
INTRODUCTION
Activation of telomerase is associated with malignancy in many human cancers [1]. Telomerase inhibition was proposed as a potential cancer therapy when telomere shortening was first described in human cells [2]. Telomerase-deficient mice have been generated via deletion of the mouse terc gene encoding the RNA component of telomerase [3]. Terc-/- mice show phenotypic abnormalities only after successive generations of terc-/- intrecrosses [4, 5]. Strong evidence has accumulated that short telomeres limit tumor growth. Crosses of terc-/- mice to tumor prone models demonstrate that the short telomere response significantly limits tumor formation [6]. However, in contrast to these findings in differentiated tumor cells, the contribution of telomerase to the biology of cancer stem cells (CSC) has not been previously investigated.
Recently we have shown that the restricted expression of the BCR-ABL oncogene, linked to chronic myeloid leukemia (CML) disease, to the hematopoietic stem cell compartment is capable of generating a full-blown tumour with all its differentiated cellular components, showing a hands-off role for BCR-ABL in regulating CML formation [7-12]. BCR-ABL oncogene inactivation could not change this tumor reprogramming fate at the CSC level, in agreement with the common occurrence of tumor relapse by which human CML evolves to escape BCR-ABL pharmacological inactivation [13-22]. Thus, it seems important to know how to eradicate and/or prevent this BCR-ABLp210-induced reprogramming of stem cells [10-12]. In order to identify the genes that are associated with BCR-ABLp210-induced reprogramming of stem cells we performed a supervised analysis of the transcriptional profiles of CSCs purified from Sca1-BCR-ABLp210 mice versus hematopoietic stem cells from control mice. The data identified that terc gene was expressed in CSCs in the Sca1-BCR-ABLp210 mice, although it was not differentially regulated in CSCs versus control hematopoietic stem cells [7, 8]. Now, to assess the functional importance of telomerase and telomere status in this Sca1-BCR-ABLp210 model, the terc gene knockout was established in these Sca1-BCR-ABLp210 mice, and tumor phenotype assessed in the first generation of telomerase heterozygous and homozygous mice. Remarkably, the data provide evidence that telomerase activity is essential for BCR-ABLp210-induced reprogramming of stem cells. Overall, our results demostrate that telomerase plays a critical role in the pathogenesis of BCR-ABL-CML, and validate telomerase and the genes it regulates as targets for therapy in CML.
RESULTS AND DISCUSSION
In order to understand the role of telomerase in leukemia stem cell (LSC) generation and maintenance, we have taken advantage of our Sca1-BCR-ABLp210 mouse model of human chronic myeloid leukaemia (CML), a paradigmatic stem-cell disorder [23, 24]. This transgenic mouse was engineered to express the human BCRABLp210 cDNA under the control of the Sca1 promoter in order to limit and determine the effect of ectopic expression of BCRABLp210 in hematopoietic stem/progenitor cells [7, 8, 25, 26]. This model not only faithfully recapitulates the human disease but also has been able to anticipate that human CML stem cells survival is Bcr-Abl kinase independent and suggest curative approaches in CML must focus on kinase-independent mechanisms of resistance [7, 27, 28]. This model represents an ideal system to analyze the contributions of telomerase activity deficiency to the LSC biology and malignant progression of CML. To this end, terc-deficient mice were crossed to Sca1-BCR-ABLp210 mice to produce and analyzed cohorts of Sca1-BCR-ABLp210 ter+/- (n=17) and of Sca1-BCR-ABLp210 ter-/- (n=17) experimental mice together with Sca1-BCR-ABLp210 ter+/+ (n=21) controls. Mice were monitored clinically and by serial peripheral blood count for evidence of CML for 20 months. As described [7], all Sca1-BCR-ABLp210 ter+/+ mice develop CML (Table I). Surprisingly, when the compound Sca1-BCR-ABLp210 telomerase-deficient mice were analysed, CML was not found in the majority of them and the survival of these Sca1-BCR-ABLp210 ter+/- and Sca1-BCR-ABLp210 ter-/- mice was significantly increased in comparison with Sca1-BCR-ABLp210 ter+/+ (Table I, Figure 1). Histologic analysis revealed only similar pathology to Sca1-BCR-ABLp210 ter+/+ mice in 4 moribund Sca1-BCR-ABLp210 ter+/- and 3 moribund Sca1-BCR-ABLp210 ter+/+ mice, respectively (Figure 2). On the contrary, the majority of old Sca1-BCR-ABLp210 ter+/- and Sca1-BCR-ABLp210 ter-/- mice do not develop CML as evidenced by the normal spleen sizes and normal leukocyte cellularity in the peripheral blood. The absence of CML disease was further confirmed by histologic analysis that revealed normal spleen in majority old Sca1-BCR-ABLp210 ter+/- and Sca1-BCR-ABLp210 ter-/- where we cannot detect the dramatic expansion of progenitors and differentiated myeloid cells that is characteristic of CML (Figure 1). Quantitative RT-PCR of BCR-ABLp210 messenger mRNA confirmed that BCR-ABL was expressed in telomerase-deficient hematopoietic stem cells (Figure 3). These results indicate that the reduction in telomerase dosage in hematopoietic stem cells does interfere with the development of CML induced by BCR-ABL.
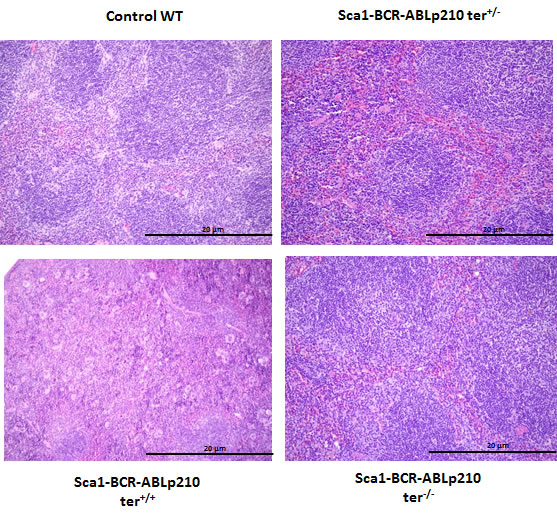
Figure 1: Telomerase is required for CML development in Sca1-BCR-ABLp210 mice. Representative histologic appearance of spleen of old Sca1-BCR-ABLp210 ter+/-, Sca1-BCR-ABLp210 ter-/-, Sca1-BCR-ABLp210 ter+/+ and control wild-type mice after hematoxylin-eosin staining. Note the organ infiltration by myeloid cells in spleen and the presence of blasts and mature myeloid cells in Sca1-BCR-ABLp210 ter+/+mice.
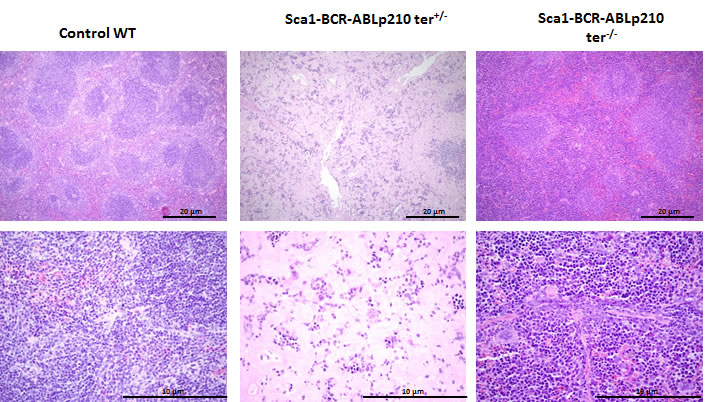
Figure 2: Some Sca1-BCR-ABLp210 ter+/- and Sca1-BCR-ABLp210 ter-/- mice develop CML. Representative histologic appearance of spleen of old Sca1-BCR-ABLp210 ter+/- Sca1-BCR-ABLp210 ter-/-, and control wild-type mice after hematoxylin-eosin staining. Note the organ infiltration by myeloid (blasts and mature) cells in spleen.
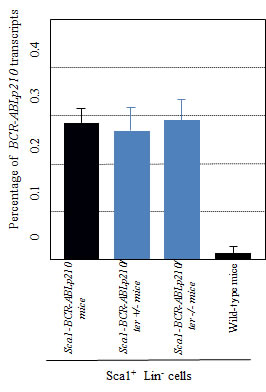
Figure 3: Quantification of Sca1-CR-ABLp210 expression in Sca1+Lin- cells of Sca1-BCR-ABLp210 ter+/- and Sca1-BCR-ABLp210 ter-/- mice. Quantification of BCR-ABLp210 expression in Sca1-BCRABLp210 ter+/- and Sca1-BCRABLp210 ter-/- mice by real-time PCR in Sca1+Lin- cells. Percentage of BCRABLp210 transcripts with reference to βactin is shown.
Table I: Incidence of CML in Sca1-BCR-ABLp210 ter+/- and ter-/- mice.
Sca1-BCR-ABLp210 ter+/- |
Sca1-BCR-ABLp210 ter-/- |
Sca1-BCR-ABLp210 ter+/+ |
|
Mice with CML |
4 |
3 |
21 |
Mice without CML |
13 |
14 |
0 |
Total mice |
n=17 |
n=17 |
n=21 |
Overall, our work demonstrates for the first time that telomerase is required for the generation and maintenance of LSCs. These results demonstrate that telomerase is essential for oncogene-induced reprogramming of hematopoietic stem cells in CML development and validate telomerase and the genes it regulates as targets for therapy in CML. This evidence that telomerase inactivation could modify LSC indicates that the reduction/absence of telomerase does not have an instructive role in the genesis of tumor CML cells, but just a permissive one, preventing cells with damage from being successfully terminally reprogrammed. This indicates that the driving force of the reprogramming process is the BCR-ABL oncogene itself. However, targeting of BCR-ABL does not affect CML stem cells. The search for ways to eliminate leukemic stem cells has became a priority due to their resistance to kinase inhibitors and the likely role they play in disease recurrence [29]. A number of strategies have yielded promissing results. Our results add the telomerase pathway as a potential therapeutic target whose inactivation can lead to CML stem cell eradication.
Acknowledgments
We thank all members of Lab 7 and L13 at IBMCC for their helpful comments and constructive discussions on this project. We are grateful to Dr. M. Blasco for the telomerase deficient mice. Research in ISG group was partially supported by FEDER and by MICINN (SAF2009-08803 to ISG), by Junta de Castilla y León (REF. CSI007A11-2 and Proyecto Biomedicina 2009-2010), by MEC OncoBIO Consolider-Ingenio 2010 (Ref. CSD2007-0017), by NIH grant (R01 CA109335-04A1), by Sandra Ibarra Foundation, and the ARIMMORA project (FP7-ENV-2011, European Union Seventh Framework Program) and by Proyecto en Red de Investigación en Celulas Madre Tumorales en Cancer de Mama, supported by Obra Social Kutxa y Conserjería de Sanidad de la Junta de Castilla y Leon. All Spanish funding is co-sponsored by the European Union FEDER program. ISG is an API lab of the EuroSyStem project.
MATERIALS AND METHODS
Ethics statement
All animal work has been conducted according to relevant national and international guidelines and it has been approved by the Bioethics Subcommittee of Consejo Superior de Investigaciones Cientificas (CSIC).
Mice
The Sca1-BCR-ABLp210 mice [7] and terc gene targeted mice [3] have both been described previously. Southern blot-based genotyping for Sca1-BCR-ABLp210 [7, 30] and terc [3], respectively, were performed as described. Heterozygous ter+/- mice were bred to Sca1-BCR-ABLp210 mice to generate compound heterozygotes. F1 animals were crossed to obtain null ter-/- mice heterozygous for Sca1-BCR-ABLp210 mice. Tumor phenotype was assessed in the first generation of telomerase heterozygous and homozygous mice.
Histological analysis
All mice included in this study were subjected to standard necropsy. All major organs were examined under the dissecting microscope, and samples of each organ were processed into paraffin, sectioned and examined histologically. All tissue samples were taken by the pathologist from homogenous and viable portions of the resected sample and fixed within 2-5 min. of excision. Hematoxylin- and eosin-stained sections of each tissue were reviewed by a single pathologist (T.F.). For comparative studies, age-matched mice were used.
Real-time PCR quantification
cDNA for use in quantitative PCR studies was synthesized using reverse transcriptase (Access RT-PCR System; Promega, Madison, WI). Two µl of second round amplified RNA was transcribed. Primers and probes used for quantitative PCR are commercially available (TaqMan Assays-on-Demand Gene Expression Products, Applied Biosystems, Foster City, CA). In addition the probes were designed so that genomic DNA would not be detected during the PCR. The sequences of the specific primers and probes were as follow: BCR-ABLp210, sense primer 5’-TTCTGAATGTCATCGTCCACTCA-3’, antisense primer 5’-AGATGCTACTGGCCGCTGA-3’ and probe 5´-CCACTGGATTTAAGCAGAGTTCAAAAGCCC-3’; c-Abl, sense primer 5’-CACTCTCAGCATCACTAAAGGTGAA-3’, antisense primer 5’-CGTTTGGGCTTCACACCATT-3’, and probe 5’-CCGGGTCTTGGGTTATAATCACAATG-3’.
Analysis and monitoring of disease
Peripheral blood was collected from retro-orbital plexus with a heparinized capillary tube, and total white blood cell and differential counts were performed twice a week. The number of white blood cells was determined with a hemocytometer after lysis of enucleated red blood cells with RCLB lysis buffer (0.15 M NH4Cl; 1 mM KHCO3; 0.1 mM Na2-EDTA, pH 7.4).
Flow cytometry
Nucleated cells were obtained from total bone marrow (flushing from the long bones), peripheral blood, thymus, liver and spleen. In order to prepare cells for flow cytometry, contaminating red blood cells were lyzed with RCLB lysis buffer and the remaining cells were then washed in PBS with 2% FCS. After staining, all cells were washed once in PBS with 2% FCS containing 2 mg/mL propidium iodide (PI) to allow dead cells to be excluded from both analyses and sorting procedures. Monoclonal antibodies were obtained from Pharmingen and included: Lineage markers (CD45R/B220, for B lineage staining; CD4, CD8 and CD3 for T cell lineage; CD11b and Gr1 for myeloid lineage and TER119 for erythroid lineage) and Sca1 (E13-161.7) for stem cells. Single cell suspensions from the different tissue samples obtained by routine techniques were incubated first with purified anti-mouse CD32/CD16 (Pharmingen) prior to the addition of other antibodies, to block binding via Fc receptors and then with an appropriate dilution of the different antibodies at room temperature or 4 oC, respectively. The samples and the data were analysed in a FACSCalibur using CellQuest software (Becton Dickinson). Specific fluorescence of FITC and PE excited at 488 nm (0.4 W) and 633 nm (30 mW), respectively, as well as known forward and orthogonal light scattering properties of mouse cells were used to establish gates. For each analysis a total of at least 5.000 viable (PI-) cells were assessed.
Cell purification
For cell sorter separation, bone marrow cells were incubated with anti-Sca1 and anti-lineage markers antibodies (CD3, CD4, CD8, B220, TER119, Gr1, and Mac1). Sca1+Lin- and Sca1-Lin+ cells were isolated and highly purified from the BM of leukemic primary mice or control mice by fluorescence-activated cell sorting (FACS) (FACSVANTAGE; Becton Dickinson). c-kit (CD117) was not used for stem cell isolation as previous studies of human and mouse specimens have described down-regulation of c-kit as a feature of leukemia stem cells [7]. Sorted cells were then re-analyzed for purity with the FACS and determined to be over 98%.
Statistical analysis
The X2 test was used to compare leukemia incidence in Sca1-BCR-ABLp210 ter-/- mice and Sca1-BCR-ABLp210 ter+/- versus Sca1-BCR-ABLp210 and wild-type mice.
References
1. Greaves M. Is telomerase activity in cancer due to selection of stem cells and differentiation arrest? Trends in genetics : TIG. 1996; 12: 127-128.
2. Harley CB, Futcher AB,Greider CW. Telomeres shorten during ageing of human fibroblasts. Nature. 1990; 345: 458-460.
3. Blasco MA, Lee HW, Hande MP, Samper E, Lansdorp PM, DePinho RA,Greider CW. Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell. 1997; 91: 25-34.
4. Lee HW, Blasco MA, Gottlieb GJ, Horner JW, 2nd, Greider CW,DePinho RA. Essential role of mouse telomerase in highly proliferative organs. Nature. 1998; 392: 569-574.
5. Rudolph KL, Chang S, Lee HW, Blasco M, Gottlieb GJ, Greider C,DePinho RA. Longevity, stress response, and cancer in aging telomerase-deficient mice. Cell. 1999; 96: 701-712.
6. Feldser DM,Greider CW. Short telomeres limit tumor progression in vivo by inducing senescence. Cancer cell. 2007; 11: 461-469.
7. Perez-Caro M, Cobaleda C, Gonzalez-Herrero I, Vicente-Duenas C, Bermejo-Rodriguez C, Sanchez-Beato M, Orfao A, Pintado B, Flores T, Sanchez-Martin M, Jimenez R, Piris MA,Sanchez-Garcia I. Cancer induction by restriction of oncogene expression to the stem cell compartment. Embo J. 2009; 28: 8-20.
8. Vicente-Duenas C, Perez-Caro M, Abollo-Jimenez F, Cobaleda C,Sanchez-Garcia I. Stem-cell driven cancer: “hands-off” regulation of cancer development. Cell Cycle. 2009; 8: 1314-1318.
9. Sanchez-Garcia I. The crossroads of oncogenesis and metastasis. The New England journal of medicine. 2009; 360: 297-299.
10. Sanchez-Garcia I. Getting to the stem of cancer. Seminars in cancer biology. 2010; 20: 63-64.
11. Castellanos A, Vicente-Duenas C, Campos-Sanchez E, Cruz JJ, Garcia-Criado FJ, Garcia-Cenador MB, Lazo PA, Perez-Losada J,Sanchez-Garcia I. Cancer as a reprogramming-like disease: implications in tumor development and treatment. Semin Cancer Biol. 2010; 20: 93-97.
12. Gonzalez-Herrero I, Vicente-Duenas C, Orfao A, Flores T, Jimenez R, Cobaleda C,Sanchez-Garcia I. Bcl2 is not required for the development and maintenance of leukemia stem cells in mice. Carcinogenesis. 2010; 31: 1292-1297.
13. Graham SM, Jorgensen HG, Allan E, Pearson C, Alcorn MJ, Richmond L,Holyoake TL. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood. 2002; 99: 319-325.
14. Cortes J, O’Brien S,Kantarjian H. Discontinuation of imatinib therapy after achieving a molecular response. Blood. 2004; 104: 2204-2205.
15. Deininger M. Resistance to imatinib: mechanisms and management. Journal of the National Comprehensive Cancer Network : JNCCN. 2005; 3: 757-768.
16. Hu Y, Swerdlow S, Duffy TM, Weinmann R, Lee FY,Li S. Targeting multiple kinase pathways in leukemic progenitors and stem cells is essential for improved treatment of Ph+ leukemia in mice. Proceedings of the National Academy of Sciences of the United States of America. 2006; 103: 16870-16875.
17. Jiang X, Zhao Y, Smith C, Gasparetto M, Turhan A, Eaves A,Eaves C. Chronic myeloid leukemia stem cells possess multiple unique features of resistance to BCR-ABL targeted therapies. Leukemia : official journal of the Leukemia Society of America, Leukemia Research Fund, U.K. 2007; 21: 926-935.
18. Kuroda J, Puthalakath H, Cragg MS, Kelly PN, Bouillet P, Huang DC, Kimura S, Ottmann OG, Druker BJ, Villunger A, Roberts AW,Strasser A. Bim and Bad mediate imatinib-induced killing of Bcr/Abl+ leukemic cells, and resistance due to their loss is overcome by a BH3 mimetic. Proceedings of the National Academy of Sciences of the United States of America. 2006; 103: 14907-14912.
19. Kaufmann SH. Imatinib spells BAD news for Bcr/abl-positive leukemias. Proceedings of the National Academy of Sciences of the United States of America. 2006; 103: 14651-14652.
20. Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, Lydon NB, Kantarjian H, Capdeville R, Ohno-Jones S,Sawyers CL. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. The New England journal of medicine. 2001; 344: 1031-1037.
21. le Coutre P, Mologni L, Cleris L, Marchesi E, Buchdunger E, Giardini R, Formelli F,Gambacorti-Passerini C. In vivo eradication of human BCR/ABL-positive leukemia cells with an ABL kinase inhibitor. Journal of the National Cancer Institute. 1999; 91: 163-168.
22. Perez-Caro M, Gutierrez-Cianca N, Gonzalez-Herrero I, Lopez-Hernandez I, Flores T, Orfao A, Sanchez-Martin M, Gutierrez-Adan A, Pintado B,Sanchez-Garcia I. Sustained leukaemic phenotype after inactivation of BCR-ABLp190 in mice. Oncogene. 2007; 26: 1702-1713.
23. Koeffler HP,Golde DW. Chronic myelogenous leukemia--new concepts (first of two parts). The New England journal of medicine. 1981; 304: 1201-1209.
24. Melo JV,Barnes DJ. Chronic myeloid leukaemia as a model of disease evolution in human cancer. Nature reviews. Cancer. 2007; 7: 441-453.
25. Fodde R. The stem of cancer. Cancer cell. 2009; 15: 87-89.
26. Nerlov C. Targeting a chronic problem: elimination of cancer stem cells in CML. The EMBO journal. 2009; 28: 167-168.
27. Corbin AS, Agarwal A, Loriaux M, Cortes J, Deininger MW,Druker BJ. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. The Journal of clinical investigation. 2011; 121: 396-409.
28. Hamilton A, Helgason GV, Schemionek M, Zhang B, Myssina S, Allan EK, Nicolini FE, Muller-Tidow C, Bhatia R, Brunton VG, Koschmieder S,Holyoake TL. Chronic myeloid leukemia stem cells are not dependent on Bcr-Abl kinase activity for their survival. Blood. 2012; 119: 1501-1510.
29. Donato NJ,Peterson LF. Chronic myeloid leukemia stem cells and developing therapies. Leukemia & lymphoma. 2011; 52 Suppl 1: 60-80.
30. Vicente-Duenas C, Abollo-Jimenez F, Ruiz-Roca L, Alonso-Escudero E, Jimenez R, Cenador MB, Criado FJ, Cobaleda C,Sanchez-Garcia I. The age of the target cell affects B-cell leukaemia malignancy. Aging (Albany NY). 2010: