
Introduction
The H19 gene, located in human in 11p15.5 locus, is submitted to genomic imprinting. It is expressed only from the maternal allele [1]. It is transcribed by the RNA polymerase II and the transcript is spliced, polyadenylated, capped and exported into the cytosol. However, no protein associated to this transcript has been discovered and Brannan et al. proposed that H19 RNA functions as a riboregulator [2]. H19 is highly expressed in the extraembryonic tissues (placenta), the embryo proper, and the fetal tissues. After birth, its expression is repressed even if a basal expression subsists in several tissues including mammary gland, adrenal gland and uterus [3-6]
The role of H19 in cancer is still matter of debate. It has been proposed that H19 acts as a tumor suppressor in Wilm’s tumors, embryonic rhabdomyosarcoma, and the Beckwith-Wiedemann syndrome [7]. In addition, using in vivo mice models of tumorigenesis, a role of tumor suppressor gene has been ascribed to H19 [8]. However, numerous studies have shown that H19 is an oncogene in many types of cancers. Indeed, H19 overexpression is often correlated with poor prognosis in bladder, lung, oesophageal and gastric cancers [9-13]. H19 exerts its oncogenic activity through different mechanisms. For example, it has been reported that H19 functions as a Myc-up-regulated gene to potentiate the tumorigenic phenotype of breast and lung cancer cells [14]. More recently, H19 was described to act as a molecular sponge to regulate the let-7 family of miRNAs [15]. In addition, H19 is also a precursor for microRNA-675 (miR-675) and generates two mature miRNAs, miR-675-5p (miR-675) and miR-675-3p (miR-675*) [16].
MicroRNAs (miRNAs) are 19- to 25-nucleotide regulatory non-coding RNAs that are initially expressed as hairpin transcripts of primary miRNA under the control of RNA polymerase II. These primary miRNA hairpins are cleaved by two enzymes, Drosha and Dicer, to generate mature miRNAs. Although several mechanisms of gene expression regulation by miRNAs have been demonstrated [17], they mainly repress gene expression at the post-transciptional level by interacting with 3’UTR of target mRNA.
Recent data indicate that H19-derived miR-675 favours tumor progression by repressing the expression of several target genes, including Rb in colorectal cancer [12], Twist1 in hepatocellular carcinoma [18], and RUNX1 in gastric cancer [19].
We have previously shown that H19 is overexpressed in 70% of breast cancer [3]. H19 gene overexpression in mammary epithelial cells promotes tumorigenesis by upregulating thioredoxin, a modulator of signal transduction and potentiator of tumorigenesis [20]. H19 gene is up-regulated by growth factors such as HGF and by transcription factors such as E2F1 to enhance cell invasion and cell cycle progression [21, 22]. Altogether theses finding are in favor of a role of H19 as an oncogene in breast cancer [23].
In this study, we have examined the role of H19-derived miR-675 in controlling the properties of breast cancer cells. Using in silico prediction and functional assays, we identified c-Cbl and Cbl-b as direct targets of miR-675. H19-miR-675-Cbl increased the expression level of tyrosine kinase receptors and sustained their activation of down-stream signaling pathways. Moreover, miR-675 overexpression increased the aggressive phenotype of breast cancer cells both in vitro and in vivo. Our findings provide novel mechanistic insights into a critical role for H19 RNA in breast cancer development and reveal a previously unknown link between H19/miR-675, Cbl and tyrosine kinase receptors to enhance breast cancer cell aggressiveness.
RESULTS
H19-derived miR-675 targets c-Cbl and Cbl-b through their coding sequences in breast cancer cells
We have previously demonstrated the oncogenic role of H19 gene in breast tumorigenesis [20]. H19 is a precursor of miR-675-5p/miR-675-3p [16], and H19-derived miR-675 has been reported to promote tumorigenesis of several cancers including colon and gastric cancers [12, 19]. To investigate the molecular mechanism of oncogenic miR-675 in breast cancer cells, we performed alignment prediction and found that miR-675-5p was aligned with coding sequences of 2 proteins belonging to the ubiquitin ligase E3 protein family: c-Cbl and Cbl-b (Figure 1A and 1B). Interestingly, putative seeds are located on coding sequence of these two mRNAs and are conserved between human and mouse. Furthermore, analyses of H19 and Cbl family gene expression in breast cancer cell lines [24] showed a negative correlation between H19 and c-Cbl or Cbl-b (Figure 1C). We then verified the expression of miR-675-5p and c-Cbl/Cbl-b in breast cancer cells overexpressing H19. As shown in Figure 1D, enhanced H19 expression in MDA-MB-231 and MCF-7 breast cancer cell lines was correlated with an increased level of miR-675-5p. Moreover, the levels of c-Cbl and Cbl-b expression decreased significantly in H19-overexpressing cells (Figure 1D and 1E). Together, these data indicate that c-Cbl and Cbl-b may be negatively regulated by H19 in breast cancer cells.
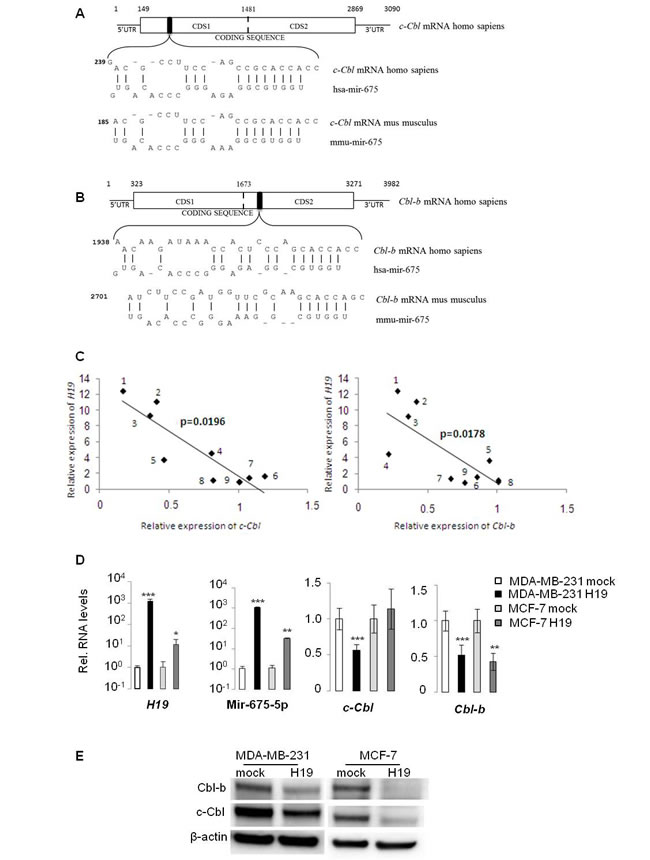
Figure 1: H19/miR675 downregulated c-Cbl and Cbl-b expression in breast cancer cells. A., B. Alignment prediction of miR-675-5p on c-Cbl and Cbl-b mRNA. Relative positions are indicated in bp. Note interaction of miR-675 on Cbl mRNA is conserved in human and mouse. Coding sequences of theses mRNA are too long, so we cloned them in pMiR-REPORT luciferase in two parts named CDS1 and CDS2. The artificial break is represented by the dotted line. C. Negative correlation between H19 and c-Cbl/Cbl-b expression in breast cancer cell lines [24]. Relative expression of H19 and c-Cbl or Cbl-b in (1) MDA-MB-361, (2) MDA-MB-134, (3) SUM225, (4) T47D, (5) S68, (6) SUM159, (7) MCF-7, (8) ZR-75-30 and (9) BT483. D. QRT-PCR analysis of expression of H19, miR-675-5p, c-Cbl and Cbl-b in breast cancer cell lines. Results are presented as relative levels compared to MDA-MB-231 mock cells (indexed to 1). Data represent mean of three independent experiments and error bar sem.*p < 0.05; **p < 0.005; ***p < 0.001. E. Western blot analysis of c-Cbl and Cbl-b levels in MDA-MB-231 and MCF-7 breast cancer cells. β actin was used as a loading control.
To confirm the direct regulation of c-Cbl and Cbl-b by H19, we cloned coding sequences (CDS) of c-Cbl and Cbl-b mRNAs, each in two fragments (CDS1 and CDS2) into the Firefly luciferase reporter vector (pMIR-REPORT) (Figure 1A, 1B). We also cloned CDS mutated on seed sequence and 3’UTR of these mRNAs in the same vector. The mutation is represented on Figure 2A. Each of these DNA constructs were transfected into various breast cancer cells together with either miR-675 mimic, anti-miR-675 (miR-675 inhibitor) or their corresponding controls. As shown in Figure 2B (left panel), miR-675 mimic decreased relative luciferase activity of pMIR-CDS1 c-Cbl of 35-40% compared to microRNA control in MCF-7 and T47D cells. This effect was abolished when seed sequence was mutated. Furthermore, miR-675 had no effect on luciferase activity of pMIR-CDS2 c-Cbl or pMIR-3’UTR c-Cbl plasmids (Figure 2B, left panel). Similar results were obtained for Cbl-b, (Figure 2B, right panel). MiR-675 mimic decreased relative luciferase activity of pMIR-CDS2 Cbl-b of 30% in the two tested cell lines. The effect of miR-675 mimic on CDS2 was abolished after mutation in seed sequence (mut CDS2). MiR-675 mimic had no effect on luciferase activity of pMIR-CDS1 Cbl-b or pMIR-3’UTR Cbl-b plasmids (Figure 2B, right panel).
Contrary to miR-675 mimic, the miR-675 inhibitor was found to increase relative luciferase activity of pMIR-CDS1 c-Cbl (50% increase in T47D, 100% increase in MCF-7 and 90% increase in MDA-MB-231 cells) (Figure 2C, left panel). The miR-675 inhibitor had no effect on luciferase activity of the other constructs including pMIR-mut CDS1, pMIR-CDS2 or pMIR-3’UTR of c-Cbl. Similar results were obtained with Cbl-b plasmid (Figure 2C, right panel). The miR-675 inhibitor increased luciferase activity of only pMIR-CDS2 plasmid in the three tested cells lines, but had no effect on luciferase activity of the other constructs (Figure 2C, right panel).
Taken together, our results confirm that miR-675 decreases both c-Cbl and Cbl-b by interacting with coding sequence of theses mRNAs and precisely, with seed sequences predicted in the alignment.
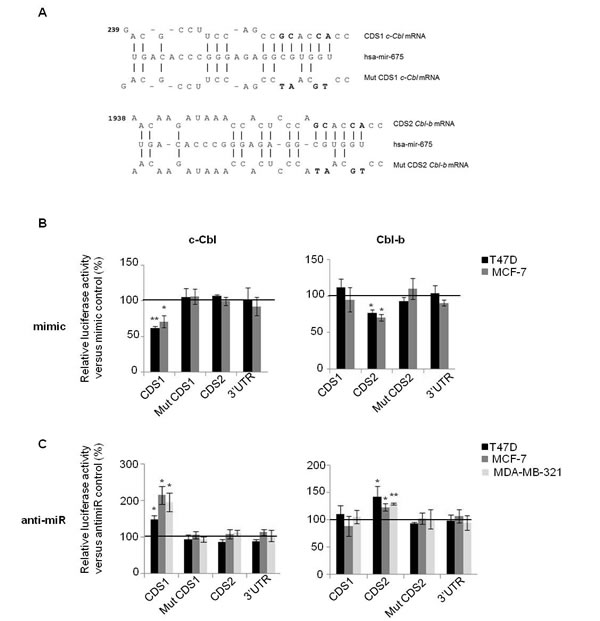
Figure 2: MiR-675 directly targeted on c-Cbl and Cbl-b through interaction with coding sequences. A. Alignment prediction of miR-675-5p on c-Cbl and Cbl-b mRNA. Mutation in seed sequence is represented in bold. Coding sequence (CDS) and 3’UTR of c-Cbl and Cbl-b mRNA were cloned into pMIR-REPORT luciferase. Coding sequence of theses mRNA are so long that we cloned them in pMIR-REPORT luciferase in two parts named CDS1 and CDS2. B. and C. The firefly luciferase activity in breast cancer cells after cotransfection with reporter construct and miR-675 mimic or miR-675 inhibitor (αmir). The luciferase activity was measured by dual-luciferase reporter assay (Promega) and was normalized to Renilla luciferase activity. Plasmids were transfected with mimic or antimir, or theirs controls in breast cancer cell lines (T47D, MCF-7 and MDA-MB-231). Data represent mean of three independent experiments versus their respective controls in percentage and error bar sem.*p < 0.05; **p < 0.005.
H19/miR-675 induce up-regulation of tyrosine kinase receptors and activation of the downstream AKT and ERK pathways
c-Cbl and Cbl-b are well known to be involved in the degradation of tyrosine kinase receptors after their activation by growth factors [25]. To evaluate the role of H19/miR-675 in this process, starved MDA-MB-231 cells overexpressing or not H19 were cultivated in the presence of EGF for different periods of time and then analyzed for the expression levels of EGF receptor (EGFR) by Western blot (Figure 3A). In control cells (mock), EGFR levels decreased progressively upon EGF treatment with nearly half (56%) of the initial EGFR expression present after 7h of culture in the presence of EGF (Figure 3A, left panel). By contrast, the expression levels of EGFR were not modified in cells overexpressing H19 (Figure 3A, right panel). This suggests that H19/miR-675 induced-down regulation of c-Cbl and Cbl-b may contribute to impair the degradation of tyrosine kinase receptors. We then evaluated cell surface expression of EGFR by flow cytometer analysis (Figure 3B). Cells overexpressing H19 clearly exhibited higher levels of cell surface EGFR compared to mock cells. Interestingly, in contrast to parental (not shown) and mock cells (Figure 3B), H19-overexpressing cells, exhibited two subpopulations expressing medium EGFR level (referred as peak a) and high EGFR level (referred as peak b) (Figure 3B). The two cell subpopulations were sorted by flow cytometry and analysed by qRT-PCR for H19 expression (Figure 3C). Cells exhibiting a higher level of cell surface EGFR (peak b) also expressed higher H19 expression levels as compared to peak a (Figure 3C) suggesting that the expression level of H19 correlated with cell surface EGFR expression. To further investigate this relationship, we delivered siRNA to knock down H19 expression in these H19-overexpressing MDA-MB-231 cells. Efficiency of siRNA was determined by qRT-PCR (Figure 3E). As shown in Figure 3D, transient H19 siRNA expression decreased cell surface EGFR level, thus confirming the ability of H19 to upregulate EGFR.
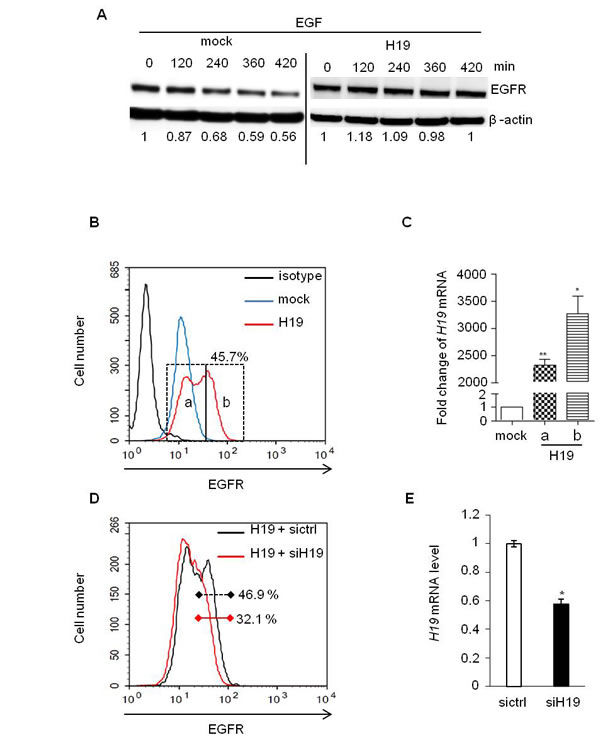
Figure 3: EGF-induced EGFR downregulation was prevented in H19 overexpressing cells. A. MDA-MB-231 control (mock) and H19-overexpressing cells (H19) were treated with EGF (50 ng/ml) and proteins were extracted at indicated times. Total EGFR levels were determined by western blot. The intensities of bands were quantified by densitometry (multigauge, Fujifilm), and the results obtained for EGFR expression during the time-course were compared to those obtained in control (indexed to 1) after normalization to actin expression. B. Flow cytometer analysis of membrane EGFR in MDA-MB-231 control cells (mock) and H19-overexpressing cells (H19). C. Subpopulations of H19 overexpressing cells with medium and high expression levels of cell surface EGFR (peaks a and b in Figure 3B) were sorted by FACS, and H19 expression was then quantified by qRT-PCR. Results are presented as relative levels in cells overexpressing H19 compared to control. *p < 0.05; **p < 0.005. D. H19-overexpressing MDA-MB-231 cells were transiently transfected with siRNA-H19, and membrane EGFR levels were then determined by flow cytometry analysis. E. Relative expression of H19 determined by qRT-PCR in breast cancer cells overexpressing H19 transfected with siRNA-H19 or siRNA-GFP as a control. *p < 0.05.
We then treated cells with EGF and studied activation of EGFR and its downstream signaling pathways over the time by Western blot and ALPHAscreen®. In MDA-MB-231 breast cancer cells stably overexpressing H19, EGF strongly activated EGFR, Akt and Erk compared to parental cells (Figure 4A and 4B). Interestingly, HGF treatment induced also a stronger activation of its receptor c-Met, Akt and Erk in MDA-MB-231 cells stably overexpressing H19 compared to parental cells (Supplementary Figure 1A-1C). Similarly, H19-overexpressing cells exhibited stronger activation of Akt and Erk upon proNGF and NGF treatments (Supplementary Figure 1D and 1E). When cells were transiently transfected with a plasmid encoding the H19 gene and then treated with EGF or HGF (Supplementary Figure 2), similar results were obtained in terms of Akt and Erk activation (Supplementary Figure 2).
To further verify if the increased activation of AKT and Erk was mediated by miR-675, we first transfected MDA-MB-231 parental cells with miR-675 mimic and cells overexpressing H19 with the miR-675 inhibitor or the corresponding controls. Then, we treated cells with EGF and studied Akt and Erk phosphorylation. As shown in Figure 4C, cells transfected with mimic and then stimulated with EGF exhibited an increase of Akt and Erk activation compared to control vector transfected cells. Conversely, in H19-overexpressing cells, the miR-675 inhibitor decreased Akt and Erk activation (Figure 4D).
All together, our results indicate that H19/mir-675-5p increase the activation of several tyrosine kinase receptors and their downstream signaling pathways in breast cancer cells.
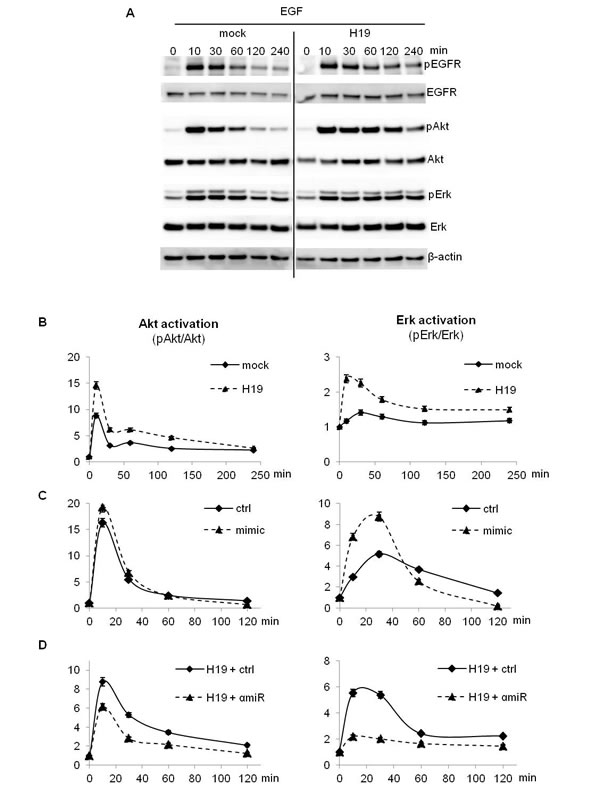
Figure 4: EGF-induced Akt and Erk phosphorylation was enhanced in H19/miR675-overexpressing cells. A., B. Control (mock) and H19-overexpressing cells (H19) were treated with 10 ng/ml EGF and proteins were extracted at indicated times. Akt and Erk activation was determined by Western blot analysis A. and ALPHAscreen® B.. C. Parental MDA-MB-231 cells (ctrl) were transfected with miR-675 (mimic) for 72 h. Then, cells were treated with 10 ng/ml EGF. Proteins were extracted at indicated times and Akt and Erk activation was performed by ALPHAscreen® analysis. D. H19-overexpressing cells (H19) were transfected with miR-675 specific inhibitor (αmiR) for 72 h. Then cells were treated with 10 ng/ml EGF. Proteins were extracted at indicated times and Akt and Erk activation was determined by ALPHAscreen® analysis. These experiments were performed three times in triplicate. Data represent one representative experiment. For ALPHAscreen® error bars represent SEM.
H19/miR-675 increase migration and proliferation of breast cancer cells and enhance the effects of growth factors
To further determine the functional impact of Akt and Erk activation in cells overexpressing H19 and miR-675, we first evaluated the migratory capacity of H19-overexpressing cells using transwell and wound-healing assays. As shown in Figure 5A and 5B, H19-overexpressing MDA-MB-231 cells exhibited an increased migratory capacity compared to control cells. Moreover, H19 overexpression further increased HGF-induced migration of MDA-MB-231 cells (Supplementary Figure 3A). Similarly, H19 gene overexpression increased also EGF-induced proliferation of MCF-7 cells (Supplementary Figure 3B).
To evaluate the role of miR-675 independently of H19 mRNA, we first evaluated the migratory ability of parental MDA-MB-231 transiently transfected with miR-675 mimic. We found that miR-675 mimic increased cell migration as revealed by wound healing assay (Figure 5C). This indicates that miR-675 per se is able to increase cell mobility. We then generated cells stably overexpressing miR-675. For this, we introduced in MDA-MB-231 cells a plasmid encoding Green Fluorescent Protein (GFP) fused with miR-675 precursor sequence. As shown in Figure 5D, the two selected clones (miR cl1 and miR cl2) expressed more than 5-fold of miR-675 when compared to mock cells. As expected, the protein levels of c-Cbl and Cbl-b were decreased in the two selected clones (Figure 5E). Importantly, these clones exhibited stronger migratory and proliferative capacities when compared to control cells (Figure 5F and 5G). Together, these data confirm that miR-675 increases cell migration and proliferation independently of H19.
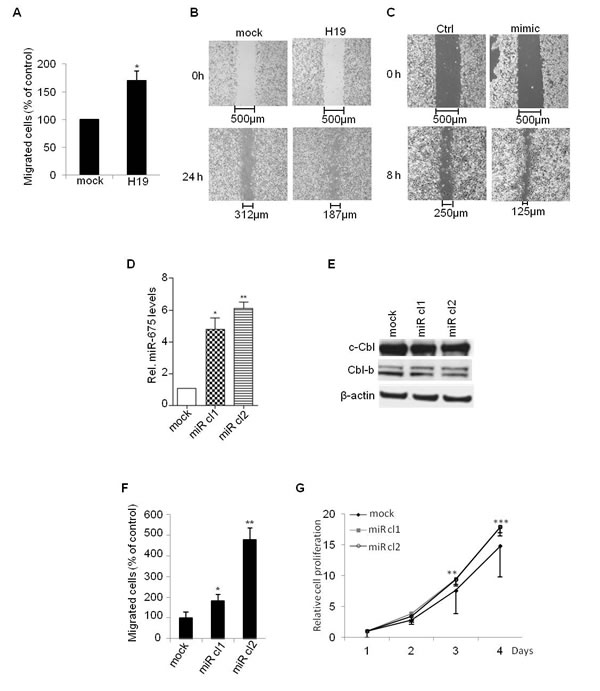
Figure 5: H19/miR675 increased migration and proliferation of MDA-MB-231 breast cancer cells. A. Control (mock) or H19-overexpressing (H19) cells were cultured in transwells for 24 h. Migrated cells were then colored with violet crystal and counted. Results are presented as the percentage of control. B. Wound healing assay performed on control (mock) and H19-overexpressing (H19) cells. C. Wound healing assay performed on control (ctrl) and transiently overexpressing miR-675 (mimic) cells. D. QRT-PCR analysis of expression of miR-675-5p in control (mock) and miR-675-overexpressing cells (miR cl1, miR cl2). Results are presented as relative levels compared to MDA-MB-231 control cells (indexed to 1). E. Western blot analysis of c-Cbl and Cbl-b levels in control and miR-675-overexpressing cells. F. Transwell migratory assay performed on control (mock) and miR-675-overexpressing cells after 6 h of culture. G. Cell proliferation determined by MTT test. Data represent mean of three independent experiments and error bar sem.*p < 0.05; **p < 0.005; ***p < 0.001.
The miR-675 enhances the tumorigenicity and metastatic potential of breast cancer cells
Since ectopic expression of miR-675 increased migration and proliferation of breast cancer cells in vitro, we examined whether overexpression of miR-675 could enhance tumor growth and metastasis in vivo by subcutaneously injecting cells into immunodeficient SCID mice. As shown in Figure 6A, tumor volume was increased in mice injected with miR-675-overexpressing clones as compared to control mice injected with parental cells. Accordingly, an increased cell proliferation (PCNA staining) and a decreased apoptosis (TUNEL detection) were found in sections of tumors formed by miR-675-overexpressing cells (Figure 6B, middle and right panels) as compared to those formed by parental cells (Figure 6B, left panels). Of note, the protein levels of c-Cbl and Cbl-b were decreased in tumor formed by miR-675-overexpressing cells compared to control (Figure 6C). We then analyzed metastases in xenografted mice thanks to GFP expression in MDA-MB-231 cells. As shown in Figure 6D and 6E, more GFP-positive cells were found in brain, liver and lungs of mice xenografted with miR-675 overexpressing cells compared to control (Figure 6D, 6E). Taken together, these results indicate that miR-675 increases primary tumor formation and promotes metastasis.
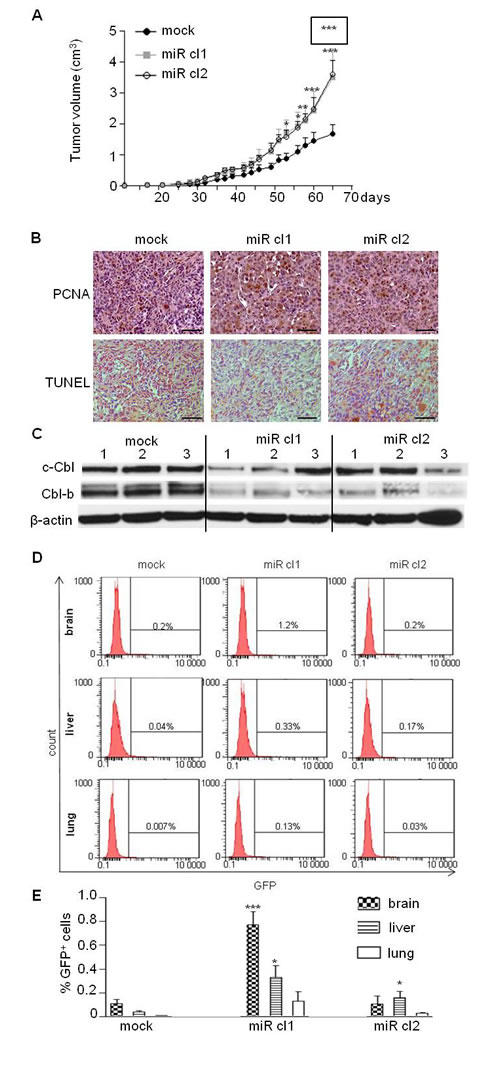
Figure 6: MiR-675 promoted tumor growth and metastasis. A. MDA-MB-231 control (mock) and overexpressing miR-675 (miR cl1, miR cl2) cells were subcutaneously injected into SCID mice (7 mice per group). Tumor growth curve represents the mean of tumor volumes in each group and error bar sem. MiR cl1 growth curve statistical analysis is surrounded. B. Detection of tumor cell proliferation (PCNA) and apoptosis (TUNEL) of paraffin-embedded sections of tumors 65 days after injections. Scale bar is 100µm. C. Western blot analysis of c-Cbl and Cbl-b levels in xenografted tumors. Tumors from three mice for each group were analyzed (mock, miR cl1, miR cl2). D. and E. Detection of GFP-positive cells by flow cytometry analysis in brain, liver and lung of mice xenografted with control or miR-675-overexpressing cells (mock, miR cl1, miR cl2). Histograms in E represent the mean of results from six xenografted mice *p < 0.05; **p < 0.005; ***p < 0.001.
DISCUSSION/CONCLUSION
It has become increasingly clear that H19 RNA plays essential role in tumor development and that the H19 gene is regulated by a complex interplay of both extrinsic and intrinsic factors. We have already demonstrated that H19 expression is positively regulated by several growth factors such as HGF, EGF and FGF-2 [21]. HGF induces H19 expression via the ERK/MAPK and phospholipase C pathways. Here, we demonstrated that H19 induced up-regulation of tyrosine kinase receptors including EGFR and c-Met as well as the activation of their downstream Akt and ERK signaling pathways. Our findings indicate that the positive feedback between H19 expression and growth factors may be of importance in promoting breast cancer development.
More recently, H19 has been found to encode miR-675. Several targets of miR-675 have been identified in cancers from different tissues. These include Twist1, CALN1, TGFβ1, and Cadherin11 [13, 18, 26, 27]. While this study was underway, it was shown that miR-675 regulates negatively Rb expression by interacting with its 3’UTR mRNA in colorectal cancer and hepatocellular carcinoma [12, 18]. However, we did not find any regulation of Rb by miR-675 in breast cancer cells (Supplementary Figure 4). Our results together with the previous identification of miR-675 targets indicate that miR-675 may function in a tissue-specific manner and that the increased proliferation of H19-overexpressing breast cancer cells cannot be explained by a negative regulation of Rb. Indeed, we demonstrated that miR-675 decreased the expression of c-Cbl and Cbl-b. MiR-675 interacted directly with c-Cbl and Cbl-b coding sequences to prevent mRNA translation. Among the reported targets of miR-675, c-Cbl and Cbl-b were the first ones found to interact with miR-675 via their coding sequences.
In mammals, three homolog of Cbl exist: c-Cbl, Cbl-b and Cbl-c. The protein structures of c-Cbl and Cbl-b are similar; they consist of a N-terminal tyrosine kinase-binding domain, a ring finger motif, a proline-rich region and a c-terminal ubiquitin-associated domain that overlap leucine zipper motif. The Cbl-c lacks the c-terminal domain (proline-rich region and ubiquitin-associated domain). Cbl proteins are involved in the regulation of actin skeleton, lymphocyte signaling and downregulation of tyrosine kinase receptors [28]. Cbl proteins negatively regulate tyrosine kinase receptors by interacting with Grb2 to prevent Grb2/SOS association. Cbl can also recognize activated receptors and induce their downregulation by lysosomal degradation.
In breast cancer cells, it has been demonstrated that c-Cbl is associated with two other proteins to form the tripartite complex Cdc42, p85Cool-1/βPix, and c-Cbl [29]. Upon EGFR activation, Cdc42 interacts with c-Cbl within the tripartite complex to repress the degradation of the activated receptor initiated by c-Cbl. In lung cancer cells, c-Cbl inactivating-mutation increases viability and migration of cells [30]. Similarly, in acute myeloid leukemia, c-Cbl mutations decrease EGFR ubiquitinylation, leading to signaling activation, cell proliferation and survival. Moreover, c-Cbl has been proposed to act as a tumor suppressor since c-Cbl null mice develop invasive cancer (notably, juvenile myelomonocytic leukemia) with complete penetrance [31]. In accordance with these data, we demonstrated that miR-675-induced decrease of c-Cbl and Cbl-b was associated with a more aggressive phenotype of breast cancer cells; miR-675-overexpressing cells presented increased levels of EGFR and c-Met (not shown), increased activation of the downstream signaling pathways as well as increased tumor growth and metastasis. In agreement with our data, Matouk et al demonstrated that miR-675 indirectly targets slug leading to increase of cell invasion and in vivo metastasis [32].
On the other hand, Berberine (an isoquinoline alkaloids used in gastroenteritis, type II diabetes, hypertension and arrhythmia) inhibits proliferation of colon cancer cells by stimulating c-Cbl activation and enhancing EGFR degradation [33]. Similarly, another drug, icotinib, activates Cbl-b protein to downregulate EGFR, and induces apoptosis and G1 phase arrest in non-small-cell lung cancer cells [34]. Thus, cancer therapeutic agents can induce downregulation of tyrosine kinase receptors by activating Cbl proteins. However, cancer resistance to chemotherapy is also frequently associated with the overexpression of tyrosine kinase receptors, which can serve as a link between tumor cells and the microenvironment [35]. The activation of tyrosine kinase receptors by specific growth factors in tumor microenvironment can protect tumors cells from drugs-induced damages. Our findings suggest that drug resistance of H19-overexpressing cancer cells could be due to Cbl downregulation by H19-derived miR-675. Supporting this hypothesis, we found that high expression of miR-675 combined with low expression of c-Cbl identified a subset of ER-negative breast cancer tumors of poorer prognosis (Supplementary Figure 5). This was found by exploiting the matched mRNA and microRNA global expression profiling of a cohort of 207 primary tumors [36] and, given the rarity of such cohort, will need to be confirmed by future similar studies.
An antisense transcript, 91H, has been found in the H19 locus [37, 39]. We verified if this long non coding RNA can act as a miR sponge and impair miR-675 effects. 91H expression does not vary in miR-675 overexpressing cells. In addition, in 91H knockdown cells, c-Cbl and Cbl-b expression is stable (Supplementary Figure 6), indicating 91H is not involved in c-Cbl and Cbl-b regulation.
In conclusion, we identified ubiquitin ligase E3 family (c-Cbl and Cbl-b) as direct targets of miR-675 in breast cancer cells. Moreover, by directly targeting c-Cbl and Cbl-b, miR-675 increased the stability and the activation of EGFR and c-Met leading to subsequent activation of Akt and Erk as well as enhanced cell proliferation and migration. Our findings provide novel mechanistic insights into a critical role for H19 RNA in breast cancer development, and reveal a link between H19/miR-675, Cbl and tyrosine kinase receptors to enhance breast cancer cell aggressiveness.
MATERIALS AND METHODS
Cells culture
The MCF-7 and T47D estrogen-sensitive and the MDA-MB-231 estrogen-insensitive breast cancer cell lines were obtained from the American Type Culture Collection and maintained routinely in Roswell Park Memorial Institute medium (RPMI, Gibco) containing 10% of foetal bovine serum (FBS) and 0.01% of Zell Shield (Minerva Biolabs). Cell lines were cultured at 37°C with 5% CO2, 95% of air in humidified atmosphere.
Migration assays
Cell migration was determined by transwell assay and wound healing assay. For transwell assay, 3x104 cells were seeded on collagen (1/100, Millipore) coated insert (0.8µm, BD Bioscience) of 6 well-plates in RPMI containing 10% FBS or 0.1 % FBS in the presence of HGF. At the end of the experiments, cells migrating to the other side of the filter were stained with 0.5% crystal violet and counted. Wound healing test was performed by using Culture-inserts for Live Cell Analysis (Ibidi). A total of 3x104 cells were plated on each compartment of insert and cultured for 5h. The insert was then removed (0h) and cells were further cultured for 24h. Cells were photographed at 0h and after 24h of culture to record the wound width.
Proliferation assays
A total of 500 cells were plated in each well of a 96-well plate. Cell proliferation was determined using MTT test every day as previously described [12].
Western blot analysis
Cells were lysed in RIPA buffer containing protease and phosphatase inhibitors (protease inhibitor P8340; phosphatase inhibitor cocktail 2 P5726, Sigma-Aldrich). Proteins were quantified with BCA protein assay (Pierce) and then reduced in NuPAGE LDS Sample buffer (Invitrogen) with NuPAGE Reducing Agent (Invitrogen) at 70°C for 10 min. Proteins were separated on SDS-PAGE 4-12% (Invitrogen) and transferred onto PolyVinylidene Fluoride (PVDF) membrane (Millipore). After saturation in PBS 0.2% of casein, membranes were incubated with primary antibodies overnight at 4°C. References of antibodies are listed in the supplemental Table S1. Membranes were washed with PBS 0.5% Tween for 30 min and incubated with secondary antibodies conjugated with Horse Radish Peroxidase (HRP) for 2 h at room temperature. Membranes were analysed with SuperSignal west Dura Chemiluminescence Substrate (Pierce).
Quantification of protein phosphorylation by ALPHAscreen®
Quantification of protein phosphorylation was performed using ALPHAscreen® SureFire technology (Perkin Elmer). This technology allows protein activation quantification in low volume of lysates. Briefly, cells were lysed in appropriate buffer completed with protease and phosphatase inhibitors and lysates were clarified by centrifugation. Lysates were incubated with different antibodies and acceptor beads for 2 h at room temperature on 384-well plates. Then donor beads were added for 2 h and light emission was measured on EnSpire Alpha (Perkin Elmer). The anti-p-Erk Thr202/Tyr204 (TGRESHV100), anti-total Erk TGRTES500, anti-p-Akt ser473 (TGRA4S500), anti-total Akt (TGRTAPS500), anti-p-Met Tyr1234/1235 (TGRCMS500), and anti-total Met (AL281C) antibodies were from Perkin Elmer.
RNA extraction, reverse transcription and Real-time RT-PCR
RNA extraction and qRT-PCR was performed as previously described [37]. Primers used for qRT-PCR are described in supplemental Table S2.
MiRNA was extracted with mirVANATM miRNA isolation kit (Life Technologies). For the detection, a total of 50 ng RNA was used in the reverse transcription reaction (miRCURY LNATM Universal RT) (Exiqon). Quantitative PCR was performed by using Exilent SYBR Green master mix (Exiqon) and hsa-miR-675-5p LNA PCR primer set, UniRT (Exiqon). Human U6 RNA was used as an internal control. Fold change of miRNA expression was calculated by the equation 2-ΔΔCt.
Construction of plasmids
Coding DNA Sequence (CDS) and 3’UTR of c-Cbl and Cbl-b mRNA were cloned in the pMIR-REPORT luciferase vector (Ambion). Because of their length, CDS were cloned in two fragments named CDS1 and CDS2. All fragments of Cbl-b and 3’UTR of c-Cbl were cloned between SpeI and HindIII enzyme site. CDS1 of c-Cbl was cloned between SpeI and PmeI and CDS2 between SpeI and MluI (New England Biolabs). Fragments were generated by PCR (primers are listed in supplemental Table S2). Point mutations in seed sequence were generated by PCR.
H19 gene was cloned in pcDNA3.1 (-) (Invitrogen) between NotI and BamHI (New England Biolabs). The normalizing vector pRL-null has no promoter sequence to drive expression of the Renilla luciferase gene and was purchased from Promega.
MiR-675-5p was amplified by PCR and cloned in pEGFP-C1 plasmid (Clontech) between HindIII and BamHI (New England Biolabs). Plasmid productions were performed in E. Coli TOP 10 (Invitrogen). Plasmid extraction was performed by using Nucleobond PC100 (Macherey-Nagel).
H19 gene and siH19 transient transfection
For transient transfection, a total of 1.3 x105 cells were plated on 6-well plates. After 24h, cells were transfected with 1µg of pcDNA3.1-H19 DNA using Exgen-500 (Euromedex) according to the manufacturer’s instructions. Cells were incubated at 37°C for 6h, and then cultured in medium used routinely. H19 siRNA (Table S2) were transfected with DharmaFECT Duo according to manufacturer’s guidelines (Thermo Fischer Scientific Dharmacon).
Establishment of cells overexpressing H19 or miR675
To establish cells overexpressing H19, MDA-MB-231 and MCF-7 breast cancer cell lines were transfected with 1µg of pcDNA3.1-H19 or pcDNA3.1 empty vector as a control using Exgen-500 and cells were allowed to recover for 48h. Cells were then selected in the presence of 1mg/ml G418 (Sigma) for at least one month. To establish cells overexpressing miR675, MDA-MB-231 cells were transfected with 1 µg of pEGFP-C1 /miR675 or pEGFP-C1 empty vector using Exgen-500 for 48h. Cells were then selected in the presence of 1mg/ml G418 (Sigma) for at least one month. Among numerous clones obtained, two of them were randomly chosen for in vitro and in vivo experiments.
MicroRNA transfection and luciferase activity assay
A total of 1x105 cells were plated on 12-well plates for 24h. Hsa-miR-675 mimic or its hairpin inhibitor (Thermo Fischer Scientific Dharmacon) were transfected with DharmaFECT-Duo according to manufacturer’s guidelines (Thermo Fischer Scientific Dharmacon). Briefly, 75 nM mimic or hairpin inhibitor (anti-miR) were transfected with 500 ng of Firefly luciferase plasmid and 12 ng of pRL-null plasmid expressing Renilla luciferase to monitor the transfection efficiency. The luciferase activity was measured 24h after transfection by using the dual-luciferase reporter assay system according to manufacturer’s instructions (Promega). Firefly luciferase was normalized with Renilla luciferase.
Flow cytometer analysis of membrane EGFR
A total of 2x105 cells were plated on 6 well-plates and cultured in RPMI containing 1% FBS. After 48h, cells were isolated by trypsinization using trypsin-versene EDTA solution, then incubated with an anti-EGFR antibody (EGFR-AF488, 1/100, SC-120, Santa Cruz) for 30 min at 4°C before analysis on flow cytometer (Calibur II or ARIA II, Becton Dickinson).
Tumorigenesis in SCID mice
Female SCID mice (8-weeks-old) were purchased from Pasteur Institute, Lille and kept under pathogen-free condition. Animals were handled in accordance with the European Communities Recommendations for Animals Experimentation. Exponentially growing cells were harvested, resuspended in PBS, and subcutaneously (2x106 cells in 150 µl) injected into the flank fat pad of each mouse. Tumor volume and metastasis were monitored as previously described [38].
Analysis of cell proliferation and apoptosis in xenografted tumors
Tumors were fixed, paraffin embedded and cut on 5 µm tumor sections. Cell proliferation was measured by immunostaining with an anti-PCNA antibody (Santa Cruz, sc-56 HRP). Apoptosis was measured using in situ cell death detection kit, POD (Roche), according to manufacturer’s instructions.
Statistical analysis
Data are expressed as mean values ± standard error of the mean of at least 3 independent experiments. The statistical analysis was done by using Student’s t-test and p value < 0.05 was considered significant.
ACKNOWLEDGMENTS
We thank Helene Bauderlique-Le Roy from the flow cytometry platform of the BioImaging Center of Lille (BICeL). We also thank the animal facility at the Pasteur Institute of Lille (PLETHA, Dir: J.P. de Cavel) for animal housing and technical advices.
CONFLICTS OF INTEREST
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
GRANT SUPPORT
This work was supported by INSERM and grant from INCA (PLBio 2010-180). C.V. was supported by doctoral fellowships from the University of Lille2 and by Fondation ARC fellowships. F.D. was supported by fellowships from INCA (PLBio 2010-180). S.J. was supported by post-doctoral fellowships from the “Région Nord-Pas de Calais”.
REFERENCES
1. Bartolomei MS, Zemel S, Tilghman SM. Parental imprinting of the mouse H19 gene. Nature 1991; 351: 153-155.
2. Brannan CI, Dees EC, Ingram RS, Tilghman SM. The product of the H19 gene may function as an RNA. Mol Cell Biol 1990; 10: 28-36.
3. Adriaenssens E, Dumont L, Lottin S, Bolle D, Leprêtre A, Delobelle A, Bouali F, Dugimont T, Coll J, Curgy JJ. H19 overexpression in breast adenocarcinoma stromal cells is associated with tumor values and steroid receptor status but independent of p53 and Ki-67 expression. Am J Pathol 1998; 153: 1597-1607.
4. Adriaenssens E, Lottin S, Dugimont T, Fauquette W, Coll J, Dupouy JP, Boilly B, Curgy JJ. Steroid hormones modulate H19 gene expression in both mammary gland and uterus. Oncogene 1999; 18: 4460-4473.
5. Liu J, Kahri AI, Heikkilä P, Ilvesmäki V, Voutilainen R. H19 and insulin-like growth factor-II gene expression in adrenal tumors and cultured adrenal cells. J Clin Endocrinol Metab 1995; 80: 492-496.
6. Ariel I, Weinstein D, Voutilainen R, Schneider T, Lustig-Yariv O, de Groot N, Hochberg A. Genomic imprinting and the endometrial cycle. The expression of the imprinted gene H19 in the human female reproductive organs. Diagn Mol Pathol 1997; 6: 17-25.
7. Soejima H, Higashimoto K. Epigenetic and genetic alterations of the imprinting disorder Beckwith-Wiedemann syndrome and related disorders. J Hum Genet 2013; 58:402-9
8. Yoshimizu T, Miroglio A, Ripoche MA, Gabory A, Vernucci M, Riccio A, Colnot S, Godard C, Terris B, Jammes H, Dandolo L. The H19 locus acts in vivo as a tumor suppressor. Proc Natl Acad Sci U S A. 2008; 105: 12417-12422.
9. Ariel I, Lustig O, Schneider T, Pizov G, Sappir M, De-Groot N, Hochberg A. The imprinted H19 gene as a tumor marker in bladder carcinoma. Urology 1995; 45: 335-338.
10. Kondo M, Suzuki H, Ueda R, Osada H, Takagi K, Takahashi T, Takahashi T. Frequent loss of imprinting of the H19 gene is often associated with its overexpression in human lung cancers. Oncogene 1995; 10: 1193-1198.
11. Hibi K, Nakamura H, Hirai A, Fujikake Y, Kasai Y, Akiyama S, Ito K, Takagi H. Loss of H19 imprinting in esophageal cancer. Cancer Res 1996; 56: 480-482.
12. Tsang WP, Ng EKO, Ng SSM, Jin H, Yu J, Sung JJY, Kwok TT. Oncofetal H19-derived miR-675 regulates tumor suppressor RB in human colorectal cancer. Carcinogenesis 2010; 31: 350-358.
13. Li H, Yu B, Li J, Su L, Yan M, Zhu Z, Liu B. Overexpression of lncRNA H19 enhances carcinogenesis and metastasis of gastric cancer. Oncotarget 2014; 5: 2318-29.
14. Barsyte-Lovejoy D, Lau SK, Boutros PC, Khosravi F, Jurisica I, Andrulis IL, Tsao MS, Penn LZ. The c-Myc oncogene directly induces the H19 noncoding RNA by allele-specific binding to potentiate tumorigenesis. Cancer Res 2006; 66: 5330-7.
15. Kallen AN, Zhou XB, Xu J, Qiao C, Ma J, Yan L, Lu L, Liu C, Yi JS, Zhang H, Min W, Bennett AM, Gregory RI, et al.The imprinted H19 lncRNA antagonizes let-7 microRNAs. Mol Cell 2013; 52: 101-12.
16. Cai X, Cullen BR. The imprinted H19 noncoding RNA is a primary microRNA precursor. RNA 2007; 13: 313-316.
17. Cipolla GA. A non-canonical landscape of the microRNA system. Front Genet. 2014 5; 337.
18. Hernandez JM, Elahi A, Clark CW, Wang J, Humphries LA, Centeno B, Bloom G, Fuchs BC, Yeatman T, Shibata D. miR-675 Mediates Downregulation of Twist1 and Rb in AFP-Secreting Hepatocellular Carcinoma. Ann Surg Oncol 2013. doi:10.1245/s10434-013-3106-3.
19. Zhuang M, Gao W, Xu J, Wang P, Shu Y. The long non-coding RNA H19-derived miR-675 modulates human gastric cancer cell proliferation by targeting tumor suppressor RUNX1. Biochem Biophys Res Commun 2014; 448: 315-322.
20. Lottin S, Adriaenssens E, Dupressoir T, Berteaux N, Montpellier C, Coll J, Dugimont T, Curgy JJ. Overexpression of an ectopic H19 gene enhances the tumorigenic properties of breast cancer cells. Carcinogenesis 2002; 23: 1885-1895.
21. Adriaenssens E, Lottin S, Berteaux N, Hornez L, Fauquette W, Fafeur V, Peyrat JP, Le Bourhis X, Hondermarck H, Coll J, Dugimont T, Curgy JJ. Cross-talk between mesenchyme and epithelium increases H19 gene expression during scattering and morphogenesis of epithelial cells. Exp Cell Res 2002; 275: 215-229.
22. Berteaux N, Lottin S, Monté D, Pinte S, Quatannens B, Coll J, Hondermarck H, Curgy JJ, Dugimont T, Adriaenssens E. H19 mRNA-like noncoding RNA promotes breast cancer cell proliferation through positive control by E2F1. J Biol Chem 2005; 280: 29625-29636.
23. Vennin C, Dahmani F, Spruyt N, Adriaenssens E. Role of long non-coding RNA in cells: Example of the H19/IGF2 locus. Advances in Bioscience and Biotechnology 2013; 04: 34-44.
24. Charafe-Jauffret E, Ginestier C, Monville F, Finetti P, Adélaïde J, Cervera N, Fekairi S, Xerri L, Jacquemier J, Birnbaum D, Bertucci F. Gene expression profiling of breast cell lines identifies potential new basal markers. Oncogene 2006; 25: 2273-2284.
25. Mohapatra B, Ahmad G, Nadeau S, Zutshi N, An W, Scheffe S, Dong L, Feng D, Goetz B, Arya P, Bailey TA, Palermo N, Borgstahl GE, et al. Protein tyrosine kinase regulation by ubiquitination: critical roles of Cbl-family ubiquitin ligases. Biochim Biophys Acta 2013; 1833:122-39.
26. Zhu M, Chen Q, Liu X, Sun Q, Zhao X, Deng R, Wang Y, Huang J, Xu M, Yan J, Yu J. lncRNA H19/miR-675 axis represses prostate cancer metastasis by targeting TGFBI. FEBS J 2014; 281: 3766-75.
27. Kim NH, Choi SH, Lee TR, Lee CH, Lee AY. Cadherin 11, a miR-675 target, induces N-cadherin expression and epithelial-mesenchymal transition in melasma. J Invest Dermatol 2014; 134: 2967-76.
28. Schmidt MHH, Dikic I. The Cbl interactome and its functions. Nat Rev Mol Cell Biol 2005; 6: 907-918.
29. Hirsch DS, Shen Y, Wu WJ. Growth and motility inhibition of breast cancer cells by epidermal growth factor receptor degradation is correlated with inactivation of Cdc42. Cancer Res 2006; 66: 3523-3530.
30. Tan Y-HC, Krishnaswamy S, Nandi S, Kanteti R, Vora S, Onel K, Hasina R, Lo FY, El-Hashani E, Cervantes G, Robinson M, Hsu HS, Kales SC et al. CBL is frequently altered in lung cancers: its relationship to mutations in MET and EGFR tyrosine kinases. PLoS ONE 2010; 5: e8972.
31. Ogawa S, Shih L-Y, Suzuki T, Otsu M, Nakauchi H, Koeffler HP, Sanada M. Deregulated intracellular signaling by mutated c-CBL in myeloid neoplasms. Clin Cancer Res 2010; 16: 3825-3831.
32. Matouk IJ, Raveh E, Abu-lail R, Mezan S, Gilon M, Gershtain E, Birman T, Gallula J, Schneider T, Barkali M, Richler C, Fellig Y, Sorin V et al. Oncofetal H19 RNA promotes tumor metastasis. Biochim Biophys Acta. 2014; 1843: 1414-26.
33. Wang L, Cao H, Lu N, Liu L, Wang B, Hu T, Israel DA, Peek RM-Jr, Polk DB, Yan F. Berberine inhibits proliferation and down-regulates epidermal growth factor receptor through activation of Cbl in colon tumor cells. PLoS ONE 2013; 8: e56666.
34. Mu X, Zhang Y, Qu X, Hou K, Kang J, Hu X, Liu Y. Ubiquitin ligase Cbl-b is involved in icotinib (BPI-2009H)-induced apoptosis and G1 phase arrest of EGFR mutation-positive non-small-cell lung cancer. Biomed Res Int 2013; 2013: 726375.
35. Corso S, Giordano S. Cell-autonomous and non-cell-autonomous mechanisms of HGF/MET-driven resistance to targeted therapies: from basic research to a clinical perspective. Cancer Discov 2013; 3: 978-992.
36. Buffa FM, Camps C, Winchester L, Snell CE, Gee HE, Sheldon H, Taylor M, Harris AL, Ragoussis J. microRNA-associated progression pathways and potential therapeutic targets identified by integrated mRNA and microRNA expression profiling in breast cancer. Cancer Res 2011; 71: 5635-5645.
37. Berteaux N, Aptel N, Cathala G, Genton C, Coll J, Daccache A, Spruyt N, Hondermarck H, Dugimont T, Curgy JJ, Forné T, Adriaenssens E. A novel H19 antisense RNA overexpressed in breast cancer contributes to paternal IGF2 expression. Mol Cell Biol 2008; 28: 6731-6745.
38. Lagadec C, Meignan S, Adriaenssens E, Foveau B, Vanhecke E, Romon R, toillon RA, Oxombre B, Hondermarck H, Le Bourhis X. TrkA overexpression enhances growth and metastasis of breast cancer cells. Oncogene 2009; 28: 1960-1970.
39. Tran VG, Court F, Duputié A, Antoine E, Aptel N, Milligan L, Carbonell F, Lelay-Taha MN, Piette J, Weber M, Montarras D, Pinset C, Dandolo L et al. H19 antisense RNA can up-regulate Igf2 transcription by activation of a novel promoter in mouse myoblasts. PLoS One. 2012;7: e37923.