
INTRODUCTION
It is estimated in 2015 in the U.S. alone that 231,000 new cases of breast cancer will be diagnosed, and over 40,000 patients will die from this disease [1]. The majority of deaths caused by breast cancer are a result of metastasis [2] with bone and brain being the primary sites of tumor dissemination [3-5]. Molecular characterization of breast cancer has identified predictive gene-expression signatures in breast tumor tissues, greatly advancing our understanding of this disease [6]. Through similar expression signatures in mouse mammary tumors, Necdin (Ndn) was identified as a driver of extracellular matrix (ECM) gene expression [7], which are a class of genes that are common components of predictive expression signatures [8, 9]. Specifically, Ndn was implicated as a driver of ECM gene expression through expression quantitative trait locus (eQTL) mapping in mammary tumors derived from the transgene-positive F1 progeny of the FVB/N-Tg(MMTV-PyVT)634Mul/J (PyMT) mouse model of mammary tumorigenesis and the AKXD recombinant inbred strains of mice. Subsequent in vitro and in vivo studies demonstrated that modulation of the expression level of Ndn impacted the metastatic capacity of a highly aggressive mouse mammary tumor cell line. Most importantly, these earlier studies demonstrated that Ndn is a metastasis suppressor that exerts its influence at the germline level.
The gene encoding human NDN is located on chromosome 15q11, a region that is maternally imprinted in Prader-Willi syndrome, a neurogenetic disorder characterized by developmental delays and behavioral abnormalities [10]. Mice with mutated Ndn display neonatal lethality and behavioral phenotypes similar to those observed in Prader-Willi syndrome [11, 12]. NDN is a member of the melanoma antigen (MAGE) family which is comprised of over 60 genes that share the highly conserved MAGE homology domain (MHD) [13, 14]. Human NDN is 321 amino acids long and mouse NDN is 325 amino acids long and the two proteins overall share 82% identity. Between human and mouse NDN, the amino-terminal region is less conserved (60% identity) and higher similarity (90% identity) is observed in the functional region of NDN (amino acids 83-292) which contains the MHD [15].
Earlier studies have reported that the expression of NDN is decreased in many different cancer types in comparison to normal tissue, including breast cancer, suggesting that it acts as a tumor suppressor [16]. A gene-expression signature induced by the over-expression of Ndn was shown to predict survival in breast cancer patients [7]. While several studies implicate NDN as a possible tumor suppressor, the mechanism behind its role in metastasis is unclear. We hypothesize that NDN, a known transcription factor [17], induces metastasis-predictive gene expression by interacting with chromatin to regulate transcription, and that Ndn variants differ in their capacity to regulate the transcription of target genes. In this study using a publically available breast cancer dataset, we demonstrate that NDN harbors polymorphisms associated with patient survival. Through in vitro and in vivo methods using mouse mammary tumor cell lines stably over-expressing variants of Ndn, we show that a specific Ndn variant acts as a germline metastasis suppressor. Chromatin immunoprecipitation (ChIP) assays demonstrate that NDN achieves this effect most likely by regulating the expression of target genes, such as c-Myc, at the transcriptional level. Our work demonstrates that several polymorphisms in NDN are significantly associated with clinical outcomes in breast cancer patients, and that these polymorphisms influence the function of NDN as a transcriptional regulator and metastasis suppressor.
RESULTS
Germline variation in NDN in a well-defined human breast cancer cohort is associated with patient outcome
Since Ndn was implicated as a germline metastasis susceptibility gene through an eQTL mapping approach in tumors derived from a mouse model of mammary tumorigenesis, we hypothesized that: a) the expression of human NDN in breast cancer is influenced by similar patterns of germline variation; and b) common variants in the human NDN gene are associated with markers of disease aggressiveness and clinical outcome in breast cancer. To investigate these hypotheses, the frequencies of haplotypes in linkage disequilibrium (LD) with NDN (Supplementary Figure 1) were analyzed in 466 breast cancer patients derived from the Cancer Genome Atlas (TCGA) repository. For eQTL analysis, the frequencies of germline haplotypes in LD with NDN were correlated with NDN gene level RNA-seq expression data in all primary tumors. One haplotype of three single nucleotide polymorphisms (SNPs) (rs1722793 - rs1524843 - rs1781208 GAG), which is located approximately 17 kb downstream of NDN and has a frequency of 0.347 in the study population, was significantly associated with the tumor expression level of NDN (P = 0.001; FDR = 0.048; Figure 1A). Full results for eQTL analysis are shown in Supplementary Table S1.
In addition to demonstrating that the expression level of NDN is associated with germline variation, our analyses illustrate that haplotypes in LD with NDN are associated with disease aggressiveness and patient survival. Specifically, one haplotype 35 kb upstream and in LD with NDN (rs850815 - rs850814 AA) was associated with a more aggressive breast cancer phenotype (Table 1): first, when tumors were classified on their PAM50 status [18], the AA haplotype was more frequent in poor prognosis basal-like tumors (frequency = 0.815 vs. 0.447 for all other subtypes; P = 1.99x10-6; FDR = 1.11x10-4); and second, the AA haplotype was more frequent in estrogen receptor (ER) negative compared to ER positive tumors (frequency = 0.716 vs. 0.459 for ER+; P = 3.57x10-4; FDR = 0.020). Finally, two haplotypes in LD with NDN were associated with overall survival in this cohort: first, a two-marker haplotype 57 kb downstream of NDN (rs11632341 - rs824195 AT) was associated with a significantly better overall survival (P = 0.001; Figure 1B); and second, a two-marker haplotype 95 kb downstream of NDN (rs1717831 - rs4267267 CG) was found to be significantly associated with a poorer overall survival in breast cancer patients (P = 0.002; Figure 1C). Complete results for the association of NDN haplotypes with tumor PAM50 basal and ER phenotypes are shown in Supplementary Tables S2 and S3, respectively.
Table 1: Association of NDN haplotypes with disease-specific traits
Trait |
Haplotype Distance from Gene (bp) |
SNPs Defining Haplotype |
Haplotype |
Frequency |
t value |
P-value |
FDR |
|
PAM50 |
35,587 |
rs850815 rs850814 |
AA |
Basal |
0.815 |
4.816 |
1.99x10-6 |
1.11x10-4 |
Other |
0.447 |
|||||||
Tumor ER Status |
35,587 |
rs850815 rs850814 |
AA |
ER- |
0.716 |
-3.597 |
3.57x10-4 |
0.020 |
ER+ |
0.459 |
|||||||
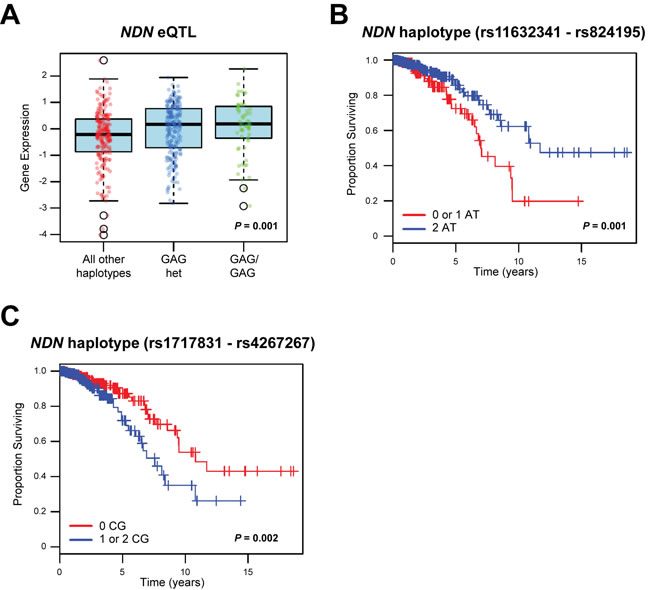
Figure 1: Haplotypes in LD with NDN are correlated with NDN expression and survival in breast cancer patients. A., haplotype analysis of 466 breast cancer patients from TCGA repository demonstrated that a three-marker haplotype (rs1722793 - rs1524843 - rs1781208 GAG) is significantly associated with the expression level of NDN. B., C., two marker haplotypes of NDN B, rs11632341 - rs824195 and C, rs1717831 - rs4267267 are significantly associated with survival (B, P = 0.001; C, P = 0.002) in this breast cancer cohort.
A non-synonymous germline variant in mouse Ndn modulates in vitro colony formation and in vivo metastasis
Having demonstrated that germline polymorphisms in human NDN are associated with patient outcome, we aimed to characterize the molecular mechanisms linking hereditary variation in this gene with metastasis. As discussed above, mouse Ndn was identified as a germline metastasis susceptibility gene using the AKXD recombinant inbred panel, which is derived from the metastasis-prone AKR/J strain and the metastasis-resistant DBA/2J strain. The DBA/2J strain contains a non-synonymous germline SNP (T50C; V17A; rs261911330) that distinguishes it from AKR/J mice. To examine the role of this SNP, we used lentiviral transduction to establish cells stably over-expressing either the wildtype Ndn 50T allele or the variant Ndn 50C allele in the highly metastatic Mvt-1 mouse mammary tumor cell line. Cells expressing Ndn at endogenous levels were used as controls. The in vitro growth properties of cell lines over-expressing Ndn variants were compared to cells stably transduced with a control lentiviral vector. Analysis of in vitro growth rates demonstrated a suppressed growth rate of cells over-expressing either the Ndn 50T allele (analysis of covariance [ANCOVA] P = 1.30x10-6) or the Ndn 50C allele (P = 3.44x10-5) compared to the control (Figure 2A). There was no significant difference in the growth rates in cells over-expressing the Ndn 50T allele and the Ndn 50C allele. However, soft agar assays performed using Mvt-1 cells stably expressing Ndn variants to examine anchorage-independent growth demonstrated a significant decrease in colony growth with the cells over-expressing the wildtype Ndn 50T allele compared to both the control (P = 0.018) and DBA/2J Ndn 50C allele (P = 2.82x10-4; Figure 2B; Supplementary Figure S2).
To investigate the in vivo effects of Ndn over-expression, these Mvt-1 cells were implanted into the mammary fat pad of 6-week old female NU/J mice, and tumor growth and pulmonary surface metastasis were quantified at 4 weeks. There was no significant difference in primary tumor burden between either of the two Ndn variants and the control. However, there was a significant increase in primary tumor burden in mice implanted with cells over-expressing the wildtype Ndn 50T allele (av. tumor burden = 0.10 g ± 0.14 g) compared to those implanted with cells over-expressing the DBA/2J Ndn 50C allele (av. tumor burden = 0.34 g ± 0.47 g; P = 0.045; Figure 2C). We observed a significant decrease in pulmonary surface metastasis with the over-expression of the wildtype Ndn 50T allele (av. pulmonary surface metastasis count = 0.7 ± 2.2; P = 0.037), and an averaged 3.5 fold increase with the over-expression of DBA/2J Ndn 50C (av. pulmonary surface metastasis count = 17.7 ± 12.0; P = 5.97x10-4) compared to that of the control (av. pulmonary surface metastasis count = 5.1 ± 8.4; Figure 2D). To confirm these results we stably over-expressed either allelic variant or a control vector in the highly metastatic 4T1 mouse mammary tumor cell line, and implanted these into the mammary fat pad of syngenic 6-week old female BALB/cJ mice. Quantification of tumor growth and pulmonary metastases was performed after 4 weeks. In 4T1 cells, there was a decrease in tumor burden with the over-expression of both the wildtype Ndn 50T allele (av. tumor burden = 1.32 g ± 0.19 g; P = 0.004) and the DBA/2J Ndn 50C allele (av. tumor burden = 1.19 g ± 0.29 g; P = 7.00x10-4) variants compared to control (av. tumor burden = 1.72 g ± 0.43 g; Figure 2E). As was the case with the Mvt-1 cell line, over-expression of wildtype Ndn 50T in 4T1 caused a significant decrease in pulmonary surface metastasis compared to control mice (av. pulmonary surface metastasis count = 4.5 ± 3.7 vs. 9.4 ± 3.9 control; P = 0.001; Figure 2F). However, with 4T1 cells there was no significant difference in pulmonary surface metastasis between controls and cells over-expressing DBA/2J Ndn 50C (av. pulmonary surface metastasis count = 8.6 ± 6.0; Figure 2F). Thus, with both cell lines, we observed a significant decrease in pulmonary metastases with the over-expression of wildtype Ndn 50T but not DBA/2J Ndn 50C compared to control (Figure 2D, 2F).
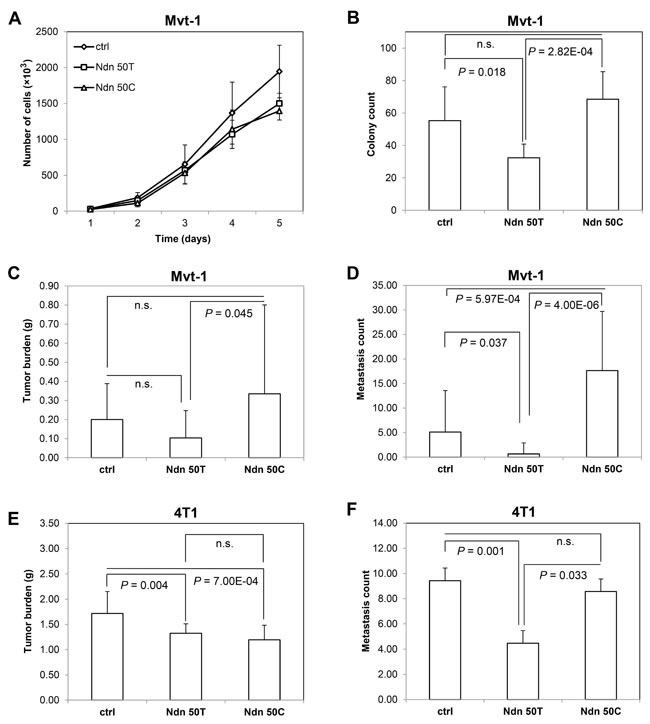
Figure 2: Ndn 50T and Ndn 50C Mvt-1 cell lines display differential growth rates in soft agar in vitro and metastases in vivo. A, cell proliferation assay. B, soft agar assay. Graphs represent the average of two independent experiments and data are represented as A, mean ± SD, and B, mean + SD. C, tumor burden and D, surface pulmonary metastases were quantified after 4 weeks of orthotopic implantation of Mvt-1 cells over-expressing Ndn 50T or Ndn 50C and control into the mammary fat pad of NU/J (n = 20). E, tumor burden and F, surface pulmonary metastases after 4 weeks of orthotopic implantation of 4T1 cells over-expressing Ndn 50T or Ndn 50C and control into the mammary fat pad of BALB/cJ (n = 30) mice. Data are represented as mean + SD.
Ndn allelic variants differentially regulate the expression of c-Myc
Previous studies have demonstrated that NDN is a multifunctional protein that can directly bind to DNA to function as a transcription factor [17] or indirectly regulate transcription by interacting with other well-known transcription factors such as E2F1 and p53 to modulate their transcriptional activities [19, 20]. Microarray analysis of Mvt-1 cells stably over-expressing wildtype Ndn 50T or DBA/2J Ndn 50C showed that the allelic variant generated a 71-gene expression signature (Figure 3A). Gene enrichment analysis using AmiGO [21] showed that one of the top biological processes in which these genes function is regulation of dendritic cell differentiation, suggesting a role in immune responses. Full gene enrichment analysis results are shown in Supplementary Table S4.
To identify specific target genes of NDN and examine the role of the Ndn T50C SNP in transcriptional regulation, we performed genome-wide ChIP-sequencing (ChIP-seq) in 4T1 cells stably over-expressing either HA-tagged wildtype Ndn 50T or DBA/2J Ndn 50C. NDN was immunoprecipitated using an antibody targeting endogenous NDN (N-20), and NDN binding peaks were called using MACS [22]. These analyses identified a total of 1039 binding peaks, of which, 339 peaks were unique to the Ndn 50T allele, 466 peaks unique to the Ndn 50C allele, and 169 peaks unique to the control (Figure 3B). A complete list of peaks identified in ChIP-seq analyses for the Ndn 50T, Ndn 50C, and control 4T1 cell lines are shown in Supplementary Tables S5, S6, and S7, respectively. For validation of ChIP-seq data, over 50 sets of primers were designed for peak regions. Of those tested through quantitative real-time PCR (qPCR), ten primer sets for ten different genes were selected for ChIP-qPCR using the NDN N-20 antibody based on the quality of amplification and dissociation curves. Overall, our ChIP-qPCR data confirm the findings of our ChIP-seq analysis (Figure 3C). To validate the specificity of the anti-NDN N-20 antibody, a second antibody recognizing the HA tag was used to immunoprecipitate ectopically expressed NDN and ChIP-qPCR analysis was performed for the same ten genes. A significant degree of overlap between the anti-NDN N-20 and anti-HA antibodies was observed in these ChIP-qPCR analyses (Figure 3D).
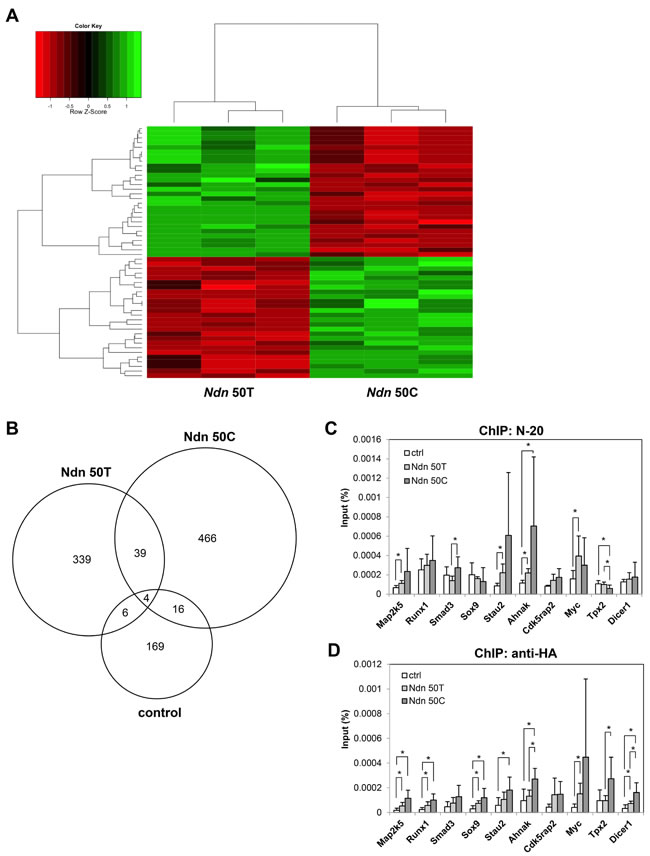
Figure 3: Ndn T50C allelic variant differentially regulates gene expression. A., microarray analysis of gene expression in Mvt-1 cells stably over-expressing either Ndn 50T or Ndn 50C. B., Venn diagram representing unique and common binding regions for NDN identified by ChIP-seq in 4T1 cells over-expressing Ndn 50T or Ndn 50C and control. C., D., verification of NDN binding regions in 4T1 cells through ChIP-qPCR in 4T1 cells over-expressing Ndn 50T or Ndn 50C and control using C., N-20 or D., anti-HA antibodies. Graphs represent the average of two independent experiments each performed in triplicate and data are represented as mean + SD. *; P ≤ 0.05.
We were particularly interested to note that our ChIP-seq data suggested that the wildtype Ndn 50T but not the DBA/2J Ndn 50C bound the coding region of the proto-oncogene c-Myc (Figure 4A), which is also known to be a metastasis suppressor [23]. Having used ChIP-qPCR to validate this differential binding of allelic variants to c-Myc in the 4T1 cell line (Figure 3C, 3D), we validated these findings in Mvt-1 cells stably over-expressing either Ndn allelic variant. As was the case in 4T1 cells, Ndn 50T, but not the Ndn 50C variant, was found to bind to c-Myc (Figure 4B). We analyzed the expression of c-Myc in various cell lines ectopically expressing either allelic variant to explore the effect of this differential binding of Ndn allelic variants. On qPCR analysis, over-expression of Ndn 50T was associated with a three-fold increase in c-Myc expression levels compared to control in both Mvt-1 and 6DT1 mammary tumor cell lines. However, over-expression of Ndn 50C did not cause a significant change in the expression level of c-Myc (Figure 4C, 4D).
Finally, having demonstrated that mouse Ndn allelic variants differentially regulate the expression of c-Myc, we examined whether germline variation in human NDN, was associated with c-MYC expression. Specifically, we characterized associations between haplotypes in LD with human NDN and the expression level of c-MYC in TCGA breast cancer cohort. One haplotype of two SNPs (rs7170719 - rs17117524) in LD with NDN was found to be associated with the level of c-MYC within primary tumors (Supplementary Figure S3).
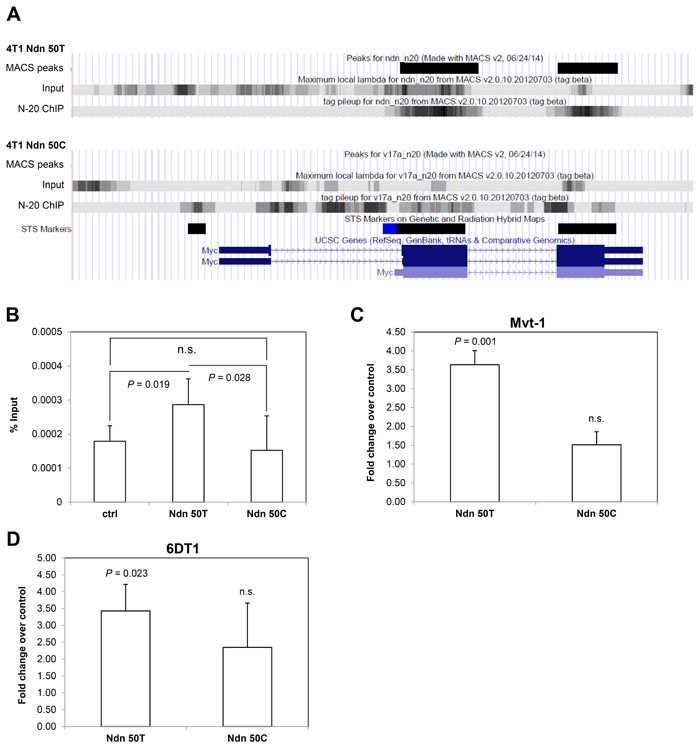
Figure 4: Ectopic expression of Ndn 50T, but not Ndn 50C, increases c-Myc expression in mouse mammary tumor cell lines. A., NDN binding peaks detected in c-Myc coding regions in ChIP-seq in 4T1 cells over-expressing Ndn 50T but not Ndn 50C. B., NDN binds to c-Myc in ChIP-qPCR in Mvt-1 cells over-expressing Ndn 50T but not Ndn 50C. C., D., expression of c-Myc detected by qPCR in three stable clonal isolates over-expressing either Ndn 50T or Ndn 50C compared to control in C., Mvt-1 cells and D., 6DT1 cells. Graphs represent the average of two independent experiments and data are represented as mean + SD. P-values shown in comparison to control.
DISCUSSION
Ndn was identified as a germline metastasis suppressor in an earlier study that sought to identify hereditary factors driving the expression of breast cancer metastasis-associated transcripts [7]. The eQTL mapping element of this previous study, which involved mapping metastasis-associated transcripts in genetically diverse PyMT mice, only indicated that Ndn was a modifier of metastasis and not the directionality of the effect. Thus, in this earlier study, the wildtype Ndn 50T was ectopically expressed in a highly aggressive mammary tumor cell line and demonstrated that Ndn suppressed metastasis. Further, it was demonstrated that a gene-expression signature induced by ectopic expression of Ndn in this cell line was associated with patient outcome in multiple breast cancer cohorts. To clarify the role of Ndn in metastasis, the aims of the current study are threefold: first, and most critically, to confirm that Ndn is a germline modifier of patient outcome in human breast cancer; second, to explore how germline variation in mouse Ndn modifies its functionality in order to more clearly understand its role in metastasis susceptibility in this model system; and third, to functionally characterize Ndn in order to define a mechanism by which it impacts metastasis. We believe that we have achieved these aims in the current work, and overall this study allows for a much clearer understanding of how Ndn acts to influence metastasis at the germline level. We have demonstrated in the small, yet well characterized TCGA human breast cancer cohort that SNPs in putative regulatory regions are not only associated with the expression of NDN in primary breast tumors, but are also associated with markers of patient outcome and survival. We have also demonstrated that germline variation in mouse Ndn is of importance with regards to metastasis, with a non-synonymous variant present in mouse strains with highly divergent metastatic capacities being strongly associated with differential tumor dissemination in both in vitro and in vivo analyses. Finally, we propose that the mechanism through which Ndn is impacting metastasis is, at least in part, through regulation of the metastasis suppressor c-Myc.
There are, however, a number of issues that warrant further explanation, the first of which is the paradoxical metastasis enhancing effect of the Ndn 50C allele, which is derived from the DBA/2J strain. This effect is paradoxical since it is well established that DBA/2J is a low metastatic capacity strain, at least when crossed with the PyMT mouse model of mammary tumorigenesis [24]. However, as noted above, the eQTL mapping approach used to identify Ndn as a metastasis modulator generally cannot be used to infer the directionality of an effect, only involvement in the phenotype of interest. The overall influence of germline variation upon metastasis in a given mouse strain (or individual human) is likely the sum of the effects of metastasis enhancing alleles minus the effects of metastasis suppressive alleles. Thus, the effect of the metastasis suppressive alleles carried by DBA/2J will outweigh the effects of metastasis enhancing alleles. Additionally, it should be noted that the identification of metastasis enhancing alleles in a metastasis suppressive strain is not unprecedented, with earlier studies demonstrating the existence of such alleles in DBA/2J [25].
The second aspect of this study that requires clarification is the equivalency of variants seen in mouse Ndn and human NDN. To be more specific, although our studies implicate the T50C coding polymorphism within mouse Ndn as being associated with metastasis, it appears that there are no coding polymorphisms in human NDN with a minor allele frequency of > 1% in any population. The most likely explanation for this observation is that the coding SNP in mouse Ndn phenocopies the regulatory variants seen in human NDN. Indeed, when one considers the wildtype mouse Ndn 50T allele in isolation, it is clear that higher levels of this have a metastasis suppressive effect (Figure 2D, 2F). Thus, variation of the expression level of mouse Ndn alone is sufficient to impact mammary tumor metastasis, at least in those strains lacking the variant 50C allele. In humans, the situation is somewhat similar, in that our eQTL analysis of TCGA breast cancer cohort demonstrates that the expression level of NDN in primary tumors is associated with germline haplotypes in LD with NDN. However, closer scrutiny of this argument reveals another limitation of this study: although we identified NDN haplotypes as being associated with the expression levels of this gene, the same haplotypes were not associated with markers of clinical outcome in the TCGA cohort. However, when one examines the results from our association and eQTL analyses, it is apparent that haplotypes significantly associated with aggressive disease clinical variables approach statistical significance in eQTL analyses, and vice versa. For example, when one considers the rs850815 - rs850814 AA haplotype associated with PAM50 basal tumors (Table 1; referred to as haplotype ID ‘X15_466.42.AA’ in Supplementary Table S2), the uncorrected P-value is statically significant for eQTL analysis for the same haplotype (P = 0.027; FDR = 0.379). This likely indicates that the current association study is underpowered, and we acknowledge the need to replicate these associations in additional breast cancer cohorts where genotype and gene expression data are available. Additionally, it is clear that haplotypes associated with different traits are all in LD with one another (Supplementary Figure 3).
In this study, we have demonstrated that over-expression of the mouse wildtype Ndn 50T allele, but not the DBA/2J 50C allele, induces increased expression of c-Myc. The outcome with the wildtype Ndn 50T allele is in accordance with previous studies demonstrating that NDN is a transcriptional regulator of c-Myc [17, 26]. c-Myc is a well-characterized proto-oncogene that promotes tumorigenesis by increasing cellular proliferation. Paradoxically, however, a prominent study by Liu and colleagues [23] demonstrated that c-MYC is in fact a metastasis suppressor, with over-expression of c-MYC suppressing both the in vitro invasiveness and in vivo metastatic capacity of human breast cancer cell lines. Surprisingly, over-expression of either Ndn allelic variant suppressed the growth rate of the Mvt-1 cell line (Figure 2A), which suggests that the increased expression of c-Myc observed with over-expression of Ndn is associated with the metastasis-suppressing properties,rather than the pro-tumorigenic properties of c-Myc. Additionally, it is plausible that Ndn influences other pathways that suppress cell growth rate, and act to counterbalance the pro-proliferative effect of c-Myc. Thus, in keeping with earlier studies [23] the decrease in metastasis observed with over-expression of Ndn 50T is similar to that observed with the over-expression of c-Myc [23]. We do, however, acknowledge that further experimentation will be required to fully understand the relationship between Ndn and c-Myc. For example, knockdown of c-Myc in conjunction with expression of the wildtype Ndn 50T could demonstrate that the metastasis suppressive effect of wildtype Ndn are solely dependent upon c-Myc. However, in our hands, knockdown of c-Myc in the Mvt-1 and 6DT1 cell lines has resulted in substantial cell death, which is presumably due to the fact that both cell lines originate from mammary tumor explant-cell cultures derived from the MMTV-c-Myc transgenic mouse model of mammary tumorigenesis [27]. Therefore, a more quantitative approach to c-Myc knockdown utilizing new technology will be required to address this issue, which we argue are beyond the scope of the current work. Thus, while we acknowledge that it is difficult to conclude that the increased expression of c-Myc is solely responsible for the inhibition of metastasis observed in cells over-expressing Ndn, it is striking that the DBA/2J Ndn 50C allele that lacks the ability to suppress metastasis also appears to lack the ability to transcriptionally regulate c-Myc.
In conclusion, this study has demonstrated that Ndn is a metastasis suppressor that acts at the germline level, and that it modulates metastasis primarily through transcriptional regulation of c-Myc. Overall, this adds to the growing body of evidence that illustrates the importance of host background on outcome in a wide variety of tumors including breast cancer [25, 28, 29], prostate cancer [30, 31], and melanoma [32]. In the era of precision medicine, it may be the case that consideration of host-specific hereditary factors will add an additional dimension of personalization of therapy, beyond that which is possible by studying the landscape of somatic mutations within a primary tumor alone. Future efforts will concentrate on identifying additional hereditary factors, which, like Ndn, play a prominent role in modulating outcome in breast cancer.
MATERIALS AND METHODS
SNP haplotype analysis
Data from 466 breast cancer patients in TCGA/BCRA cohort was used for SNP haplotype analysis. SNP Hardy-Weinberg equilibrium (HWE) P-values were estimated with PLINK. Of the 77 SNPs within 100 kb of NDN, 15 SNPs were excluded from the analysis due to being uninformative or if the HWE P-value < 10-4, and 62 were analyzed. For haplotype analysis, LD blocks were constructed using Haploview, and fastPHASE was performed to estimate haplotypes for each individual based on the LD blocks on NIH biowulf super cluster computer system (http://biowulf.nih.gov). Survival analysis was performed using the Cox model of the survival package in R [33, 34]. A generalized linear model (glm) was used to perform the association analyses between haplotypes and either NDN gene expression or clinical subtypes. Age and PC1, PC2, and PC3 were included as covariates in the glm. Correction for multiple testing was performed using the Benjamini-Hochberg FDR [35] by the MULTITEST package of R. Clinical subtypes analyzed included ER and PR status, PAM50 subtypes, P53 mutant status, PIK3CA mutation status, MAP3K1 mutation status, and MAP2K4 mutation status.
Generation of stable cell lines
Cells stably over-expressing either Ndn 50T or Ndn 50C were generated in mouse mammary tumor cell lines Mvt-1, 4T1, and 6DT1 using lentiviral transduction. For control cell lines, an empty pLenti vector was used. Cells were selected using 10 µg/mL puromycin for Mvt-1 and 6DT1, or 5 µg/mL puromycin for 4T1 cells. Clonal isolates were derived through serial dilution, and expression of Ndn 50T or Ndn 50C was confirmed by qPCR (Supplementary Figure S4).
Orthotopic mammary fat pad implantation
Six to 8-week-old female NU/J and BALB/cJ mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA). NU/J mice were injected with 106 cells of either control, Ndn 50T, or Ndn 50C Mvt-1 cells (N = 20 per group) and BALB/cJ mice were injected with 106 cells of either control, Ndn 50T, or Ndn 50C 4T1 cells (N = 30 per group) as previously described [36]. Mice were euthanized four weeks after injections, and pulmonary surface metastases were counted and tumors were weighed. All animal experiments were performed in compliance with the National Human Genome Research Institute Animal Care and Use Committee’s guidelines.
Growth curve and soft agar assay
For growth curve analysis, 2.5x104 cells for each clonal isolate were plated in 12 well plates. Cells from each clonal isolate were counted in duplicate on a daily basis for 5 days. The growth rates of each cell line were analyzed by comparing scaling parameters (i.e., slopes) between two groups (control vs. Ndn 50T, control vs. Ndn 50C, and Ndn 50T vs. Ndn 50C). In this comparison, ANCOVA was used to compare the two general linear models by testing the effect of a group on a cell count while controlling for the effect of the time (days). For soft agar assays, four clonal isolates of each of the control, Ndn 50T, or Ndn 50C Mvt-1 cells were plated in 0.33% agar in 12-well plates at 4×103 cells per well. Each isolate was plated in duplicate and allowed to grow in 37°C, 5% CO2. Cells were then stained with 0.005% crystal violet for colony counting.
RNA isolation and qRT-PCR
Total RNA was prepared from cells using RNeasy Plus Mini Kit (QIAGEN) following manufacturer’s protocol. RNA concentration was measured using NanoDrop (Wilmington, DE, USA). Reverse transcription was performed using iScript DNA Synthesis Kit (Bio-Rad, Hercules, CA, USA) and qRT-PCR reactions were performed using ABI Fast SYBR Green Master Mix (Life Technologies, Grand Island, NY, USA) as previously described [36].
Microarray and data analysis
Total RNA from Mvt-1 clonal isolates stably over-expressing Ndn 50T or Ndn 50C was extracted using RNeasy Plus Mini Kit (QIAGEN), then processed using Affymetrix Mouse Gene 1.0 ST Array (Santa Clara, CA, USA) according to the manufacturer’s protocol. Data obtained were analyzed using Partek Genomics Suite (St. Louis, MO, USA) and heatmaps were generated using R [37]. Microarray data are publicly available through Gene Expression Omnibus (GEO accession no. GSE65824).
Chromatin immunoprecipitation (ChIP)
ChIP assays were performed as previously described [38]. Briefly, after pre-clearing with Protein G Sepharose beads (GE Healthcare, Piscataway, NJ, USA), antibodies targeting endogenous NDN (N-20, Santa Cruz, Santa Cruz, CA, USA), HA (anti-HA, Roche) or IgG (12-370, Millipore, Billerica, MA, USA) was added to the sonicated cell lysates and incubated at 4 °C for 2 hours. Protein G Sepharose beads were then added and incubated overnight. The following day, immunocomplexes were washed twice with lysis buffer, once with high salt buffer (50 mM HEPES-KOH pH 7.5, 500 mM NaCl, 1 mM EDTA, 0.1% Triton X-100, 0.1% sodium deoxycholate), twice with LiCl buffer (10 mM Tris-HCl pH 8.0, 0.25 M LiCl, 0.5% NP-40, 0.5% sodium deoxycholate, 1 mM EDTA) and once with TE buffer, followed by elution in TE buffer containing 1% SDS. Samples were incubated at 65 °C overnight for reverse-crosslinking, and treated with RNAse A and proteinase K the next day. ChIP DNA was purified using QIAquick PCR Purification Kit (QIAGEN) according to manufacturer’s protocol. The DNA obtained was analyzed either by high-throughput sequencing or qPCR. All ChIP-qPCR reactions were performed in duplicate using ABI Fast SYBR Green Master Mix (Life Technologies) mentioned above.
High-throughput sequencing analysis
ChIP-seq was performed on the Illumina HiSeq2500 platform as previously described [38]. Sequence reads from the ChIP-enriched samples and corresponding input were 51 bps in length. ELAND was used to map sequences to the mouse genome (mm9), and alignments were outputted in BAM format. The software bamToBed (BedTools suite v. 2.10.1; http://code.google.com/p/bedtools), was used to convert the BAM-formatted mapping data to BED format. Reads that mapped to satellite DNA were removed using the script bed_intersect.py (bx-python 0.5.0; “-v” option). A BED file containing genomic locations of satellite DNA was obtained from mm9 RepeatMasker data available at UCSC. These filtered BED files were used as data input for the Model-based Analysis of ChIP-seq version 2 (MACS2 v. 2.0.10.20120703) software. When calculating ChIP peaks, mapping data from the ChIP input sequences were supplied to MACS2 as a control. The following parameters were used when executing MACS2 with the “macs callpeak” command: effective genome size = 1.87x109; band width = 300; model fold = [5,50]; q value cutoff = 0.05; range for calculating regional lambda: 1000 bps and 10000 bps.
ACKNOWLEDGMENTS
We would like to thank Dr. Kendra Williams for her insightful comments and suggestions. This study utilized the high-performance computational capabilities of the Biowulf Linux cluster at the National Institutes of Health, Bethesda, MD (http://biowulf.nih.gov).
FUNDING
This work was supported by the Intramural Research Program of the National Human Genome Research Institute, National Institutes of Health, USA.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
REFERENCES
1. Siegel RL, Miller KD and Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015; 65:5-29.
2. Weigelt B, Peterse JL and van ‘t Veer LJ. Breast cancer metastasis: markers and models. Nat Rev Cancer. 2005; 5:591-602.
3. Coleman RE. Clinical features of metastatic bone disease and risk of skeletal morbidity. Clin Cancer Res. 2006; 12:6243s-6249s.
4. Wong M and Pavlakis N. Optimal management of bone metastases in breast cancer patients. Breast Cancer (Dove Med Press). 2011; 3:35-60.
5. Lin NU, Bellon JR and Winer EP. CNS metastases in breast cancer. J Clin Oncol. 2004; 22:3608-3617.
6. Reis-Filho JS and Pusztai L. Gene expression profiling in breast cancer: classification, prognostication, and prediction. Lancet. 2011; 378:1812-1823.
7. Crawford NP, Walker RC, Lukes L, Officewala JS, Williams RW and Hunter KW. The Diasporin Pathway: a tumor progression-related transcriptional network that predicts breast cancer survival. Clin Exp Metastasis. 2008; 25:357-369.
8. Ramaswamy S, Ross KN, Lander ES and Golub TR. A molecular signature of metastasis in primary solid tumors. Nat Genet. 2003; 33:49-54.
9. van de Vijver MJ, He YD, van’t Veer LJ, Dai H, Hart AA, Voskuil DW, Schreiber GJ, Peterse JL, Roberts C, Marton MJ, Parrish M, Atsma D, Witteveen A, Glas A, Delahaye L, van der Velde T, et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 2002; 347:1999-2009.
10. Jay P, Rougeulle C, Massacrier A, Moncla A, Mattei MG, Malzac P, Roeckel N, Taviaux S, Lefranc JL, Cau P, Berta P, Lalande M and Muscatelli F. The human necdin gene, NDN, is maternally imprinted and located in the Prader-Willi syndrome chromosomal region. Nat Genet. 1997; 17:357-361.
11. Gerard M, Hernandez L, Wevrick R and Stewart CL. Disruption of the mouse necdin gene results in early post-natal lethality. Nat Genet. 1999; 23:199-202.
12. Muscatelli F, Abrous DN, Massacrier A, Boccaccio I, Le Moal M, Cau P and Cremer H. Disruption of the mouse Necdin gene results in hypothalamic and behavioral alterations reminiscent of the human Prader-Willi syndrome. Hum Mol Genet. 2000; 9:3101-3110.
13. Barker PA and Salehi A. The MAGE proteins: emerging roles in cell cycle progression, apoptosis, and neurogenetic disease. J Neurosci Res. 2002; 67:705-712.
14. Doyle JM, Gao J, Wang J, Yang M and Potts PR. MAGE-RING protein complexes comprise a family of E3 ubiquitin ligases. Mol Cell. 2010; 39:963-974.
15. Chapman EJ and Knowles MA. Necdin: a multi functional protein with potential tumor suppressor role? Mol Carcinog. 2009; 48:975-981.
16. De Faveri LE, Hurst CD, Platt FM, Taylor CF, Roulson JA, Sanchez-Carbayo M, Knowles MA and Chapman EJ. Putative tumour suppressor gene necdin is hypermethylated and mutated in human cancer. Br J Cancer. 2013; 108:1368-1377.
17. Matsumoto K, Taniura H, Uetsuki T and Yoshikawa K. Necdin acts as a transcriptional repressor that interacts with multiple guanosine clusters. Gene. 2001; 272:173-179.
18. Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, Davies S, Fauron C, He X, Hu Z, Quackenbush JF, Stijleman IJ, Palazzo J, Marron JS, Nobel AB, Mardis E, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol. 2009; 27:1160-1167.
19. Taniura H, Taniguchi N, Hara M and Yoshikawa K. Necdin, a postmitotic neuron-specific growth suppressor, interacts with viral transforming proteins and cellular transcription factor E2F1. J Biol Chem. 1998; 273:720-728.
20. Hasegawa K and Yoshikawa K. Necdin regulates p53 acetylation via Sirtuin1 to modulate DNA damage response in cortical neurons. J Neurosci. 2008; 28:8772-8784.
21. Carbon S, Ireland A, Mungall CJ, Shu S, Marshall B, Lewis S, Ami GOH and Web Presence Working G. AmiGO: online access to ontology and annotation data. Bioinformatics. 2009; 25:288-289.
22. Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W and Liu XS. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008; 9:R137.
23. Liu H, Radisky DC, Yang D, Xu R, Radisky ES, Bissell MJ and Bishop JM. MYC suppresses cancer metastasis by direct transcriptional silencing of alphav and beta3 integrin subunits. Nat Cell Biol. 2012; 14:567-574.
24. Lifsted T, Le Voyer T, Williams M, Muller W, Klein-Szanto A, Buetow KH and Hunter KW. Identification of inbred mouse strains harboring genetic modifiers of mammary tumor age of onset and metastatic progression. International journal of cancer Journal international du cancer. 1998; 77:640-644.
25. Winter SF, Lukes L, Walker RC, Welch DR and Hunter KW. Allelic variation and differential expression of the mSIN3A histone deacetylase complex gene Arid4b promote mammary tumor growth and metastasis. PLoS Genet. 2012; 8:e1002735.
26. Wu M, Yang C, Tao B, Bu S and Guay-Woodford LM. The ciliary protein cystin forms a regulatory complex with necdin to modulate Myc expression. PLoS One. 2013; 8:e83062.
27. Pei XF, Noble MS, Davoli MA, Rosfjord E, Tilli MT, Furth PA, Russell R, Johnson MD and Dickson RB. Explant-cell culture of primary mammary tumors from MMTV-c-Myc transgenic mice. In vitro cellular & developmental biology Animal. 2004; 40:14-21.
28. Park YG, Zhao X, Lesueur F, Lowy DR, Lancaster M, Pharoah P, Qian X and Hunter KW. Sipa1 is a candidate for underlying the metastasis efficiency modifier locus Mtes1. Nat Genet. 2005; 37:1055-1062.
29. Crawford NP, Ziogas A, Peel DJ, Hess J, Anton-Culver H and Hunter KW. Germline polymorphisms in SIPA1 are associated with metastasis and other indicators of poor prognosis in breast cancer. Breast Cancer Res. 2006; 8:R16.
30. Patel SJ, Molinolo AA, Gutkind S and Crawford NP. Germline genetic variation modulates tumor progression and metastasis in a mouse model of neuroendocrine prostate carcinoma. PLoS One. 2013; 8:e61848.
31. Williams KA, Lee M, Hu Y, Andreas J, Patel SJ, Zhang S, Chines P, Elkahloun A, Chandrasekharappa S, Gutkind JS, Molinolo AA and Crawford NP. A Systems Genetics Approach Identifies CXCL14, ITGAX, and LPCAT2 as Novel Aggressive Prostate Cancer Susceptibility Genes. PLoS Genet. 2014; 10:e1004809.
32. Ferguson B, Ram R, Handoko HY, Mukhopadhyay P, Muller HK, Soyer HP, Morahan G and Walker GJ. Melanoma susceptibility as a complex trait: genetic variation controls all stages of tumor progression. Oncogene. 2015; 34:2879-2886.
33. Therneau TM and Grambsch PM. (2000). Modeling Survival Data: Extending the Cox Model. (New York: Springer).
34. Therneau TM. (2014). A Package for Survival Analysis in S. R package version 237-7. (http://CRAN.R-project.org/package=survival)
35. Storey JD and Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci U S A. 2003; 100:9440-9445.
36. Lee M, Dworkin AM, Gildea D, Trivedi NS, Program NCS, Moorhead GB and Crawford NP. RRP1B is a metastasis modifier that regulates the expression of alternative mRNA isoforms through interactions with SRSF1. Oncogene. 2014; 33:1818-1827.
37. Warnes GR, Bolker B, Bonebakker L, Gentleman R, Huber W, Liaw A, Lumley T, Maechler M, Magnusson A, Moeller S, Schwartz M and Venables B. gplots: Various R programming tools for plotting data. R package version 2113. 2013.
38. Lee M, Dworkin AM, Lichtenberg J, Patel SJ, Trivedi NS, Gildea D, Bodine DM and Crawford NP. Metastasis-Associated Protein Ribosomal RNA Processing 1 Homolog B (RRP1B) Modulates Metastasis through Regulation of Histone Methylation. Mol Cancer Res. 2014; 12:1818-1828.