. Xu Z, Huang G, Gong W, Zhou P, Zhao Y, Zhang Y, Zeng Y, Gao M, Pan Z, He F. FXR ligands protect against hepatocellular inflammation via SOCS3 induction. Cellular signalling. 2012; 24:1658–1664.
23. Sitko JC, Yeh B, Kim M, Zhou H, Takaesu G, Yoshimura A, McBride WH, Jewett A, Jamieson CA, Cacalano NA. SOCS3 regulates p21 expression and cell cycle arrest in response to DNA damage. Cellular signalling. 2008; 20:2221–2230.
24. Barclay JL, Anderson ST, Waters MJ, Curlewis JD. SOCS3 as a tumor suppressor in breast cancer cells, and its regulation by PRL. International journal of cancer Journal international du cancer. 2009; 124:1756–1766.
25. Baltayiannis G, Baltayiannis N, Tsianos EV. Suppressors of cytokine signaling as tumor repressors. Silencing of SOCS3 facilitates tumor formation and growth in lung and liver. Journal of BUON: official journal of the Balkan Union of Oncology. 2008; 13:263–265.
26. Wei RC, Cao X, Gui JH, Zhou XM, Zhong D, Yan QL, Huang WD, Qian QJ, Zhao FL, Liu XY. Augmenting the antitumor effect of TRAIL by SOCS3 with double-regulated replicating oncolytic adenovirus in hepatocellular carcinoma. Human gene therapy. 2011; 22:1109–1119.
27. Iwahori K, Serada S, Fujimoto M, Nomura S, Osaki T, Lee CM, Mizuguchi H, Takahashi T, Ripley B, Okumura M, Kawase I, Kishimoto T, Naka T. Overexpression of SOCS3 exhibits preclinical antitumor activity against malignant pleural mesothelioma. International journal of cancer Journal international du cancer. 2011; 129:1005–1017.
28. Rigby RJ, Simmons JG, Greenhalgh CJ, Alexander WS, Lund PK. Suppressor of cytokine signaling 3 (SOCS3) limits damage-induced crypt hyper-proliferation and inflammation-associated tumorigenesis in the colon. Oncogene. 2007; 26:4833–4841.
29. Chao F, Gong W, Zheng Y, Li Y, Huang G, Gao M, Li J, Kuruba R, Gao X, Li S, He F. Upregulation of scavenger receptor class B type I expression by activation of FXR in hepatocyte. Atherosclerosis. 2010; 213:443–448.
30. Gautier T, de Haan W, Grober J, Ye D, Bahr MJ, Claudel T, Nijstad N, Van Berkel TJ, Havekes LM, Manns MP, Willems SM, Hogendoorn PC, Lagrost L, Kuipers F, Van Eck M, Rensen PC, et al. Farnesoid X receptor activation increases cholesteryl ester transfer protein expression in humans and transgenic mice. Journal of lipid research. 2013; 54:2195–2205.
31. Berger H, Vegran F, Chikh M, Gilardi F, Ladoire S, Bugaut H, Mignot G, Chalmin F, Bruchard M, Derangere V, Chevriaux A, Rebe C, Ryffel B, Pot C, Hichami A, Desvergne B, et al. SOCS3 transactivation by PPARgamma prevents IL-17-driven cancer growth. Cancer research. 2013; 73:3578–3590.
32. Xin XM, Zhong MX, Yang GL, Peng Y, Zhang YL, Zhu W. GW4064, a farnesoid X receptor agonist, upregulates adipokine expression in preadipocytes and HepG2 cells. World journal of gastroenterology: WJG. 2014; 20:15727–15735.
33. Fiorucci S, Rizzo G, Antonelli E, Renga B, Mencarelli A, Riccardi L, Morelli A, Pruzanski M, Pellicciari R. Cross-talk between farnesoid-X-receptor (FXR) and peroxisome proliferator-activated receptor gamma contributes to the antifibrotic activity of FXR ligands in rodent models of liver cirrhosis. The Journal of pharmacology and experimental therapeutics. 2005; 315:58–68.
34. Degirolamo C, Modica S, Vacca M, Di Tullio G, Morgano A, D’Orazio A, Kannisto K, Parini P, Moschetta A. Prevention of spontaneous hepatocarcinogenesis in farnesoid X receptor-null mice by intestinal-specific farnesoid X receptor reactivation. Hepatology. 2015; 61:161–170.
35. Li G, Kong B, Zhu Y, Zhan L, Williams JA, Tawfik O, Kassel KM, Luyendyk JP, Wang L, Guo GL. Small heterodimer partner overexpression partially protects against liver tumor development in farnesoid X receptor knockout mice. Toxicology and applied pharmacology. 2013; 272:299–305.
36. Yang F, Huang X, Yi T, Yen Y, Moore DD, Huang W. Spontaneous development of liver tumors in the absence of the bile acid receptor farnesoid X receptor. Cancer research. 2007; 67:863–867.
37. Meng Z, Wang X, Gan Y, Zhang Y, Zhou H, Ness CV, Wu J, Lou G, Yu H, He C, Xu R, Huang W. Deletion of IFNgamma enhances hepatocarcinogenesis in FXR knockout mice. Journal of hepatology. 2012; 57:1004–1012.
38. Kim I, Morimura K, Shah Y, Yang Q, Ward JM, Gonzalez FJ. Spontaneous hepatocarcinogenesis in farnesoid X receptor-null mice. Carcinogenesis. 2007; 28:940–946.
39. Zhang Y, Gong W, Dai S, Huang G, Shen X, Gao M, Xu Z, Zeng Y, He F. Downregulation of human farnesoid X receptor by miR-421 promotes proliferation and migration of hepatocellular carcinoma cells. Molecular cancer research: MCR. 2012; 10:516–522.
40. Wang YD, Chen WD, Wang M, Yu D, Forman BM, Huang W. Farnesoid X receptor antagonizes nuclear factor kappaB in hepatic inflammatory response. Hepatology. 2008; 48:1632–1643.
41. Jiang Y, Iakova P, Jin J, Sullivan E, Sharin V, Hong IH, Anakk S, Mayor A, Darlington G, Finegold M, Moore D, Timchenko NA. Farnesoid X receptor inhibits gankyrin in mouse livers and prevents development of liver cancer. Hepatology. 2013; 57:1098–1106.
42. Wolfe A, Thomas A, Edwards G, Jaseja R, Guo GL, Apte U. Increased activation of the Wnt/beta-catenin pathway in spontaneous hepatocellular carcinoma observed in farnesoid X receptor knockout mice. The Journal of pharmacology and experimental therapeutics. 2011; 338:12–21.
43. Liu X, Zhang X, Ji L, Gu J, Zhou M, Chen S. Farnesoid X receptor associates with beta-catenin and inhibits its activity in hepatocellular carcinoma. Oncotarget. 2015; 6:4226–4238.
44. Deuschle U, Schuler J, Schulz A, Schluter T, Kinzel O, Abel U, Kremoser C. FXR controls the tumor suppressor NDRG2 and FXR agonists reduce liver tumor growth and metastasis in an orthotopic mouse xenograft model. PloS one. 2012; 7:e43044.
45. He X, Xu Z, Wang B, Zheng Y, Gong W, Huang G, Zhang L, Li Y, He F. Upregulation of thrombomodulin expression by activation of farnesoid X receptor in vascular endothelial cells. European journal of pharmacology. 2013; 718:283–289.
46. Mauro L, Catalano S, Bossi G, Pellegrino M, Barone I, Morales S, Giordano C, Bartella V, Casaburi I, Ando S. Evidences that leptin up-regulates E-cadherin expression in breast cancer: effects on tumor growth and progression. Cancer research. 2007; 67:3412–3421.
47. Chen K, Li Z, Jiang P, Zhang X, Zhang Y, Jiang Y, He Y, Li X. Co-expression of CD133, CD44v6 and human tissue factor is associated with metastasis and poor prognosis in pancreatic carcinoma. Oncology reports. 2014; 32:755–763.