
MULTIPLE MYELOMA
Multiple Myeloma (MM) is a hematological malignancy characterized by a malignant proliferation of bone marrow (BM) post-germinal center plasma cells (PCs) and release of monoclonal protein in blood or urine. MM accounts for 1% of all neoplastic disease and represents 13% of hematologic cancers. In Western countries, its annual incidence is approximately 5.6 cases per 100.000 individuals. The median age at diagnosis is about 70 years [1, 2].
MM represents a highly biologically and clinically heterogeneous neoplasia; it shows four distinguishable clinical phases that, however, may not be discernible in each patient (Figure 1). MM may be preceded by a monoclonal gammopathy of undetermined significance (MGUS), an indolent, asymptomatic, premalignant phase characterized by a small clonal population (<10%) of PCs within the BM. MGUS may progress to MM at a rate of 1% per year. Intramedullary MM may present as an asymptomatic, smoldering, multiple myeloma (SMM), or associated with organ dysfunction including hypercalcemia, renal failure, anemia, and bone disease. SMM has an average risk of progression to MM of 10% per year [3, 4]. The final stage of MM is represented by the plasma cell leukemia (PCL), defined as at least 20% of PCs or an absolute PCs count of more than 2 × 109/L in the peripheral blood. PCL is a rapidly progressive and fatal disease which may occur as secondary (sPCL) in the context of a preexisting refractory MM, or primary (pPCL) if presenting de novo in leukemic phase [5, 6].
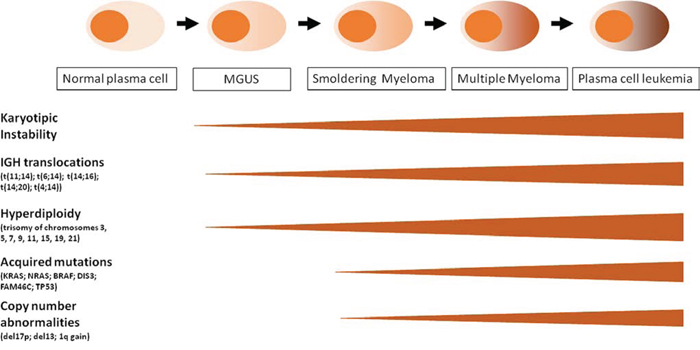
Figure 1: Schematic representation of MM progression and oncogenic events along the four clinical phases: MGUS, SMM, MM, PCL. See details in the text.
In the last decade, important advances in molecular cytogenetics and global genomic studies of myeloma cells and their normal counterparts have allowed a significant progress in understanding MM pathogenesis, providing the basis for a molecular prognostic classification and the identification of novel potential therapeutic targets. MM is characterized by a profound genomic instability that involves both ploidy and structural rearrangements. Nearly half of MM tumors are defined as hyperdiploid (HD) associated with trisomies of odd chromosomes (including 3, 5, 7, 9, 11, 15, 19, and 21). The remaining tumors are referred as non-hyperdiploid and are frequently associated with the constitutive activation of CCND1 (11q13), CCND3 (6p21), MAF (16q23), MAFB (20q11), or FGFR3/MMSET (4p16.3) genes as a result of IGH translocations. Generally, HD patients have a better prognosis [7, 8]. Recent data based on whole exome/genome sequencing indicated a heterogeneous pattern of gene mutations in MM, frequently involving member of the ERK pathway (NRAS, KRAS or BRAF) and, at a lesser extent, other genes such as DIS3 or FAM46c [8–11].
MM is associated with bone disease in more than 80% of MM patients, due to osteoclast-mediated bone destruction which causes hypercalcemia, osteoporosis, bone pain and fractures [12]. In particular, up to 70% of patients have vertebral fractures, which are associated with a high impairment of quality of life, morbidity and mortality [12]. Bone resorption is not only a relevant issue for patients quality of life, but represents also a critical step in the development of this disease, since it supports tumor growth and survival and finally contributes to the development of drug resistance [13, 14].
High incidence of bone lesions in MM patients is due to the ability of malignant PCs to alter the ratio between osteoclasts (OCLs) and osteoblasts (OBLs) in favor of the first [13, 15]. This effect is mediated by an increase of BM-associated anti-osteoblastogenic factors, such as DKK1, IL3, IL7 and TGF-β [11], or pro-osteoclastogenic factors, such as TNFα and RANKL [16, 17, 18]. Importantly, MM cells play a key role in inducing bone disease directly or indirectly, i.e. MM cells may autonomously produce RANKL [16] or stimulate the surrounding BM cells to release RANKL and other soluble factors that promote OCL differentiation [18]. OCLs directly support MM cell proliferation and survival, leading to disease progression [19].
Thus, malignant transformation in MM represents a multistep process due to accumulating genetic and epigenetic alterations of PCs as well as to their aberrant interactions with BM microenvironment.
The use of novel therapeutic agents such as immunomodulators (i.e. thalidomide and lenalidomide) and proteasome inhibitors (bortezomib), as well as the incorporation of high-dose chemotherapy followed by autologous stem cell transplantation represents the current therapy for MM patients up to 65 years old, without comorbidities and organ dysfunction [1, 20, 21]. Conventional chemotherapy (such as melphalan) combined with novel therapeutic drugs is generally administered in patients older than 65 years or unfit [22]. Recently, two different groups of new generation drugs have been developed; these include novel proteasome inhibitors (carfilzomib, ixazomib and marizomib) and drugs with novel mechanisms of action such as monoclonal antibodies, specific inhibitors of signaling pathways and kinases, deacetylase inhibitors and agents activating the unfolded protein response, especially Hsp90 inhibitors [23]. Nowadays, the median overall survival of MM patients is 7–8 years [1].
However, despite the recent remarkable improvements in the treatment of patients and the development of investigational platforms, MM remains still incurable mainly because of intrinsic or acquired drug resistance. MM cells localization in the BM milieu allows the direct interaction with non-tumor BM cells that provide several stimuli promoting neoplastic cell growth and drug resistance, and consequently patient’s relapse [24]. In addition, approximately twenty percent of patients at diagnosis are at high-risk of treatment failure defined by prognostic markers or due to presentation as PCL. The following chromosomal aberrations have been associated with an adverse outcome: i) translocation t(4;14) in 15% patients, which deranges the expression of FGFR3 (a receptor tyrosine kinase) and multiple myeloma SET domain (MMSET; a histone methyltransferase acting as a transcriptional corepressor); ii) translocations t(14;16) and t(14;20) which affects approximately 6% patients resulting in the upregulation of oncogenes c-MAF and MAFB, respectively; iii) the deletion of the short arm of chromosome 17 (del17p) observed in 10% of newly diagnosed MM patients and increasing with MM progression, which results in the loss of TP53 hampering its role in the control of cell cycle and survival; iv) the gain of the long arm of chromosome 1 which is found in approximately 40% of patients at onset and results in gene amplification of PDZK1, CKS1B and ADAR1 [7–9]. It has been shown that PDZK1 plays a role in conferring drug resistance to MM cells, whereas CSK1B promotes MM cell proliferation and drug resistance through JAK/STAT3 and MEK/ERK [25]. ADAR1 may play a role in tumor progression through its activities of RNA editing, microRNA processing and RNA-induced gene silencing, possibly resulting in malignant reprogramming [26–27].
NOTCH SIGNALING PATHWAY
A strain of Drosophila characterized by wings with irregular, “notched” margins was discovered in 1919 and lately associated to haploinsufficiency in the Notch gene [28]. In mammals, the Notch family of genes is composed by four transmembrane receptors, characterized by highly homologue sequences (Notch1–4), and two closely related families of membrane-bound ligands: the Delta-like ligands (DLL1, 3, 4) and the Serrate-like ligands (Jagged1 and 2) [29]. The interaction between receptors and ligands induces two proteolytic cleavages, the release of the cytoplasmic portion and the translocation to the nucleus of the active form of Notch (intra-cellular Notch, ICN) [29].
The Notch pathway regulates cell differentiation, apoptosis, proliferation, morphogenesis and it is essential for embryonic development of multicellular organisms [30]. Specifically, in mammals Notch is able to regulate various processes such as vasculogenesis, myogenesis, gliogenesis, neurogenesis and hematopoiesis [29]. Moreover, the Notch pathway is also involved in the homeostasis of adult tissues by regulating cell differentiation [30], promoting stem cells self-renewal [31] and determining cell fate choice in tissue development, including the commitment to T and B cell lineages [32]. The involvement of Notch signaling in the regulation of these important processes may explain the occurrence of the deregulation of Notch receptors or ligands in several types of cancer, including solid (i.e. breast cancer, melanoma, colorectal cancer, glioblastoma, pancreatic cancer) [33] and hematologic tumors (i.e. T-ALL, B-ALL, AML, B-CLL, MM) [34–37].
THE NOTCH PATHWAY: A KEY MEDIATOR OF MM PROGRESSION
Notch signaling dysregulation in MM can be ascribed to the overexpression of both receptors and ligands (Table 1). In particular, immunohistochemical analyses revealed that Notch1, Notch2 and Jagged1 are highly expressed in primary MM cells compared to low/undetectable levels in non-neoplastic counterparts [38]. Furthermore the increase of Notch1 and Jagged1 expression was reported upon disease progression from MGUS to MM [39]. NOTCH2 gene expression levels and activity were reported to be increased in the group of MM patients (approximately 6%) carrying the translocations t(14;16)(q32;q23) and t(14;20)(q32;q11) [40]. These translocations result in the activation of two transcription factors (C-MAF and MAFB respectively), responsible of NOTCH2 transcription [40]. Jagged2 deregulation seems an even more essential step in MM pathogenesis since its overexpression is an early event occurring in the benign MGUS phase [41]. The mechanisms involved in Jagged2 dysregulation are complex and include Jagged2 promoter hypomethylation [41], the aberrant expression of Skeletrophin, an Ubiquitin-ligase necessary for Jagged2 activity [42], and the loss of SMRT/NCoR2 corepressor which results in Jagged2 promoter acetylation and increased transcription [43].
Table 1: Alterations of Notch pathway in multiple myeloma
Deregulation type |
Phase |
Mechanism |
Reference |
Notch1/Jagged1 expression |
Progression from MGUS to MM |
unknown |
(39) |
Notch2 overexpression |
MM |
Transactivation by MAF genes due to t(14;16) (q32;q23) and t(14;20)(q32;q11) |
(40) |
Jagged2 overexpression |
Since MGUS |
Gene expression deregulation due to promoter hypomethylation or loss of SMRT/NCoR2 corepressor. Increased ligand ubiquitination and activity due to aberrant expression of Skeletrophin |
|
HES5 overexpression |
MM (LB subgroup) |
unknown |
(44) |
Increased copy number of Notch pathway members |
MM (HY subgroup) |
Possible mechanism: trisomies of chromosomes 3, 5, 7, 9, 11, 15, 19 and 21 |
(44) |
Finally, with regard to Notch signaling dysregulation in MM, it can be noted that “hyperdiploid” cases are associated with trisomies of different chromosomes [44] at which genes belonging to Notch pathway, such as NOTCH1 (chr.9q34.3), NOTCH3 (19p13.2–p13.1), DLL3 (19q13), DLL4 (15q14), MAML1 (5q35), and MAML2 (11q21), are located. Moreover, high levels of HES5, a direct Notch transcriptional target [45], have been reported in the subgroup of LB patients (low bone disease) [44]. Although these evidences suggest a possible increase of Notch activity in these tumors, they should be further corroborated by a thorough molecular analysis of the expression of Notch signaling members in MM patients subgroups.
The first outcome of Notch receptors and ligands dysregulation in MM is the activation of Notch signaling within tumor cell due to homotypic interaction among nearby myeloma cells or to the engagement of Notch receptors by ligands expressed on the surrounding stromal cells (see scheme in Figure 2). Several reports indicate that Notch inhibition in MM cells induces apoptosis, decreases proliferation rate [46, 47] and increases their sensitivity to pro-apoptotic compounds such as Bcl-2/Bcl-XL inhibitors [48]. Moreover, Notch blockade causes an increase of MM cells sensitivity to standard chemotherapics such as doxorubicin and melphalan both in vitro and in vivo, finally preventing the development of BM-derived drug resistance [47]. We also showed a role for Notch in the directional migration of MM cell [46] in analogy to other healthy and neoplastic cell types [49–52]. In particular, Notch signaling controls the expression and function of the CXCR4/SDF1α chemokine system, which is crucial in malignant PC growth, survival and migration [46]. Accordingly, in vivo inhibition of Notch activity in a xenograft murine model of MM results in the reduction of CXCR4 expression in MM cells and in a consequent significant decrease of MM cell localization to the BM, the primary source of SDF1α chemokine [46]. The evidence that Notch signaling dysregulation may drive MM cell localization at the BM strongly suggests that the increase of Notch signaling activity during MM progression may promote the continuous migration of MM cells from the initially infiltrated BM site to different bone districts, resulting in the formation of multiple bone lesions.
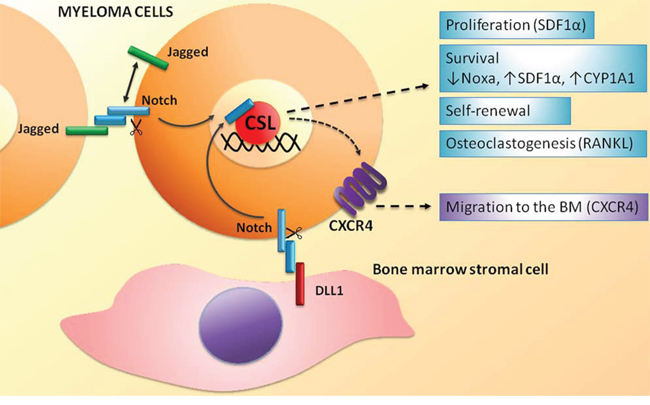
Figure 2: Homotypic and heterotypic activation of the Notch signaling in MM cells. Biological effects and molecular effectors activated by Notch signaling in MM cell. See text for details.
In the last years, another crucial role for Notch signaling has been reported in cancer stem cell self-renewal [53]. The proposed involvement of MM stem cells (MMSCs) in drug resistance, tumor dormancy and relapse has drawn the attention of the scientific community on the identification of specific MMSC markers. Unfortunately, up to now, there are not univocal MMSC markers. Indeed, it is still a matter of debate whether MMSCs originate from B cells, and therefore can be identified as CD138−CD19+CD20+ cells [53], or if they derive from mature plasma cells, which can be found in the CD138+ population [55]. Moreover it cannot be excluded that different MMSC subpopulations rise during MM progression. Even though the lack of unambiguous markers makes the study of MMSCs difficult, a role of Notch signaling in MMSCs has been described on the basis of functional assays [56, 57]. Xu and colleagues reported that MS5 stromal cell line, genetically modified to have a high constitutive expression of human DLL1, increased human and murine MM cells clonogenic growth in vitro and accelerated disease development in the 5T33MM murine model [56]. Chiron et al. reported that Jagged2 is critical for MM cell self-renewal, showing that spontaneous clonogenic growth of MM cell lines correlated with the expression of Jagged2, whereas on the other end, clonogenic and in vivo growth was impaired by Jagged2 silencing [57]. Overall these results suggest that the activation of Notch signaling pathway, mediated by the overexpression of Notch receptors or ligands, may have a key role in promoting MM progression and maintenance.
On the other side, Notch signaling in MM cells can be also activated by Notch ligands expressed on the surface membrane of surrounding BM cells (see scheme in Figure 2). Xu et al. demonstrated that BMSCs expressed DLL1 and were able to engage Notch2 in MM cells. In turn, Notch2 activation caused the upregulation of CYP1A1 (cytochrome P450, family 1, subfamily A, polypeptide 1), contributing to the development of resistance to treatment with bortezomib [58]. Interestingly, the combined treatment with a Notch-blocking agent and bortezomib leads to an increase sensitivity to bortezomib resulting in a significant improvement of the overall survival in an in vivo model of MM [58].
A second and important outcome of Notch receptors and ligands dysregulation in MM concerns the ability of MM cells to shape the BM niche (see scheme in Figure 3). Jagged2 increases the release of soluble factors including interleukin 6 (IL6), vascular endothelial growth factor (VEGF) and insulin-like growth factor 1 (IGF1) from BMSCs [41]. The released factors have a recognized promoting activity for MM. Indeed, IL6 is the major growth factor for MM cells [59], also involved in the development of resistance to dexamethasone in vitro [60]. This is further confirmed by the evidence that Siltuximab, an anti-IL6 monoclonal antibody, gives promising results alone or in combination with dexamethasone in a phase 2 clinical trial on patients with refractory MM [61]. VEGF promotes MM cell growth [62] as well as neo-angiogenesis [63], thereby directly and indirectly promoting tumor burden and progression of MM [64]. IGF1 promotes survival in MM cell and development of bortezomib resistance [65].
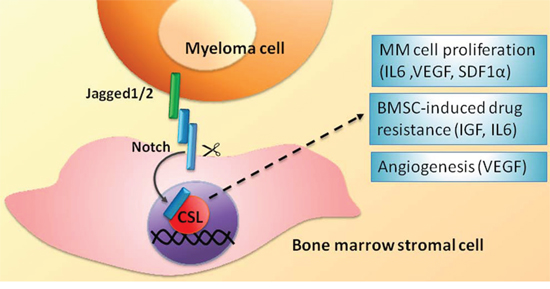
Figure 3: BMSC-mediated heterotypic activation of the Notch signaling in MM cells. Biological effects and molecular effectors involved. See text for details.
Notch signaling dysregulation in MM plays also a role in MM-associated bone disease, contributing to the unbalance between OCLs and OBLs in favor of the first (see scheme in Figure 4). We recently dissected the different effects due to Notch signaling dysregulation in MM-induced osteoclastogenesis and bone resorption. In particular we showed that: i) high Notch signaling in MM cells stimulates the release of the major osteoclastogenic soluble factor, RANKL; ii) MM cell-derived Notch ligands (Jagged1 and 2) activate Notch signaling in surrounding BMSCs, boosting the secretion of RANKL; iii) RANKL engages RANK on OCL progenitors, thereby activating the osteoclastogenic NF-kB pathway, which in turn stimulates the osteoclastogenic Notch signaling by promoting Notch2 expression; and iv) MM cell-derived Jagged ligands further boost Notch signaling in OCL progenitors by engaging Notch2. Notably, Jagged1/2 silencing is able to revert these effects [66].
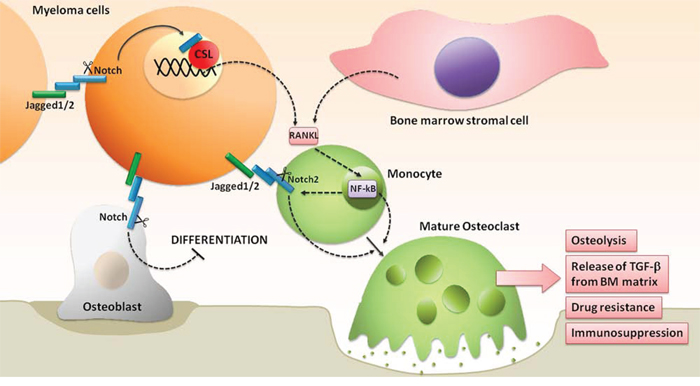
Figure 4: Notch hyperactivation drives the unbalancing of OBLs and OCLs activity, promoting the development of MM-associated bone disease. See text for details.
Notch hyperactivity in MM may also affect OBL development. Zanotti and co-workers demonstrated in transgenic mice that the selective Notch activation in OBL progenitors inhibits their differentiation [67]. This suggests that MM cell-derived Notch ligands can also be responsible for the decrease of OBLs by stimulating Notch activity that hampers OBL progenitors differentiation.
These results, confirmed in vivo by the experiments of Schwarzer and colleagues [68] with γ-secretase inhibitors (GSIs), suggest that the Notch pathway may represent a suitable target for the treatment of MM-associated bone disease. Moreover, the tight cooperation of Notch and NF-kB pathways (mediated by RANKL) in MM-induced osteoclastogenesis suggests that a combination treatment of Notch and proteasome inhibitors could be even more effective. Interestingly, this combined approach could not only affect osteolysis, but also overcome MM-associated drug resistance. Indeed, as reported by Xu and coworkers, GSI-mediated inhibition of Notch2 in MM cell increases the sensitivity to bortezomib [58]. These findings provide a rationale for a combined treatment with Notch and proteasome inhibitors that could synergistically prevent bone disease and drug resistance. Taken together the data reported above suggest that the Notch pathway may represent a rational target for MM therapy, alone or in combination with standard of care chemotherapics.
NOTCH TARGETED THERAPIES: STATE OF THE ART AND FUTURE PERSPECTIVES
As reported, Notch signaling alterations described in MM result in an increased expression of Notch pathway members including receptors and ligands [42]. These alterations cause a moderate increase of Notch signaling in comparison to Notch pathway hyperactivation occurring in other types of cancer characterized by gain-of-function Notch mutations, i.e. T-ALL [36], other lymphoid malignancies [34] and solid tumors, including breast cancer [69], ovarian cancer [70] and esophageal squamous cell carcinoma [71]. It must also be noted that if a moderate activation of Notch has a pro-tumor effect in MM cells, a stronger activation due to a mutated constitutively active Notch results in MM cell growth inhibition and apoptosis [72]. Recently, Kannan and colleagues [73] provided a possible explanation for this behavior, revealing that the transcription of the Notch target Hairy/Enhancer of Split1 (HES1), occurring upon Notch activation, may have a different outcome depending on the relative levels of HES1 and Poly ADP-Ribose Polymerase1 (PARP1). Indeed, the interaction between HES1 and PARP1 inhibits HES1 growth promoting function and induces PARP1 activation resulting in consequent apoptosis. Consistently, T-ALL cells that carry mutated hyperactive Notch1 and low levels of PARP1 display high levels of cell proliferation and do not undergo apoptosis. Concerning MM cells, we can speculate that, similarly to B-ALL cells, they cannot bear high Notch signaling activation due to the relatively high expression of PARP1 [73].
Although MM cell growth is affected not only by Notch withdrawal but also by its hyperactivation, a therapeutic approach based on Notch inhibition appears to be safer in consideration of the oncogenic activity of Notch in several cell types. A therapeutic approach directed to inhibit Notch signaling in MM relies on previously detailed in vitro and in vivo evidences of the outcomes of Notch signaling inhibition, including decreased MM cell proliferation [46, 47], stemness [56, 57], migration and BM infiltration [46], MM-associated bone disease [66–68] and an increased sensibility to pharmacological treatments [47, 48, 58].
On the other side, despite the critical role of Notch signaling in MM, the wide genetic heterogeneity might give origin to molecular MM subtypes with different sensibilities to an anti-Notch treatment.
Although a more comprehensive analysis should be addressed to define the putative MM patients who could benefit from a Notch-targeted approach by correlating the molecular characterization and the outcome of Notch withdrawal in primary cells, we can speculate that a suitable subgroup could be MM patients with MAF translocations and consequent high Notch2 levels [40]. A complete gene expression analysis of Notch pathway members could help in evaluating if other patients subgroups (i.e. LB and HY; ref. 44) could be possible candidates for a Notch tailored therapy.
Another aspect that should be considered is the possible resistance of MM cells to anti-Notch treatment. GSI-resistance, caused by mutations occurring in downstream Notch mediators or by epigenetic mechanisms, is well described in T-ALL [74, 75, 76], nonetheless up to now it has not been reported in MM cells.
A Notch-directed therapy in MM can get advantage by the recent development of several compounds targeting the different components of the Notch pathway (Table 2). The most widely used drugs inducing Notch withdrawal are the GSIs. The γ-secretase complex is responsible for the proteolytic cleavage that allows the intracellular portion of the Notch receptor to translocate to the nucleus and activate the transcription by the CSL (CBF1/RBP-J, Su(H), Lag1) nuclear factor [77]. We report the outcome of different GSIs in clinical trials for cancer therapy.
Table 2: Clinical trials of Notch pathway inhibitors
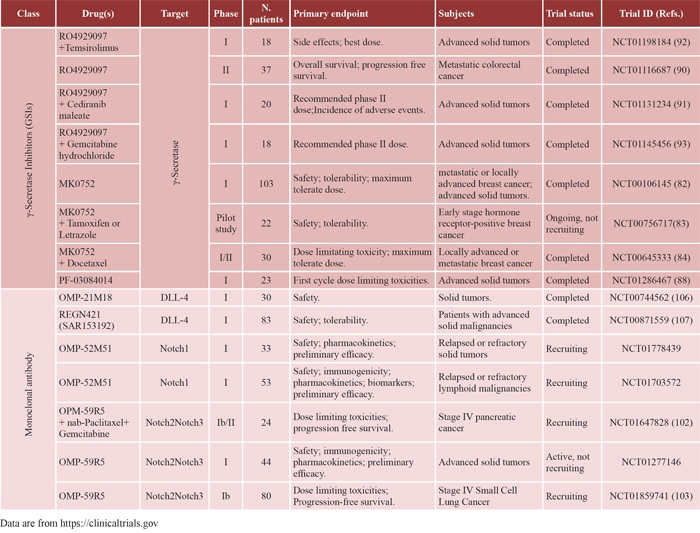
MK0752 (Merck Chemicals Ltd) is a powerful non-competitive oral GSI and represents the clinical analogues of the compound MRK003. MRK003 has cytotoxic and pro-apoptotic effects on non-Hodgkin lymphoma and MM cells in vitro and ex vivo and is able to overcome the protective effects of BMSCs [78]; it gave also promising results on in vivo xenograft models of uterine serous carcinoma and breast cancer [79, 80]. In the last years, MK0752 has been clinically tested for T-ALL treatment with poor results [81], while a trial on patients with metastatic or locally advanced breast cancer or advanced solid tumors, using intermittent MK0752 dosage, resulted in Notch pathway inhibition associated to good tolerability and clinical benefits (ref. 82; Trial ID NCT00106145).
Recently, MK0752 has been studied in combination with other chemotherapics. A combination with tamoxifen or letrozole gave promising results in the treatment of early stage hormone receptor-positive breast cancer (ref. 83; Trial ID NCT00756717), while a combination with docetaxel has been used for the treatment of patients with locally advanced or metastatic breast cancer and resulted effective in the depletion of the cancer stem cell population (ref. 84; Trial ID NCT00645333).
PF-03084014 (Pfizer Oncology) is a small non-competitive and reversible GSI. This compound has shown a significant antitumor activity in several T-ALL cell lines [85] and in breast cancer xenograft mice [86]. Moreover, PF-03084014 in combination with fludarabine displayed a synergistic effect on cell proliferation and chemotactic response of primary chronic lymphocytic leukemia cells [87]. PF-03084014 is currently tested in phase 1 trial in patients with advanced solid tumors (ref. 88; Trial ID NCT01286467).
RO4929097 (Roche) was tested in phase I study with positive results in single cases of metastatic melanoma, colorectal adenocarcinoma, epithelioid sarcoma out of 92 patients with advanced solid tumors [89]. In phase II clinical trial involving patients with metastatic colorectal cancer, RO4929097 did not change radiographic response and time to progression at the used dose and schedule (ref. 90; Trial ID NCT01116687).
RO4929097 was also tested in combination with different chemotherapics or targeting agents like cediranib (VEGF inhibitor; ref. 91; Trial ID NCT01131234), temsirolimus (a mTOR inhibitor; ref. 92; Trial ID NCT01198184) and gemcitabine (ref. 93; Trial ID NCT01145456). These combination studies, despite the presence of a high percentage (~50–70%) of patients with stable disease, resulted in the reciprocal interference of the used drugs causing the interruption of trials [94].
Independently from their efficacy, the most relevant objection for the use of GSIs is that these drugs do not exclusively target the Notch pathway. Indeed, the GSIs affect γ-secretase which regulates the functions of several substrates such as E-cadherin, N-cadherin [95] and syndecan-3, ErbB4 and CD44 [96]. Moreover, GSIs inhibit the activation of all the four Notch isoforms, thereby affecting all the physiologic functions mediated by the Notch pathway. This may explain the presence of relevant adverse events as fatigue, skin disorders, headache, hypophosphatemia and severe gastrointestinal toxicity [89]. Gut toxicity is the most serious GSI-associated side effect, due to goblet cell metaplasia in the intestinal crypts [97].
Attempts to decrease GSIs toxicity without affecting their efficacy had some successful results using an intermitting dosage schedule in combination with corticosteroids [94]. Additionally, in the last years, new generation drugs were developed for a more specific Notch directed therapy with the purpose of overcoming GSI toxicity. Indeed, recent findings encourage the use of approaches directed to selectively inhibit Notch receptors or ligands. As a matter of fact, whereas it is recognized that the inhibition of both Notch1 and Notch2 induces intestinal goblet cell metaplasia [98] resulting in a major gastrointestinal toxicity, a recent report indicates that this side effect can be avoided through the selective inhibition of Notch signaling activation mediated only by one Notch receptor or one family of Notch ligands (Jagged or DLL). Indeed, antibodies against Notch1 or Notch2 [99], or Notch decoy molecules that selectively disrupt Notch/Jagged or Notch/DLL interaction [100], did not or mildly affect intestine differentiation.
In general, compounds for Notch directed-therapy may be divided into two classes: monoclonal antibodies and molecular drugs.
Monoclonal antibodies
Monoclonal antibodies (mAbs) were recently developed against both Notch receptors and ligands. mAbs are specific for single members of the Notch family; nonetheless their delivery in solid tumors can be highly problematic, due to their high molecular weight. Thus, anti-Notch antibodies are more frequently used in studies on hematopoietic malignancies [101]. Notch targeting mAbs have entered into early phase clinical development as reported below.
OMP-59R5 (Tarextumab, OncoMed Pharmaceuticals-GlaxoSmithKline) is a fully humanized antibody that targets Notch-2 and 3 receptors [102]. It was tested in combination with nab-paclitaxel and gemcitabine in a still ongoing trial on patients with untreated metastatic pancreatic cancer and other epithelial tumors giving preliminary promising results (Trial ID NCT01647828). Phase I clinical studies are also conducted on advanced solid tumors (Trial ID NCT01277146) and untreated extensive-stage small-cell lung cancer (ref. 103; Trial ID NCT01859741).
OMP-52M51 (Anti-Notch1, OncoMed Pharmaceuticals-GlaxoSmithKline) is a novel antibody specific for Notch-1 that blocks the activation of this receptor by binding its negative regulatory region. It was shown to improve the survival of T-ALL xenograft mice [104] and it is currently under study in two Phase I clinical trials in advanced lymphoid malignancies (NCT01703572) and in solid cancers (NCT01778439). OMP-52M51 and OMP-59R5 have not been tested yet for MM treatment, although they promise to be effective on patients characterized by high levels of Notch1 and Notch2 expression.
OMP-21M18 (Demcizumab, OncoMed Pharmaceuticals), REGN421/SAR153192 (Enoticumab, Regeneron Pharmaceuticals) are humanized antibody-based drugs against the Notch ligand DLL4, which is key in vessels formation [105]. These novel drugs have been developed and successfully tested to contrast tumor-associated angiogenesis giving promising results in phase I clinical trials on advanced solid tumors, causing a reduction in tumor size with a general good tolerability (ref. 106, Trial ID NCT00744562; ref. 107, Trial ID NCT00871559).
The evidence that angiogenesis is increased in the BM of MM patients in correlation with MM cell infiltration, growth and survival [64], provides the rational for a DLL4-based anti-angiogenic therapy in this tumor; nonetheless it must be noted that trials with bevacizumab in MM [108] gave limited results and do not encourage an anti-angiogenic approach in MM.
Recent data in the literature highlight the possibility of affecting other biological features including drug resistance and bone disease by targeting Notch ligands in MM. Several evidences justify a therapeutic approach directed to Notch ligands. Indeed, BMSC-derived DLL1 promotes MM cells growth and resistance to bortezomib [56, 58], Jagged2 overexpression in MM may induce the expression of proliferative and survival factors such as IL6, VEGF and IGF1 [41]; and MM-derived Jagged1 and 2 play a crucial role in MM-induced osteolysis [66]. Currently, the relative antibodies have been generated and now are in preclinical studies [109].
Molecular drugs
In comparison to antibody-based drugs, molecular drugs are more easily deliverable. SAHM1 is a stapled peptide derived from Mastermind-like (MAML) protein, able to block the canonical Notch signaling and has a therapeutic potential in hematopoietic tumors, indeed it is able to induce apoptosis in MM and T-ALL cells [46, 110].
Recently, Kangsamaksin et al. developed new decoy peptides based on Notch1 EGF-like repeats fused to human IgGγ heavy chain (Fc). These decoys may selectively inhibit DLL4 or Jagged1 without affecting the signal mediated by other ligands. The authors showed their efficacy against melanoma, breast, lung and pancreatic cancer both in vitro and in vivo, including a significantly reduced renal, gastrointestinal and hepatic toxicity, compared to GSI treatment [100].
CONCLUSIONS
Current studies on Notch pathway in MM indicate that the dysregulated Notch receptors and ligands increase the activation of Notch signaling in MM cells promoting their progression and the ability to shape a supportive surrounding microenvironment. The contribution of aberrantly activated Notch signaling in drug resistance and development of MM associated bone disease provides a strong rational for a therapeutic approach in MM directed to control Notch activity in both malignant plasma cells and the surrounding BM stroma. In our opinion, among all the Notch-targeted therapeutic approaches available or in development, those specifically directed to Jagged1 and 2 promise to be effective and prevent the side effects associated to GSIs.
ACKNOWLEDGMENTS
RC was supported from Università degli Studi di Milano (Transition Grant); AN from Associazione Italiana Ricerca sul Cancro (Grant IG 10136); NP’s post-doctoral fellowship is sponsored by Fondazione Umberto Veronesi; MC by the post-doctoral program of the University of Milano as temporary research associate; SGar and SGal respectively by the PhD programs in Molecular and Translational Medicine and in Experimental Medicine and Medical Biotechnology (University of Milano).
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Palumbo A, Anderson K. Multiple myeloma. N Engl J Med. 2011; 364:1046–1060.
2. Rajkumar SV, Dimopoulos MA, Palumbo A, Blade J, Merlini G, Mateos MV, Kumar S, Hillengass J, Kastritis E, Richardson P, Landgren O, Paiva B, Dispenzieri A, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014; 15:e538–548.
3. International Myeloma Working Group. Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: a report of the International Myeloma Working Group. Br J Haematol. 2003; 121:749–757.
4. Kyle RA, Durie BG, Rajkumar SV, Landgren O, Blade J, Merlini G, Kröger N, Einsele H, Vesole DH, Dimopoulos M, San Miguel J, Avet-Loiseau H, Hajek R, Chen WM, et al. Monoclonal gammopathy of undetermined significance (MGUS) and smoldering (asymptomatic) multiple myeloma: IMWG consensus perspectives risk factors for progression and guidelines for monitoring and management. Leukemia. 2010; 24:1121–1127.
5. Albarracin F, Fonseca R. Plasma cell leukemia. Blood Rev. 2011; 25:107–112.
6. Fernández de Larrea C, Kyle RA, Durie BG, Ludwig H, Usmani S, Vesole DH, Hajek R, San Miguel JF, Sezer O, Sonneveld P, Kumar SK, Mahindra A, Comenzo R, et al. Plasma cell leukemia: consensus statement on diagnostic requirements, response criteria and treatment recommendations by the International Myeloma Working Group. Leukemia. 2013; 27:780–791.
7. Fonseca R1 Bergsagel, PL Drach, J Shaughnessy, J Gutierrez, N Stewart, AK Morgan, G Van Ness, B Chesi, M Minvielle, S Neri, A Barlogie, B Kuehl, WM et al, et al. International Myeloma Working Group molecular classification of multiple myeloma: spotlight review. Leukemia. 2009; 23:2210–2221.
8. Kuehl WM, Bergsagel PL. Molecular pathogenesis of multiple myeloma and its premalignant precursor. J Clin Invest. 2012; 122:3456–3463.
9. Morgan GJ, Walker BA, Davies FE. The genetic architecture of multiple myeloma. Nat Rev Cancer. 2012; 12:335–348.
10. Lohr JG, Stojanov P, Carter SL, Cruz-Gordillo P, Lawrence MS, Auclair D, Sougnez C, Knoechel B, Gould J, Saksena G, Cibulskis K, McKenna A, Chapman MA, Straussman R, Levy J, Perkins LM, Keats JJ, Schumacher SE, Rosenberg M; Multiple Myeloma Research Consortium Getz, G Golub. Widespread genetic heterogeneity in multiple myeloma: implications for targeted therapy. Cancer Cell. 2014; 25:91–101.
11. Lionetti M, Barbieri M, Todoerti K, Agnelli L, Marzorati S, Fabris S, Ciceri G, Galletti S, Milesi G, Manzoni M, Mazzoni M, Greco A, Tonon G, Musto P, Baldini L, Neri A. Molecular spectrum of BRAF, NRAS, and KRAS gene mutations in plasma cell dyscrasias: implication for MEK-ERK pathway activation. Oncotarget. 2015 Jun 10. PMID: 26090869 [Epub ahead of print].
12. Borggrefe J, Giravent S, Thomsen F, Pena J, Campbell G, Wulff A, Günther A, Heller M, Glüer CC. Association of QCT bone mineral density and bone structure with vertebral fractures in patients with multiple myeloma. J Bone Miner Res. 2015; 30:1329–1337.
13. Abe M. Targeting the interplay between myeloma cells and the bone marrow microenvironment in myeloma. Int J Hematol. 2011; 94:334–343.
14. Anderson KC, Shaughnessy JD, Barlogie B, Harousseau J-L, Roodman GD. Multiple Myeloma. Hematology Am Soc Hematol Educ Program. 2002; 2002:214–240.
15. Bataille R, Chappard D, Marcelli C, Dessauw P, Sany J, Baldet P, Alexandre C. Mechanisms of bone destruction in multiple myeloma: the importance of an unbalanced process in determining the severity of lytic bone disease. J Clin Oncol. 1989; 7:1909–1914.
16. Jurišić V M. Correlation of sera TNF-α with percentage of bone marrow plasma cells, LDH, β2-microglobulin, and clinical stage in multiple myeloma. Med Oncol. 2002; 19:133–139.
17. Lai FP, Cole-Sinclair M, Cheng WJ, Quinn JM, Gillespie MT, Sentry JW, Schneider HG. Myeloma cells can directly contribute to the pool of RANKL in bone bypassing the classic stromal and osteoblast pathway of osteoclast stimulation. Br J Haematol. 2004; 126:192–201.
18. Giuliani N, Colla S, Rizzoli V. New insight in the mechanism of osteoclast activation and formation in multiple myeloma: focus on the receptor activator of NF-kappaB ligand (RANKL). Exp Hematol. 2004; 32:685–91.
19. Yaccoby S. Advances in the understanding of myeloma bone disease and tumour growth. Br J Haematol. 2010; 149:311–21.
20. Avigan D, Rosenblatt J. Current treatment for multiple myeloma. N Engl J Med. 2014; 371:961–2.
21. Cavo M, Rajkumar SV, Palumbo A, Moreau P, Orlowski R, Bladé J, et al. International Myeloma Working Group consensus approach to the treatment of multiple myeloma patients who are candidates for autologous stem cell transplantation. Blood. 2011; 117:6063–73.
22. Palumbo A, Rajkumar SV, San Miguel JF, Larocca A, Niesvizky R, Morgan G, Landgren O, Hajek R, Einsele H, Anderson KC, Dimopoulos MA, Richardson PG, Cavo M, et al. International Myeloma Working Group consensus statement for the management, treatment, and supportive care of patients with myeloma not eligible for standard autologous stem-cell transplantation. J Clin Oncol. 2014; 32:587–600.
23. Ocio EM, Richardson PG, Rajkumar SV, Palumbo A, Mateos MV, Orlowski R, Kumar S, Usmani S, Roodman D, Niesvizky R, Einsele H, Anderson KC, Dimopoulos MA, et al. New drugs and novel mechanisms of action in multiple myeloma in 2013. a report from the International Myeloma Working Group (IMWG). Leukemia. 2014; 28:525–542.
24. Mitsiades CS, McMillin DW, Klippel S, Hideshima T, Chauhan D, Richardson PG, Munshi NC, Anderson KC, et al. The role of the bone marrow microenvironment in the pathophysiology of myeloma and its significance in the development of more effective therapies. Hematol Oncol Clin North Am., vii–viii. 2007; 21:1007–1034.
25. Abdi J, Chen G., Chang H. Drug resistance in multiple myeloma: latest findings and new concepts on molecular mechanisms. Oncotarget. 2013; 4:2186–2206.
26. Ota H, Sakurai M, Gupta R, Valente L, Wulff BE, Ariyoshi K, Iizasa H, Davuluri RV, Nishikura K. ADAR1 forms a complex with Dicer to promote microRNA processing and RNA-induced gene silencing. Cell. 2013; 153:575–589.
27. Jiang Q, Crews LA, Barrett CL, Chun HJ, Court AC, Isquith JM, Zipeto MA, Goff DJ, Minden M, Sadarangani A, Rusert JM, Dao KH, Morris SR, et al. ADAR1 promotes malignant progenitor reprogramming in chronic myeloid leukemia. Proc Natl Acad Sci U S A. 2013; 110:1041–1046.
28. Grimwade BG, Muskavitch MA, Welshons WJ, Yedvobnick B, Artavanis-Tsakonas S. The molecular genetics of the Notch locus in Drosophila melanogaster. Dev Biol. 1985; 107:503–519.
29. Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009; 137:216–233.
30. Lai EC. Notch signaling: control of cell communication and cell fate. Development. 2004; 131:965–973.
31. Bigas A, D’Altri T, Espinosa L. The Notch pathway in hematopoietic stem cells. Curr Top Microbiol Immunol. 2012; 360:1–18.
32. Pui JC, Allman D, Xu L, DeRocco S, Karnell FG, Bakkour S, Lee JY, Kadesch T, Hardy RR, Aster JC, Pear WS. Notch1 expression in early lymphopoiesis influences B versus T lineage determination. Immunity. 1999; 11:299–308.
33. Yin L, Velazquez OC, Liu ZJ. Notch signaling: emerging molecular targets for cancer therapy. Biochem Pharmacol. 2010; 80:690–701.
34. Mirandola L, Comi P, Cobos E, Kast WM, Chiriva-Internati M, Chiaramonte R. Notch-ing from T-cell to B-cell lymphoid malignancies. Cancer Lett. 2011; 308:1–13.
35. Chiaramonte R, Basile A, Tassi E, Calzavara E, Cecchinato V, Rossi V, Biondi A, Comi P. A wide role for NOTCH1 signaling in acute leukemia. Cancer Lett. 2005; 219:113–120.
36. Weng AP, Ferrando AA, Lee W, Morris JPt Silverman, LB Sanchez-Irizarry, C Blacklow, SC Look, AT Aster. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004; 306:269–271.
37. Colombo M, Mirandola L, Platonova N, Apicella L, Basile A, Figueroa AJ, Cobos E, Chiriva-Internati M, Chiaramonte R. Notch-directed microenvironment reprogramming in myeloma: a single path to multiple outcomes. Leukemia. 2013; 27:1009–1018.
38. Jundt F, Probsting KS, Anagnostopoulos I, Muehlinghaus G, Chatterjee M, Mathas S, Bargou RC, Manz R, Stein H, Dörken B. Jagged1-induced Notch signaling drives proliferation of multiple myeloma cells. Blood. 2004; 103:3511–3515.
39. Skrtic A, Korac P, Kristo DR, Ajdukovic Stojisavljevic R, Ivankovic D, Dominis M. Immunohistochemical analysis of NOTCH1 and JAGGED1 expression in multiple myeloma and monoclonal gammopathy of undetermined significance. Hum Pathol. 2010; 41:1702–10.
40. van Stralen E, van de Wetering M, Agnelli L, Neri A, Clevers HC, Bast BJ. Identification of primary MAFB target genes in multiple myeloma. Exp Hematol. 2009; 37:78–86.
41. Houde C, Li Y, Song L, Barton K, Zhang Q, Godwin J, Nand S, Toor A, Alkan S, Smadja NV, Avet-Loiseau H, Lima CS, Miele L, Coignet LJ. Overexpression of the NOTCH ligand JAG2 in malignant plasma cells from multiple myeloma patients and cell lines. Blood. 2004; 104:3697–3704.
42. Takeuchi T, Adachi Y, Ohtsuki Y. Skeletrophin, a novel ubiquitin ligase to the intracellular region of Jagged-2, is aberrantly expressed in multiple myeloma. Am J Pathol. 2005; 166:1817–1826.
43. Ghoshal P1 Nganga, AJ Moran-Giuati, J Szafranek, A Johnson, TR Bigelow, AJ Houde, CM Avet-Loiseau, H Smiraglia, DJ Ersing, N Chanan-Khan, AA Coignet. Loss of the SMRT/NCoR2 corepressor correlates with JAG2 overexpression in multiple myeloma. Cancer Res. 2009; 69:4380–4387.
44. Zhan F, Huang Y, Colla S, Stewart JP, Hanamura I, Gupta S, Epstein J, Yaccoby S, Sawyer J, Burington B, Anaissie E, Hollmig K, Pineda-Roman M, et al. The molecular classification of multiple myeloma. Blood. 2006; 108:2020–2028.
45. Varnum-Finney B1 Dallas, MH Kato, K Bernstein. Notch target Hes5 ensures appropriate Notch induced T-versus B-cell choices in the thymus. Blood. 2008; 111:2615–2620.
46. Mirandola L, Apicella L, Colombo M, Yu Y, Berta DG, Platonova N, Lazzari E, Lancellotti M, Bulfamante G, Cobos E, Chiriva-Internati M, Chiaramonte R. Anti-Notch treatment prevents multiple myeloma cells localization to the bone marrow via the chemokine system CXCR4/SDF-1. Leukemia. 2013; 27:1558–1566.
47. Nefedova Y, Sullivan DM, Bolick SC, Dalton WS, Gabrilovich DI, Nefedova Y. Inhibition of Notch signaling induces apoptosis of myeloma cells and enhances sensitivity to chemotherapy. Blood. 2008; 111:2220–2229.
48. Li M, Chen F, Clifton N, Sullivan DM, Dalton WS, Gabrilovich DI, Nefedova Y. Combined inhibition of Notch signaling and Bcl-2/Bcl-xL results in synergistic anti-myeloma effect. Mol Cancer Ther. 2010; 9:3200–9.
49. Mirandola L, Chiriva-Internati M, Montagna D, Locatelli F, Zecca M, Ranzani M, Basile A, Locati M, Cobos E, Kast WM, Asselta R, Paraboschi EM, Comi P, Chiaramonte R. Notch1 regulates chemotaxis and proliferation by controlling the chemokine receptors 5 and 9 in T-cell acute lymphoblastic leukemia. J Pathol. 2012; 226:713–722.
50. Buonamici S, Trimarchi T, Ruocco MG, Reavie L, Cathelin S, Mar BG, Klinakis A, Lukyanov Y, Tseng JC, Sen F, Gehrie E, Li M, Newcomb E, et al. CCR7 signalling as an essential regulator of CNS infiltration in T-cell leukaemia. Nature. 2009; 459:1000–1004.
51. Hoshino N1 Katayama, N Shibasaki, T Ohishi, K Nishioka, J Masuya, M Miyahara, Y Hayashida, M Shimomura, D Kato, T Nakatani, K Nishii, K Kuribayashi, K Nobori, T Shiku. A novel role for Notch ligand Delta-1 as a regulator of human Langerhans cell development from blood monocytes. J Leukoc Biol. 2005; 78:921–929.
52. Maerki S, Ceredig R, Rolink A. Induction of chemokine receptor expression during early stages of T cell development. Immunol Lett. 2006; 104:110–107.
53. Takebe N, Miele L, Harris PJ, Jeong W, Bando H, Kahn M, Yang SX, Ivy SP. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nat Rev Clin Oncol. 2015; 12:445–464.
54. Matsui W, Wang Q, Barber JP, Brennan S, Smith BD, Borrello I, McNiece I, Lin L, Ambinder RF, Peacock C, Watkins DN, Huff CA, Jones RJ. Clonogenic multiple myeloma progenitors, stem cell properties, and drug resistance. Cancer Res. 2008; 68:190–197.
55. Yata K, Yaccoby S. The SCID-rab model: a novel in vivo system for primary human myeloma demonstrating growth of CD138-expressing malignant cells. Leukemia. 2004; 18:1891–1897.
56. Xu D, Hu J, Xu S, De Bruyne E, Menu E, Van Camp B, Vanderkerken K, Van Valckenborgh E. Dll1/Notch activation accelerates multiple myeloma disease development by promoting CD138+ MM-cell proliferation. Leukemia. 2012; 26:1402–1405.
57. Chiron D, Maiga S, Descamps G, Moreau P, Le Gouill S, Marionneau S, Ouiller T, Moreaux J, Klein B, Bataille R, Amiot M, Pellat-Deceunynck C. Critical role of the NOTCH ligand JAG2 in self-renewal of myeloma cells. Blood Cells Mol Dis. 2012; 48:247–253.
58. Xu D, Hu J, De Bruyne E, Menu E, Schots R, Vanderkerken K, Van Valckenborgh E. Dll1/Notch activation contributes to bortezomib resistance by upregulating CYP1A1 in multiple myeloma. Biochem Biophys Res Commun. 2012; 428:518–524.
59. Anderson KC, Carrasco RD. Pathogenesis of myeloma. Annu Rev Pathol. 2011; 6:249–274.
60. Liu T, Fei Z, Gangavarapu KJ, Agbenowu S, Bhushan A, Lai JC, Daniels CK, Cao S. Interleukin-6 and JAK2/STAT3 signaling mediate the reversion of dexamethasone resistance after dexamethasone withdrawal in 7TD1 multiple myeloma cells. Leuk Res. 2013; 37:1322–1328.
61. Voorhees PM, Manges RF, Sonneveld P, Jagannath S, Somlo G, Krishnan A, Lentzsch S, Frank RC, Zweegman S, Wijermans PW, Orlowski RZ, Kranenburg B, Hall B, et al. A phase 2 multicentre study of siltuximab, an anti-interleukin-6 monoclonal antibody, in patients with relapsed or refractory multiple myeloma. Br J Haematol. 2013; 161:357–366.
62. Podar K, Tai YT, Davies FE, Lentzsch S, Sattler M, Hideshima T, Lin BK, Gupta D, Shima Y, Chauhan D, Mitsiades C, Raje N, Richardson P, Anderson KC. Vascular endothelial growth factor triggers signaling cascades mediating multiple myeloma cell growth and migration. Blood. 2001; 98:428–435.
63. Liu Z, Fan F, Wang A, Zheng S, Lu Y. Dll4-Notch signaling in regulation of tumor angiogenesis. J Cancer Res Clin Oncol. 2014; 140:525–536.
64. Giuliani N, Storti P, Bolzoni M, Palma BD, Bonomini S. Angiogenesis and Multiple Myeloma. Cancer Microenviron. 2011; 4:325–337.
65. Kuhn DJ, Berkova Z, Jones RJ, Woessner R, Bjorklund CC, Ma W, Davis RE, Lin P, Wang H, Madden TL, Wei C, Baladandayuthapani V, Wang M, et al. Targeting the insulin-like growth factor-1 receptor to overcome bortezomib resistance in preclinical models of multiple myeloma. Blood. 2012; 120:3260–3270.
66. Colombo M, Thümmler K, Mirandola L, Garavelli S, Todoerti K, Apicella L, Lazzari E, Lancellotti M, Platonova N, Akbar M, Chiriva-Internati M, Soutar R, Neri A, Goodyear CS, Chiaramonte R. Notch signaling drives multiple myeloma induced osteoclastogenesis. Oncotarget. 2014; 5:10393–10406.
67. Zanotti S, Smerdel-Ramoya A, Stadmeyer L, Durant D, Radtke F, Canalis E. Notch inhibits osteoblast differentiation and causes osteopenia. Endocrinology. 2008; 149:3890–3899.
68. Schwarzer R, Nickel N, Godau J, Willie BM, Duda GN, Cirovic B, Leutz A, Manz R, Bogen B, Dörken B, Jundt F. Notch pathway inhibition controls myeloma bone disease in the murine MOPC315.BM model. Blood Cancer J. 2014; 4:e217.
69. Wang K, Zhang Q, Li D, Ching KA, Zhang CC, Zheng X, Ozeck M, Shi S, Li X, Wang H, Rejto P, Christensen J, Olson P. PEST Domain Mutations in Notch Receptors Comprise an Oncogenic Driver Segment in Triple Negative Breast Cancer Sensitive to a gamma-Secretase Inhibitor. Clin Cancer Res. 2015; 2:1487–1496.
70. Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011; 474:609–615.
71. Gao YB, Chen ZL, Li JG, Hu XD, Shi XJ, Sun ZM, Zhang F, Zhao ZR, Li ZT, Liu ZY, Zhao YD, Sun J, Zhou CC, Yao R, et al. Genetic landscape of esophageal squamous cell carcinoma. Nat Genet. 2014; 46:1097–1102.
72. Zweidler-McKay PA, He Y, Xu L, Rodriguez CG, Karnell FG, Carpenter AC, Aster JC, Allman D, Pear WS. Notch signaling is a potent inducer of growth arrest and apoptosis in a wide range of B-cell malignancies. Blood. 2005; 106:3898–3906.
73. Kannan S, Fang W, Song G, Mullighan CG, Hammitt R, McMurray J, Zweidler-McKay PA. Notch/HES1-mediated PARP1 activation: a cell type-specific mechanism for tumor suppression. Blood. 2011; 117:2891–2900.
74. Hales EC, Taub JW, Matherly LH. New insights into Notch1 regulation of the PI3K-AKT-mTOR1 signaling axis: targeted therapy of γ-secretase inhibitor resistant T-cell acute lymphoblastic leukemia. Cell Signal. 2014; 26:149–161.
75. O’Neil J, Grim J, Strack P, Rao S, Tibbitts D, Winter C, Hardwick J, Welcker M, Meijerink JP, Pieters R, Draetta G, Sears R, Clurman BE, Look AT. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to gamma-secretase inhibitors. J Exp Med. 2007; 204:1813–1824.
76. Knoechel B, Roderick JE, Williamson KE, Zhu J, Lohr JG, Cotton MJ, Gillespie SM3 Fernandez D5, Ku M6 Wang H7, Piccioni F8 Silver SJ8, Jain M, et al. An epigenetic mechanism of resistance to targeted therapy in T cell acute lymphoblastic leukemia. Nat Genet. 2014; 46:364–370.
77. Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999; 284:770–776.
78. Ramakrishnan V, Ansell S, Haug J, Grote D, Kimlinger T, Stenson M, Timm M, Wellik L, Halling T, Rajkumar SV, Kumar S. MRK003, a gamma-secretase inhibitor exhibits promising in vitro pre-clinical activity in multiple myeloma and non-Hodgkin’s lymphoma. Leukemia. 2012; 26:340–348.
79. Kondratyev M, Kreso A, Hallett RM, Girgis-Gabardo A, Barcelon ME, Ilieva D, Ware C, Majumder PK, Hassell JA. Gamma-secretase inhibitors target tumor-initiating cells in a mouse model of ERBB2 breast cancer. Oncogene. 2012; 31:93–103.
80. Groeneweg JW, Hall TR, Zhang L, Kim M, Byron VF, Tambouret R, Sathayanrayanan S, Foster R, Rueda BR, Growdon WB. Inhibition of gamma-secretase activity impedes uterine serous carcinoma growth in a human xenograft model. Gynecol Oncol. 2014; 133:607–615.
81. Deangelo D, Stone R, Silverman L, Stock W, Attar E, Fearen I, Dallob A, Matthews C, Stone J, Freedman SJ, Aster J. A phase I clinical trial of the notch inhibitor MK-0752 in patients with T-cell acute lymphoblastic leukemia/lymphoma (T-ALL) and other leukemias. J Clin Oncol. 2006; 2:4. (18S).
82. Krop I, Demuth T, Guthrie T, Wen PY, Mason WP, Chinnaiyan P, Butowski N, Groves MD, Kesari S, Freedman SJ, Blackman S, Watters J, Loboda A, et al. Phase I pharmacologic and pharmacodynamic study of the gamma secretase (Notch) inhibitor MK-0752 in adult patients with advanced solid tumors. J Clin Oncol. 2012; 30:2307–2313.
83. Albain K CC, Zlobin A, Covington K, Rajan P, Godellas C, Bova D, Lo SS, Robinson P, Sarker S, Gaynor ER, Cooper R, Aranha G, Czaplicki K, et al. Modulation of cancer and stem cell biomarkers by the Notch Inhibitor MK-0752 added to endocrine therapy for early stage ER+ breast cancer. Cancer Res. 2011; S1–S5.
84. Schott AF, Landis MD, Dontu G, Griffith KA, Layman RM, Krop I, Paskett LA, Wong H, Dobrolecki LE, Lewis MT, Froehlich AM, Paranilam J, Hayes DF, Wicha MS, Chang JC. Preclinical and Clinical Studies of Gamma Secretase Inhibitors with Docetaxel on Human Breast Tumors. Clinical Cancer Research. 2013; 19:1512–1524.
85. Samon JB, Castillo-Martin M, Hadler M, Ambesi-Impiobato A, Paietta E, Racevskis J, Wiernik PH, Rowe JM, Jakubczak J, Randolph S, Cordon-Cardo C, Ferrando AA. Preclinical analysis of the gamma-secretase inhibitor PF-03084014 in combination with glucocorticoids in T-cell acute lymphoblastic leukemia. Mol Cancer Ther. 2012; 11:1565–1575.
86. Zhang CC, Pavlicek A, Zhang Q, Lira ME, Painter CL, Yan Z, Zheng X, Lee NV, Ozeck M, Qiu M, Zong Q, Lappin PB, Wong A, Rejto PA, Smeal T, Christensen JG. Biomarker and pharmacologic evaluation of the gamma-secretase inhibitor PF-03084014 in breast cancer models. Clin Cancer Res. 2012; 18:5008–5019.
87. Lopez-Guerra M, Xargay-Torrent S, Rosich L, Montraveta A, Roldan J, Matas-Cespedes A, Villamor N2, Aymerich M2, López-Otín C3, Pérez-Galán P1, Roué G1, Campo E2, Colomer D4. The [gamma]-secretase inhibitor PF-03084014 combined with fludarabine antagonizes migration, invasion and angiogenesis in NOTCH1-mutated CLL cells. Leukemia. 2015; 29:96–106.
88. Messersmith WA, Shapiro GI, Cleary JM, Jimeno A, Dasari A, Huang B, Shaik MN, Cesari R, Zheng X, Reynolds JM, English PA, McLachlan KR, Kern KA, LoRusso PM. A Phase, I, Dose-finding Study in Patients With Advanced Solid Malignancies of the Oral Gamma-Secretase Inhibitor PF-03084014. Clin Cancer Res. 2015; 21:60–67.
89. Tolcher AW, Messersmith WA, Mikulski SM, Papadopoulos KP, Kwak EL, Gibbon DG, Patnaik A, Falchook GS, Dasari A, Shapiro GI, Boylan JF, Xu ZX, Wang K, et al. Phase I study of RO4929097, a gamma secretase inhibitor of Notch signaling, in patients with refractory metastatic or locally advanced solid tumors. J Clin Oncol. 2012; 30:2348–2353.
90. Strosberg JR1, Yeatman T, Weber J, Coppola D, Schell MJ, Han G, Almhanna K, Kim R, Valone T, Jump H, Sullivan D. A phase II study of RO4929097 in metastatic colorectal cancer. Eur J Cancer. 2012; 48:997–1003.
91. Sahebjam S, Bedard PL, Castonguay V, Chen Z, Reedijk M, Liu G, Cohen B, Zhang WJ, Clarke B, Zhang T, Kamel-Reid S, Chen H, Ivy SP, et al. A phase I study of the combination of ro4929097 and cediranib in patients with advanced solid tumours (PJC-004/NCI 8503). Br J Cancer. 2013; 109:943–949.
92. Diaz-Padilla I, Hirte H, Oza AM, Clarke BA, Cohen B, Reedjik M, Zhang T, Kamel-Reid S, Ivy SP, Hotte SJ, Razak AA, Chen EX, Brana I, et al. A phase Ib combination study of RO4929097, a gamma-secretase inhibitor, and temsirolimus in patients with advanced solid tumors. Invest New Drugs. 2013; 31:1182–1191.
93. Richter S, Bedard PL, Chen EX, Clarke BA, Tran B, Hotte SJ, Stathis A, Hirte HW, Razak AR, Reedijk M, Chen Z, Cohen B, Zhang WJ, et al. A phase I study of the oral gamma secretase inhibitor R04929097 in combination with gemcitabine in patients with advanced solid tumors (PHL-078/CTEP 8575). Invest New Drugs. 2014; 32:243–249.
94. Takebe N, Nguyen D, Yang SX. Targeting Notch signaling pathway in cancer: Clinical development advances and challenges. Pharmacol Ther. 2014; 141:140–149.
95. Maetzel D, Denzel S, Mack B, Canis M, Went P, Benk M, Kieu C, Papior P, Baeuerle PA, Munz M, Gires O. Nuclear signalling by tumour-associated antigen EpCAM. Nat Cell Biol. 2009; 11:162–171.
96. Kopan R, Ilagan MXG. Gamma-secretase: proteasome of the membrane?. Nat Rev Mol Cell Biol. 2004; 5:499–504.
97. Fre S, Huyghe M, Mourikis P, Robine S, Louvard D, Artavanis-Tsakonas S. Notch signals control the fate of immature progenitor cells in the intestine. Nature. 2005; 435:964–968.
98. Riccio O, van Gijn ME, Bezdek AC, Pellegrinet L, van Es JH, Zimber-Strobl U, Strobl LJ, Honjo T, Clevers H, Radtke F. Loss of intestinal crypt progenitor cells owing to inactivation of both Notch1 and Notch2 is accompanied by derepression of CDK inhibitors p27Kip1 and p57Kip2. EMBO Rep. 2008; 9:377–383.
99. Wu Y, Cain-Hom C, Choy L, Hagenbeek TJ, de Leon GP, Chen Y, Finkle D. Therapeutic antibody targeting of individual Notch receptors. Nature. 2010; 464:1052–1057.
100. Kangsamaksin T, Murtomaki A, Kofler NM, Cuervo H, Chaudhri RA, Tattersall IW, Rosenstiel PE, Shawber CJ, Kitajewski J. NOTCH Decoys That Selectively Block DLL/NOTCH or JAG/NOTCH Disrupt Angiogenesis by Unique Mechanisms to Inhibit Tumor Growth. Cancer Discov. 2015; 5:182–197.
101. Purow B. Notch inhibition as a promising new approach to cancer therapy. Adv Exp Med Biol. 2012; 727:305–319.
102. Yen W-C, Fischer MM, Axelrod F, Bond C, Cain J, Cancilla B, Henner WR, Meisner R, Sato A, Shah J, Tang T, Wallace B, Wang M, et al. Targeting Notch Signaling with a Notch2/Notch3 Antagonist (Tarextumab) Inhibits Tumor Growth and Decreases Tumor-Initiating Cell Frequency. Clin Cancer Res; 21:2084–2095.
103. Spigel DR, Spira AI, Jotte RM, Gadgeel SM, Mita AC, Hart LL, Kapoun A, Xu L, Hill D, Zhou L, Dupont J, Pietanza MC. Phase 1b of anticancer stem cell antibody OMP-59R5 (anti-Notch2/3) in combination with etoposide and cisplatin (EP) in patients (pts) with untreated extensive-stage small-cell lung cancer (ED-SCLC). J Clin Oncol. 2014; 32:5s. (suppl; abstr 7601).
104. Agnusdei V, Minuzzo S, Frasson C, Grassi A, Axelrod F, Satyal S, Gurney A, Hoey T, Seganfreddo E, Basso G, Valtorta S, Moresco RM, Amadori A, Indraccolo S. Therapeutic antibody targeting of Notch1 in T-acute lymphoblastic leukemia xenografts. Leukemia. 2014; 28:278–288.
105. Li JL, Sainson RC, Shi W, Leek R, Harrington LS, Preusser M, Biswas S, Turley H, Heikamp E, Hainfellner JA, Harris AL. Delta-like 4 Notch ligand regulates tumor angiogenesis, improves tumor vascular function, and promotes tumor growth in vivo. Cancer Res. 2007; 67:11244–11253.
106. Smith DC, Eisenberg PD, Manikhas G, Chugh R, Gubens MA, Stagg RJ, Kapoun AM, Xu L, Dupont J, Sikic B. A Phase I Dose Escalation and Expansion Study of the Anticancer Stem Cell Agent Demcizumab (Anti-DLL4) in Patients with Previously Treated Solid Tumors. Clin Cancer Res. 2014; 20:6295–6303.
107. Jimeno A LP, Strother RM, Robinson Diamond J, Plato L, Younger A, Messersmith WA, Kittaneh M, Sawyer D, Adriaens L, Liu L, Kao RJ, DiCioccio AT, Brownstein CM, Lowy I, Trail P, Chiorean EG. Phase I study of REGN421 (R)/SAR153192, a fully-human delta-like ligand 4 (Dll4) monoclonal antibody (mAb), in patients with advanced solid tumors. J Clin Oncol 31. 2013. (suppl; abstr 2502).
108. White D, Kassim A, Bhaskar B, Yi J, Wamstad K, Paton VE. Results from AMBER, a randomized phase 2 study of bevacizumab and bortezomib versus bortezomib in relapsed or refractory multiple myeloma. Cancer. 2013; 119:339–347.
109. Ersvaer E, Hatfield KJ, Reikvam H, Bruserud O. Future perspectives: therapeutic targeting of notch signalling may become a strategy in patients receiving stem cell transplantation for hematologic malignancies. Bone Marrow Res. 2011; 2011:570796.
110. Moellering RE, Cornejo M, Davis TN, Del Bianco C, Aster JC, Blacklow SC, Kung AL, Gilliland DG, Verdine GL, Bradner JE. Direct inhibition of the NOTCH transcription factor complex. Nature. 2009; 462:182–188.