
Introduction
Glioblastomas (GBMs) are the most common and deadly type of malignant gliomas. These tumors are highly aggressive, diffusively infiltrative, and exhibit a high degree of intra- and intertumor heterogeneity [1-4]. GBMs are characterized as Proneural (PN), Neural (N), Classical (C) or Mesenchymal (Mes) in nature, based on chromosomal number variations (CNV) [5, 6], gene expression patterns [1, 2] and abnormalities in DNA methylation [3]. However, most GBM contain more than one subtype [3], suggesting subtypes may evolve from one type into another [4, 7].
NF-κB, a family of transcription factors, mediates immune and inflammatory signaling [8]. Classical NF-κB is a cytoplasmic inactive heterodimer of p65 and p50 bound to Inhibitor of NF-κB (IκB) proteins [9]. NF-κB is activated by TNF-α and other proinflammatory cytokines. TNF-α binds to TNF Receptor 1 (TNFR1), which causes the receptor to oligomerize and recruit TNF receptor associated death domain (TRADD). TRADD recruits additional proteins that indirectly activate IκB kinase (IKK). IKK phosphorylates IκB, which promotes its degradation. Once released from IκB, NF-κB migrates to the nucleus and activates numerous target genes [9]. While NF-κB is tightly regulated, in many cancers including GBMs, NF-κB is constitutively activated, its target genes are overexpressed, and these events promote the formation and/or progression of cancer [10, 11]. As such, it is important to understand the role of NF-κB signaling in GBM.
MicroRNAs (miRs) are short, endogenous, single-stranded RNAs that inhibit gene expression [12]. MiRs are often dysregulated in cancer, which contributes to tumor development and/or progression by disrupting gene expression [13, 14]. The MIR31HG gene, which encodes miR-31, lies adjacent to the CDKN2A/B locus, the most frequently deleted loci in GBM [5, 6]. MiR-31 expression is reduced or absent in many cancers including ovarian, breast, osteosarcoma, prostate and adult T cell leukemia [15]. In GBM, the significance of miR-31 remains poorly characterized. Herein, we demonstrate MIR31HG is deleted in most GBM, inhibits TRADD expression, and limits NF-κB activity. Reconstitution of miR-31 inhibits the migration and proliferation of GBM cells, while loss of miR-31 significantly enhances NF-κB activity and increases tumor growth. In patients with GBM, the loss of miR-31 correlates with reduced survival times. Our data identify miR-31 as a novel non-coding tumor suppressor in GBM.
Results
MicroRNA-31 is deleted in glioblastoma
We analyzed all cancers in the TCGA repository for status of MIR31HG CNVs. MIR31HG is deleted in numerous cancers but deletions occur most frequently in GBM, exceeding 73% of all GBMs (Figure 1A). In GBM, 30.92% were homozygous null (-/-), 42.68% were heterozygous (+/-) and 26.40% were wildtype for miR-31 (+/+) (Figure 1B). Loss of one or both copies of MIR31HG significantly reduced the levels of miR-31 compared to tumors with no MIR31HG deletions (Figure 1B, blue asterisks; Supp. Figure 1A), indicating loss of one or both copies of MIR31HG is sufficient to diminish miR-31 levels in GBM. In low grade gliomas (LGG), 6.96% were MIR31HG-/-, 27.04% were MIR31HG+/-, and 66.00% were MIR31HG+/+ (Figure 1B). Chi-square analyses indicated homozygous MIR31HG deletions predominantly associated with Mes- and C-GBMs (Figure 1C, blue asterisks).
MIR31HG lies adjacent to CDKN2B, CDKN2A and MTAP, and the IFNA loci at 9p21.3 (Figure 1D). CDKN2A is the most frequently deleted gene in GBM [16]. We found most 9p21.3 deletions (71.5%) in GBM spanned CDKN2B, CDKN2A, MTAP, MIR31HG and the IFNA loci (Figure 1D). Focal deletions of 9p21.3 were present but rare (<8% of GBM). One GBM exhibited homozygous deletion of MIR31HG without deletions in CDKN2A/B (TCGA-27-1832-01) (Supp. Figures 1B, 1C). Nearly 21% of all GBM harbor no deletions at 9p21.3. We also found miR-31 expression was absent in established human GBM cell lines (Supp. Figure 1D, indicated by “-“) and human GBM xenografts (Supp. Figure 1E, indicated by “-“).
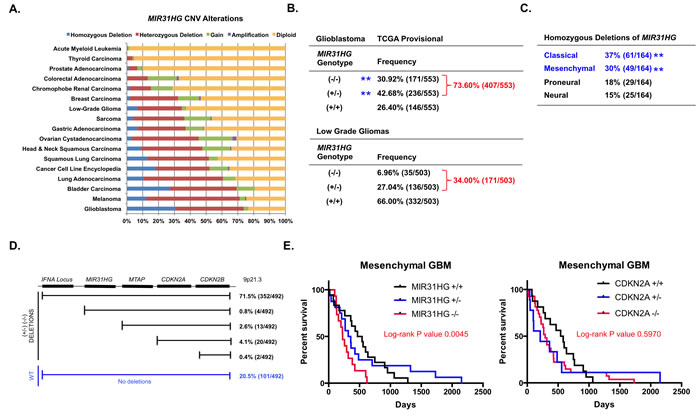
Figure 1: MIR31HG is deleted in GBM. A. MIR31HG CNVs from non-embargoed cancers in the TCGA. B. Frequency of MIR31HG deletions in Glioblastoma and Low Grade Gliomas (LGG). Blue asterisks indicate that the levels of MIR31HG are significantly reduced compared to MIR31HG+/+ tumors. (**, p < 0.005). C. Homozygous MIR31HG deletions stratified by GBM subtype. Blue indicates significance when compared to PN and N subtypes. D. Diagram of chromosome 9p21.3. Indicated below are the frequencies the indicated region is deleted in GBM. E. Kaplan-Meier survival curve for Mes-GBM stratified by MIR31HG (left) or CDKN2A (right) genotypes.
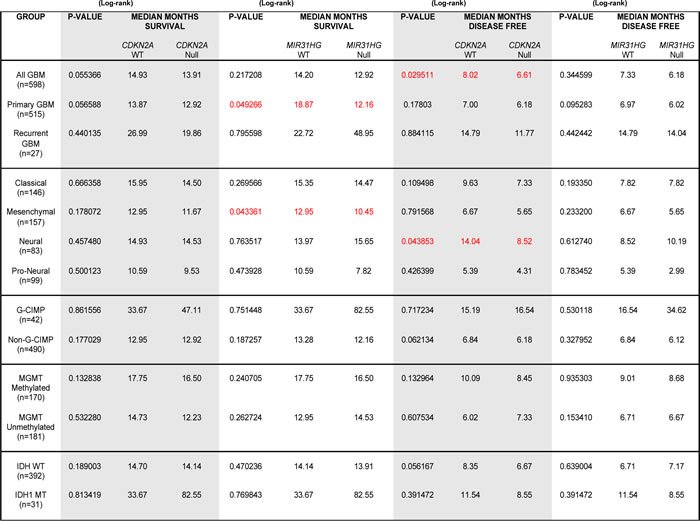
MIR31HG deletions predict poor prognosis in human GBM
We used the Mantel-Cox test to assess the significance of homozygous deletions of CDKN2A or MIR31HG on overall patient survival (Median Months Survival (MMS), left 2 columns) and Disease Free Survival (DFS) (median months disease free, right two columns) in GBM (). CDKN2A deletions were associated with diminished DFS times in all GBM, and patients with N-GBM. MIR31HG deletions predicted shorter MMS in patients with primary GBM (Supp. Figure, Table I) and patients with Mes-GBM (Figure 1E, left). Importantly, deletions in CDKN2A, which lies adjacent to MIR31HG, did not predict shorter MMS in patients with Mes-GBM (Figure 1E, right) or primary GBM (Supp. Figure, Table I). MIR31HG deletions were not correlated with diminished DFS (Supp. Figure, Table I).
Table I: Patient data from GBMs stratified according to CDKN2A or MIR31HG status.
MiR-31 expression inversely correlates with TRADD expression
In GBM, TRADD is overexpressed (Supp. Figure 2) and NF-κB is constitutively activated [10, 11, 17-19], particularly in Mes-GBM [3]. We identified a consensus miR-31 binding element within the TRADD 3’ UTR (Figure 2A). In established malignant glioma cells, TRADD levels were inversely correlated with miR-31 status (Figure 2B). We used the luciferase reporter assay to assess the validity of the TRADD miR-31 element. U87 miR-31 (-) cells were transfected with negative control miRNA (Neg CT) or miR-31 for 48 h and then transfected with TRADD-Luc, which contains the TRADD 3’ UTR, or a control plasmid (CT-Luc) for 24 h. MiR-31 significantly inhibited luciferase activity in cells transfected with TRADD-Luc, but not in cells transfected with CT-Luc (Figure 2C). AntagomiRs are chemically modified oligonucleotides that bind to and inhibit the functions of miRs [20]. U251 miR-31 (+) cells were transfected with Neg CT or antagomiR-31 for 48 h and the plasmids described above. AntagomiR-31 significantly increased luciferase activity in cells containing TRADD-Luc, but not in cells containing CT-Luc (Figure 2D). These data indicate that the TRADD miR-31 element is sensitive to miR-31.
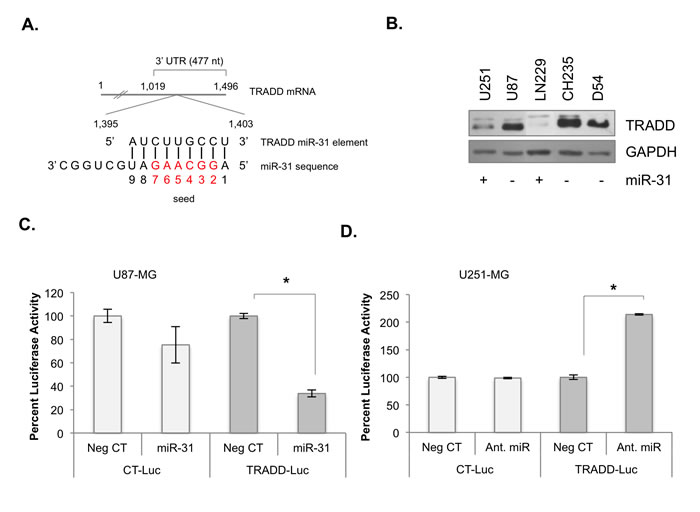
Figure 2: TRADD is a target of miR-31. A. Diagram depicting TRADD mRNA, and its putative miR-31 seed sequence (red). B. The levels of miR-31 and TRADD protein in human glioma cell lines of known miR-31 status. C. U87-MG cells were transfected with luciferase expression plasmid containing a control 3’ UTR (CT-Luc) or the 3’ UTR of TRADD (TRADD-Luc), and either control miR (Neg CT) or miR-31, and luciferase activity was analyzed. (*, p < 0.05). D. U251-MG cells were transfected with either CT-Luc or TRADD-Luc, and then with either Neg CT or antagomiR-31. Luciferase activity was analyzed. (*, p < 0.05).
MiR-31 regulated TRADD inhibits NF-κB signaling
Next, we assessed the impact of miR-31 on TRADD expression and NF-κB activation. We found U87-MG miR-31 (-) cells expressed significantly more TRADD RNA and protein than U251-MG miR-31 (+) cells, and this was not altered by TNF-α stimulation (Supp. Figures 3A-3C). Additionally, the levels of activated NF-κB (Supp. Figure 3D), and its target genes IκBα (Supp. Figure 3E), IL-6, IL-8, A20, SOCS3 and c-Myc (Supp. Figure 3F) were significantly greater in U87 cells compared to U251 cells. Herein, we chose to use the IL-6 gene as a reporter of miR-31 mediated effects on TRADD/NF-κB signaling. As shown in Supp. Figures. 3G and 3H, U87-MG cells expressed higher levels of IL-6 mRNA and protein in response to TNF-α stimulation compared to U251-MG cells.
Next, U87-MG cells were transfected with either a control miRNA (CT) or a miR-31 mimic (miR-31). MiR-31 reduced the levels of TRADD mRNA and protein (Figure 3A), and IL-6 mRNA and protein (Figure 3B, 3C, 3D). We confirmed miR-31 inhibited E2F2 (Supp. Figure 4), a validated target of miR-31 [21, 22]. Above, our data suggest that miR-31 inhibits TRADD and consequently NF-κB signaling. To test this, we assessed whether TRADD overexpression would rescue NF-κB signaling. U87 cells were first transfected with CT or miR-31 for 48 h. Next, cells were transfected with either a control plasmid (CT) or a plasmid encoding TRADD. As the TRADD plasmid lacks the 3’ UTR, it restores TRADD expression but is insensitive to miR-31. We found miR-31 reduced the levels of IL-6, but not if TRADD were co-expressed (Figure 3F). These data were also confirmed in U251-MG cells (Supp. Figure 5) and in patient-derived GBM xenograft cells (PDGXs). X1016 miR-31 (+) cells transfected with miR-31 antagomiR displayed higher levels of TRADD mRNA (Figure 4A) and protein (Figure 4B) than did X1016 cells transfected with CT miRNA. Finally, the levels of the NF-κB regulated genes IL-6 (Figure 4C), A20 (Figure 4D), IκBα (Figure 4E) and IL-8 (Figure 4F) were higher in cells with miR-31 antagomiR compared to cells transfected with CT miRNA. These data confirm that miR-31 limits NF-κB signaling by regulating TRADD expression.
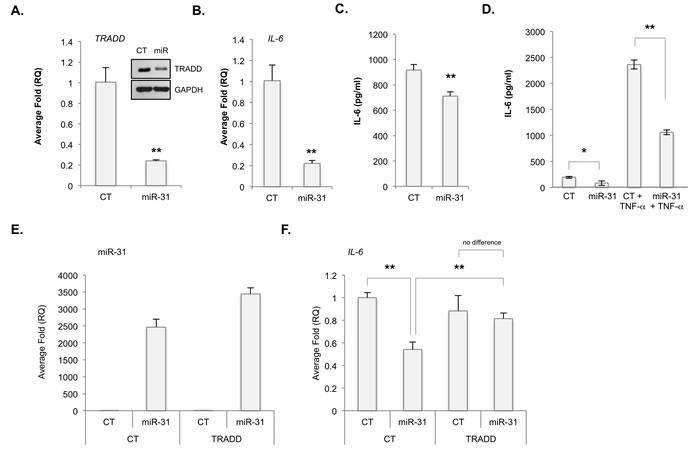
Figure 3: MiR-31 inhibits TRADD expression and NF-κB-induced gene expression. A.-D. U87-MG cells were transfected with control (CT) miR or miR-31. A. TRADD mRNA and protein levels were evaluated by qRT-PCR and immunoblot analyses, respectively. (**, p < 0.005). B., C. The levels of IL-6 mRNA B. and protein C. in U87-MG cells as described in A., were evaluated by qRT-PCR and ELISA, respectively. (**, p < 0.005). D. U87-MG cells were transfected with CT or miR-31, and grown in the absence or presence of TNF-α (10 ng/ml). IL-6 protein levels were evaluated by ELISA. (*, p < 0.05; **, p < 0.005). E., F. U87-MG cells were transfected with CT or miR-31, and either a control plasmid (CT) or a plasmid expressing TRADD, but lacking the 3’ UTR. MiR-31 E. and IL-6 F. RNA levels were measured by qRT-PCR. (**, p < 0.005).
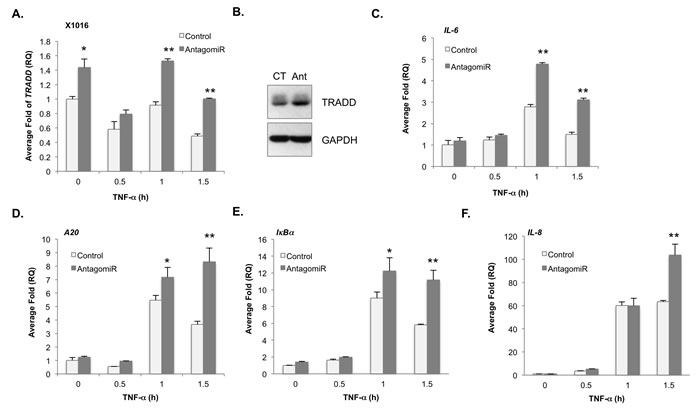
Figure 4: Loss of miR-31 enhances TRADD and NF-κB target gene expression in human GBM xenografts. A., B. Human GBM xenograft X1016 was transfected with control or AntagomiR-31 (AntagomiR) and the levels of TRADD mRNA A. and protein B. evaluated by qRT-PCR or immunoblotting. (*, p < 0.05; **, p < 0.005). C.- F. The levels of IL-6 mRNA C., A20 mRNA D., IκBα mRNA E. and IL-8 mRNA F. in X1016 cells transfected as described in (A) were evaluated by qRT-PCR. (*, p < 0.05; **, p < 0.005).
MIR31HG and NFKBIA deletions are mutually exclusive in GBM
NF-κB is constitutively activated in GBM [10, 11, 17, 18]. Mono-allelic deletions in NFKBIA, which encodes an inhibitor of NF-κB, are predominantly found in Non-C-GBM[11]. We found MIR31HG deletions also enhanced NF-κB activation (Figures 3, 4), but this event predominated in C- and Mes-GBM (Figure 1C). Therefore we reasoned that NFKBIA and MIR31HG deletions might stratify to distinct GBM subtypes. We found NFKBIA deletions were mutually exclusive with MIR31HG deletions (p = 0.0029) (Supp. Figure 6A), but not with deletions of a gene adjacent to MIR31HG (CDKN2A) (Supp. Figure 6B). These data suggest that there may be selective pressure to delete MIR31HG in GBMs that do not harbor mono-allelic NFKBIA deletions as these CNVs may be functionally redundant with respect to NF-κB activation.
NF-κB induces miR-31
Because NF-κB regulates IκBα, its own inhibitor, we hypothesized that NF-κB also regulates miR-31 expression. Indeed, the MIR31HG promoter [21, 23] contains three putative NF-κB binding sites (Figure 5A). To assess whether NF-κB induces miR-31, normal murine astrocytes were grown in the absence or presence of TNF-α for various times and miR-31 levels were measured. In response to TNF-α stimulation, miR-31 expression levels were elevated and displayed an oscillation pattern consistent with NF-κB activation patterns[24, 25] (Figure 5B). These data were confirmed in human glioma cells where miR-31 levels were elevated in response to TNF-α stimulation (Figure 5C). We used chromatin immunoprecipitation (ChIP) assays to assess whether TNF-α induced miR-31 expression required NF-κB. In the absence of TNF-α, minimal NF-κB p65 binding was detected at the MIR31HG promoter (Figure 5D). After TNF-α stimulation, the levels of NF-κB p65 were significantly elevated, but only at the distal -1000 bp site, which contains an NF-κB response element. We also evaluated acetylated histone 4 (AcH4) and tri-methylated histone 3 at lysine 4 (H3K4Me3) levels as mark areas of active transcription [26]. In resting cells, the MIR31HG promoter exhibits low levels of AcH4 and H3K4Me3, but these levels were significantly increased upon TNF-α stimulation (Figure 5D). These data indicate that NF-κB induces the expression of miR-31, likely to limit its own activities.
MiR-31 inhibits the migration and growth of GBM cells in vitro
Next, we assessed the functional effects of miR-31 in glioma cells. We analyzed cell migration using U251-MG cells transfected with either CT or AntagomiR. Cells were grown to 100% confluence and then wounded (initial percent open area = ~30%) (Supp. Figure 7A, Initial). After 8 h, ~16% of the wound remained present in cells transfected with CT, while cells with reduced miR-31 expression displayed only ~4% of the wound at 8 h (Supp. Figure 7A, Final), indicating miR-31 inhibits cell migration. To evaluate the impact of miR-31 on GBM cell growth, we used the WST-1 assay on U87-MG cells transfected with CT or miR-31. Glioma cells expressing miR-31 displayed reduced cell growth compared to cells transfected with CT (Supp. Figure 7B).
MiR-31 inhibits GBM growth in vivo
To assess the impact of miR-31 on tumor growth, we used JX12 PDGXs, which are characterized as Classical GBM and are miR-31 (+). These cells were transfected with a control miRNA (CT) or antagomiR-31 ex vivo for 48 h, and then injected s.c. and tumor growth was monitored. JX12 cells with reduced miR-31 expression grew faster and larger than JX12 tumors expressing miR-31 (Figure 6A & 6B). Tumors with reduced miR-31 levels (AntagomiR) appeared heterogeneous, showed blood vessel (BV) formation (Figure 6B, 6H & 6E, α-CD31), and expressed higher levels of TRADD and activated NF-κB (P-p65) (Supp. Figure 8A). Additionally, tumors with antagomiR-31 showed elevated levels of genes associated with NF-κB activation (IL-6), angiogenesis (VEGF), proliferation (Cyclin D1) and Mesenchymal GBMs (TAZ, YKL40), and reduced levels of E-cadherin, which correlates with epithelial to mesenchymal transition (EMT) (Supp. Figure 8B). These data were confirmed using an intracranial GBM model, wherein the loss of miR-31 in Classical GBM correlated with a switch to a more Mesenchymal GBM profile (Supp. Figures 8C).
We repeated these experiments using stable U87 cells that express CT miR (-miR-31) or miR-31 (+miR-31) or U251 cells that express CT (+ miR-31) or miR-31 sponge, which inhibits miR-31 function (-miR-31). At the end of these experiments, cells with no or reduced miR-31 formed more tumors (3/5 in U87, 4/5 in U251) than cells with miR-31 (0/5 in U87, 2/5 in U251) (Supp. Figure 9A). Unfortunately, tumors formed at different rates, making parallel measurements difficult. However, at euthanasia, tumors with no or reduced miR-31 tended to be larger than tumors expressing miR-31 (Supp. Figure 9B). H&E analyses indicated that U251 cells with reduced miR-31 activity had increased cellularity and showed BV formation (Supp. Figure 9C, α-CD31).
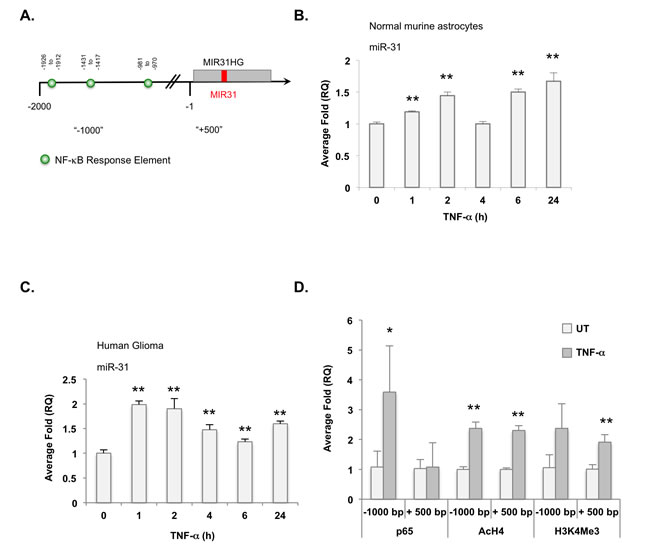
Figure 5: TNF-α activated NF-κB induces miR-31 expression. A. Diagram of the MIR31HG promoter and putative NF-κB response elements. Positions marked are relative to transcriptional start site. B., C. Normal murine astrocytes B. or U251-MG cells C. were stimulated with TNF-α (10 ng/ml) for the times indicated and miR-31 levels evaluated by qRT-PCR. (**, p < 0.005). D. U251-MG cells were left untreated (UT) or stimulated with TNF-α (10 ng/ml) for 4 h, and ChIP analyses were performed using the antibodies specified. Primers targeting regions upstream (“-1000”) and downstream of the TSS (“+500”) were used. (*, p < 0.05; **, p < 0.005).
MiR-31 prolongs survival in vivo in an intracranial GBM model
Finally, we evaluated the role of miR-31 on intracranial GBM growth in mice using stable U87-MG cells expressing CT (- miR-31) or miR-31 (+ miR-31). Within 30 days, all mice with (-) miR-31 cells were moribund and had to be euthanized (Figure 6C). However, mice receiving (+) miR-31 cells survived significantly longer (p = 0.008) (Figure 6C). These data indicate that restoring miR-31 expression inhibits GBM growth and prolongs survival in vivo. Upon resection, we assessed miR-31 levels (Supp. Figure 10A), and confirmed that tumors with miR-31 cells displayed lower levels of TRADD and activated P-p65 compared to tumors lacking miR-31 (Supp. Figure 10B). We also confirmed that miR-31 inhibited E2F2 (Supp. Figure 10C), which regulates cell growth and is inhibited by miR-31 [21, 22].
Discussion
Herein we characterize miR-31 as a novel tumor suppressor encoded by the MIR31HG gene. MIR31HG is dysregulated in many cancers but MIR31HG deletions are most profound in GBM (>73% all GBM). MIR31HG is situated at 9p21.3, adjacent to CDKN2A, and statistically, MIR31HG deletions coincide with CDKN2A deletions (p < 0.005). Most studies have focused on the consequences of CDKN2B and CDKN2A deletions, and rarely acknowledge the impact of other deletions at 9p21.3. However, we find nearly all deletions at 9p21.3 impact CDKN2B, CDKN2A, and MIR31HG. Using GBM cells and PDGXs, we find that altering the levels of endogenous miR-31 alone is sufficient to significantly alter GBM behavior in vitro and in vivo. Finally, we show that loss of MIR31HG predicts poor prognosis in patients with primary GBM and/or Mes-GBM. Based on our findings, we propose MIR31HG is a novel non-coding tumor suppressor gene in GBM (Figure 6D).
We believe miR-31 suppresses tumor formation, in part, by targeting TRADD to reduce NF-κB activation. Our studies focus on NF-κB for several reasons. NF-κB is a critical mediator of inflammation, which has been linked to all phases of tumor development. We and others show that NF-κB is constitutively activated in GBM and correlated with poor patient prognosis [10, 17, 27]. Additionally, NF-κB induces the expression of proteins (IL-6, PDGFB, EGFR, HGF), which activate pathways (STAT3, PDGFR, EGFR, MET) and processes (angiogenesis, migration, invasion) critical for GBM maintenance [28]. Finally, as Mes-GBM are characterized by elevated levels of TRADD and NF-κB activity [1, 2, 7, 29], the loss of miR-31 may explain, in part, the consequences of activated NF-κB in this GBM subtype. For example, NF-κB induces IL-6 expression to ensure the activation of STAT3 [30], a master regulator of Mesenchymal GBMs [31, 32]. However, we acknowledge that additional, previously identified targets of miR-31 (E2F2 and radixin) as well as the loss of additional genes at 9p21.3 also impact GBM growth [33-35].
In this study, we demonstrate that NF-κB induces miR-31 expression and that in turn, miR-31 inhibits the levels of TRADD, an upstream activator of NF-κB. Therefore, we propose that the degree of NF-κB activation is regulated, in part, by the relationship between TRADD/NF-κB and miR-31. Unfortunately in GBM, NF-κB is constitutively activated, perhaps as a consequence of loss of proper regulation. Indeed, MIR31HG and/or NFKBIA are frequently deleted, while ING4, which inhibits NF-κB, is mutated or absent [10, 11, 17], and Pin1, an enhancer of NF-κB activity, is often overexpressed [19]. Consequently, GBMs utilize numerous mechanisms to ensure constitutive NF-κB activation.
MicroRNA-mediated therapy to inhibit NF-κB in GBM is enticing for several reasons. In particular, miRs readily cross cell membranes [36] and the blood-brain barrier[37]. Moreover, miRs are not immunogenic, and because they lack poly-A tail, these molecules are fairly stable both in vitro and in vivo [38, 39], although they can be further modified to prolong their biological half-life [40]. Presently, numerous mechanisms to restore tumor suppressive miR expression are being considered, including cationic lipid complexes, magnetic lipid complexes and viral hosts [36, 41]. Therefore, we are hopeful that this study provides enticing data to support the use of miRs, specifically miR-31, for the treatment of GBM and other cancers.
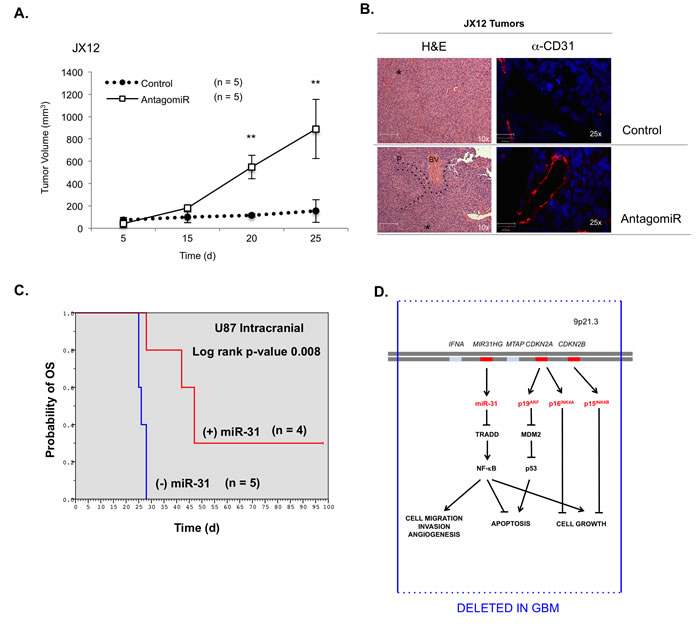
Figure 6: The levels of miR-31 inversely correlate with tumor growth in vivo. A. Human GBM xenograft JX12 was transfected with control or AntagomiR-31 (AntagomiR) and injected into the flanks of nude mice (n = 5) and volume measured. (**, p < 0.005). B. Mouse bearing JX12 miR-31 positive tumor (green circle) and JX12 miR-31 reduced tumor (red circle). Tumors resections were analyzed with H&E staining (x, y-scale of 42 μm). Blood vessel formation indicated with BV, and areas of necrosis are indicated with an (*). Blood vessels were stained using α-CD31 antibodies (x, y-scale of 42 μm). C. Stable U87-MG cells that express CT miR (-) or miR-31 (+) were injected intracranially, and survival times recorded as probability of overall survival (OS). D. Chr 9p21.3 is deleted in most GBM. This locus harbors three tumor suppressor genes (red lines), which encode four tumor suppressors (red type) in GBM: miR-31, p16INK4A, p19ARF and p15INK4B. Loss of these genes increases cell migration, invasion, angiogenesis and cell growth, and apoptosis is inhibited.
Materials and Methods
Malignant glioma cell lines and human GBM xenografts
Established human glioma cell lines were obtained from ATCC and maintained as previously described [10]. Patient derived GBM xenografts (PDGXs) were maintained as previously described [30]. The UAB Brain Tumor Animal Model Core Facility performed short tandem repeat DNA profiling on all human glioma cell lines and xenografts.
In silico data
Data was obtained from The Cancer Genome Atlas (TCGA) pilot project established by the NCI and NHGRI. Information about TCGA and the investigators and institutions who constitute the TCGA research network can be found at http://cancergenome.nih.gov/. Analyses were performed using cbioportal, Oncomine and microrna.org, and all references therein [42, 43]. The TCGA Provisional data set was analyzed using “miR-31/31” “MIR31HG” or “MIR31” as a search parameter.
Reagents
TNF-α was from R & D Systems. Secondary antibodies and enhanced chemiluminescence reagents were from Amersham. Anti-TRADD and anti-p65 antibodies were from Santa Cruz Biotechnology. Anti-phosphorylated p65 (p-p65) and anti-AcH3 antibodies were from Cell Signaling, and anti-GAPDH and anti-H3K4Me3 antibodies were from Abcam. Real time qRT primers and Taqman qRT-PCR reagents were from Applied Biosystems. Sybr green qRT-PCR reagents were from Qiagen. MicroRNA-31 mimic, miR-31 antagomiR and negative control (-CT) miR were purchased from Ambion/Life Technologies. MicroRNAs were transfected using Lipofectamine RNAiMax Transfection reagent (Life Technologies).
Scratch wound assay
Scratch wound assays were performed as previously described [19] and quantitated using Tscratch software [44].
In vivo tumor models, tumor fixation, processing and immunofluorescence
Tumor injections were performed as previously described [30, 45]. In s.c. flank experiments, JX12, U87-MG or U251-MG cells (5 x 105) were injected into the flanks of athymic nude mice. Tumors were measured on the indicated days using digital calipers and tumor volume was calculated using the following equation: v = (0.5 x longest diameter x shortest diameter2). In intracranial GBM experiments, U87 cells (5 x 105) or JX12 cells (3 x 105) in 5 µl of methylcellulose were injected 2 mm anterior and 1 mm lateral to bregma at a depth of 2 mm over 2 minutes as previously described [30, 45]. Mice were monitored for survival. After euthanasia, tumors were excised and snap frozen for RNA, miRNA, protein and/or IHC analyses.
Statistical analysis
Student t tests, Mann-Whitney rank sum tests, Mantel-Cox tests, and LogRank tests for Kaplan-Meier survival curves were used as previously described [11, 46]. p < 0.05 was considered statistically significant.
Ethics statement
In vivo experiments in female athymic nude mice were performed with the approval of UAB IACUC.
Acknowledgments
We thank Stephanie Robert, Cathy Langford, Dr. G. Yancey Gillespie and the UAB Brain Tumor Animal Models Core Facility (P20CA151129) for human GBM xenografts. Funding for this work was provided in part by grants from the National Institutes of Health (T32NS048039 to B.C.M., R01CA158534 to E.N.B., and R01CA138517 to S.E.N.); American Brain Tumor Association Basic Research Fellowship in Honor of Paul Fabbri to B.C.M. (www.abta.org); Southeastern Brain Tumor Foundation funding to E.N.B. (www.sbtf.org); and a Faculty Development Grant from the UAB Comprehensive Cancer Center to S.E.N. and M.B.
Conflicts of Interest
No potential conflicts of interest were disclosed by the authors.
Editorial note
This paper has been accepted based in part on peer-review conducted by another journal and the authors' response and revisions as well as expedited peer-review in Oncotarget.
References
1. Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, Misra A, Nigro JM, Colman H, Soroceanu L, Williams PM, Modrusan Z, Feuerstein BG and Aldape K. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006; 9:157-173.
2. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, Alexe G, Lawrence M, O’Kelly M, Tamayo P, Weir BA, Gabriel S, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010; 17:98-110.
3. Sottoriva A, Spiteri I, Piccirillo SG, Touloumis A, Collins VP, Marioni JC, Curtis C, Watts C and Tavare S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc Natl Acad Sci U S A. 2013; 110:4009-4014.
4. Ozawa T, Riester M, Cheng YK, Huse JT, Squatrito M, Helmy K, Charles N, Michor F and Holland EC. Most human non-GCIMP glioblastoma subtypes evolve from a common proneural-like precursor glioma. Cancer Cell. 2014; 26:288-300.
5. Cancer Genome Atlas Research N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008; 455:1061-1068.
6. Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, Olivi A, McLendon R, Rasheed BA, Keir S, Nikolskaya T, Nikolsky Y, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008; 321:1807-1812.
7. Li B, Senbabaoglu Y, Peng W, Yang ML, Xu J and Li JZ. Genomic estimates of aneuploid content in glioblastoma multiforme and improved classification. Clin Cancer Res. 2012; 18:5595-5605.
8. Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006; 441:431-436.
9. Ben-Neriah Y and Karin M. Inflammation meets cancer, with NF-kappaB as the matchmaker. Nat Immunol. 2011; 12:715-723.
10. Nozell S, Laver T, Moseley D, Nowoslawski L, De Vos M, Atkinson GP, Harrison K, Nabors LB and Benveniste EN. The ING4 tumor suppressor attenuates NF-kappaB activity at the promoters of target genes. Mol Cell Biol. 2008; 28:6632-6645.
11. Bredel M, Scholtens DM, Yadav AK, Alvarez AA, Renfrow JJ, Chandler JP, Yu IL, Carro MS, Dai F, Tagge MJ, Ferrarese R, Bredel C, Phillips HS, Lukac PJ, Robe PA, Weyerbrock A, et al. NFKBIA deletion in glioblastomas. N Engl J Med. 2011; 364:627-637.
12. Lujambio A and Lowe SW. The microcosmos of cancer. Nature. 2012; 482:347-355.
13. Calin GA, Sevignani C, Dumitru CD, Hyslop T, Noch E, Yendamuri S, Shimizu M, Rattan S, Bullrich F, Negrini M and Croce CM. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci U S A. 2004; 101:2999-3004.
14. Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, Aldler H, Rattan S, Keating M, Rai K, Rassenti L, Kipps T, Negrini M, Bullrich F and Croce CM. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 2002; 99:15524-15529.
15. Valastyan S and Weinberg RA. miR-31: a crucial overseer of tumor metastasis and other emerging roles. Cell Cycle. 2010; 9:2124-2129.
16. Ichimura K, Schmidt EE, Yamaguchi N, James CD and Collins VP. A common region of homozygous deletion in malignant human gliomas lies between the IFN alpha/omega gene cluster and the D9S171 locus. Cancer Res. 1994; 54:3127-3130.
17. Garkavtsev I, Kozin SV, Chernova O, Xu L, Winkler F, Brown E, Barnett GH and Jain RK. The candidate tumour suppressor protein ING4 regulates brain tumour growth and angiogenesis. Nature. 2004; 428:328-332.
18. Zhao X, Laver T, Hong SW, Twitty GB, Jr., Devos A, Devos M, Benveniste EN and Nozell SE. An NF-kappaB p65-cIAP2 link is necessary for mediating resistance to TNF-alpha induced cell death in gliomas. J Neurooncol. 2011; 102:367-381.
19. Atkinson GP, Nozell SE, Harrison DK, Stonecypher MS, Chen D and Benveniste EN. The prolyl isomerase Pin1 regulates the NF-kappaB signaling pathway and interleukin-8 expression in glioblastoma. Oncogene. 2009; 28:3735-3745.
20. Krutzfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M and Stoffel M. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. 2005; 438:685-689.
21. Lin PC, Chiu YL, Banerjee S, Park K, Mosquera JM, Giannopoulou E, Alves P, Tewari AK, Gerstein MB, Beltran H, Melnick AM, Elemento O, Demichelis F and Rubin MA. Epigenetic repression of miR-31 disrupts androgen receptor homeostasis and contributes to prostate cancer progression. Cancer Res. 2013; 73:1232-1244.
22. Li T, Luo W, Liu K, Lv X and Xi T. miR-31 promotes proliferation of colon cancer cells by targeting E2F2. Biotechnol Lett. 2014; 37:523-532.
23. Vire E, Curtis C, Davalos V, Git A, Robson S, Villanueva A, Vidal A, Aparicio S, Esteller M, Caldas C and Kouzarides T. The breast cancer oncogene EMSY represses transcription of antimetastatic microRNA miR-31. Mol Cell. 2014; 53:806-818.
24. Nelson DE, Ihekwaba AE, Elliott M, Johnson JR, Gibney CA, Foreman BE, Nelson G, See V, Horton CA, Spiller DG, Edwards SW, McDowell HP, Unitt JF, Sullivan E, Grimley R, Benson N, et al. Oscillations in NF-kappaB signaling control the dynamics of gene expression. Science. 2004; 306:704-708.
25. Ashall L, Horton CA, Nelson DE, Paszek P, Harper CV, Sillitoe K, Ryan S, Spiller DG, Unitt JF, Broomhead DS, Kell DB, Rand DA, See V and White MR. Pulsatile stimulation determines timing and specificity of NF-kappaB-dependent transcription. Science. 2009; 324:242-246.
26. Gu B and Lee M. Histone H3 lysine 4 methyltransferases and demethylases in self-renewal and differentiation of stem cells. Cell Biosci. 2013; 3:39.
27. Wang H, Wang H, Zhang W, Huang HJ, Liao WS and Fuller GN. Analysis of the activation status of Akt, NFkappaB, and Stat3 in human diffuse gliomas. Lab Invest. 2004; 84:941-951.
28. Nogueira L, Ruiz-Ontanon P, Vazquez-Barquero A, Lafarga M, Berciano MT, Aldaz B, Grande L, Casafont I, Segura V, Robles EF, Suarez D, Garcia LF, Martinez-Climent JA and Fernandez-Luna JL. Blockade of the NFkappaB pathway drives differentiating glioblastoma-initiating cells into senescence both in vitro and in vivo. Oncogene. 2011; 30:3537-3548.
29. Huse JT, Phillips HS and Brennan CW. Molecular subclassification of diffuse gliomas: seeing order in the chaos. Glia. 2011; 59:1190-1199.
30. McFarland BC, Hong SW, Rajbhandari R, Twitty GB, Jr., Gray GK, Yu H, Benveniste EN and Nozell SE. NF-kappaB-induced IL-6 ensures STAT3 activation and tumor aggressiveness in Glioblastoma. PLoS One. 2013; 8:e78728.
31. Carro MS, Lim WK, Alvarez MJ, Bollo RJ, Zhao X, Snyder EY, Sulman EP, Anne SL, Doetsch F, Colman H, Lasorella A, Aldape K, Califano A and Iavarone A. The transcriptional network for mesenchymal transformation of brain tumours. Nature. 2010; 463:318-325.
32. Bhat KP, Salazar KL, Balasubramaniyan V, Wani K, Heathcock L, Hollingsworth F, James JD, Gumin J, Diefes KL, Kim SH, Turski A, Azodi Y, Yang Y, Doucette T, Colman H, Sulman EP, et al. The transcriptional coactivator TAZ regulates mesenchymal differentiation in malignant glioma. Genes Dev. 2011; 25:2594-2609.
33. Wang S, Jiao B, Geng S, Song J, Liang Z and Lu S. Concomitant microRNA-31 downregulation and radixin upregulation predicts advanced tumor progression and unfavorable prognosis in patients with gliomas. J Neurol Sci. 2014; 338:71-76.
34. Hua D, Ding D, Han X, Zhang W, Zhao N, Foltz G, Lan Q, Huang Q and Lin B. Human miR-31 targets radixin and inhibits migration and invasion of glioma cells. Oncol Rep. 2012; 27:700-706.
35. Ballarino M, Cazzella V, D’Andrea D, Grassi L, Bisceglie L, Cipriano A, Santini T, Pinnaro C, Morlando M, Tramontano A and Bozzoni I. Novel long noncoding RNAs (lncRNAs) in myogenesis: a miR-31 overlapping lncRNA transcript controls myoblast differentiation. Mol Cell Biol. 2015; 35:728-736.
36. Boon RA and Vickers KC. Intercellular transport of microRNAs. Arterioscler Thromb Vasc Biol. 2013; 33:186-192.
37. Kouri FM, Hurley LA, Daniel WL, Day ES, Hua Y, Hao L, Peng CY, Merkel TJ, Queisser MA, Ritner C, Zhang H, James CD, Sznajder JI, Chin L, Giljohann DA, Kessler JA, et al. miR-182 integrates apoptosis, growth, and differentiation programs in glioblastoma. Genes Dev. 2015; 29:732-745.
38. Bail S, Swerdel M, Liu H, Jiao X, Goff LA, Hart RP and Kiledjian M. Differential regulation of microRNA stability. RNA. 2010; 16:1032-1039.
39. Bauman Y and Mandelboim O. MicroRNA based immunoevasion mechanism of human polyomaviruses. RNA Biol. 2011; 8:591-594.
40. Lennox KA, Owczarzy R, Thomas DM, Walder JA and Behlke MA. Improved performance of anti-miRNA oligonucleotides using a novel non-nucleotide modifier. Mol Ther Nucleic Acids. 2013; 2:e117.
41. Lai CP and Breakefield XO. Role of exosomes/microvesicles in the nervous system and use in emerging therapies. Front Physiol. 2012; 3:228.
42. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C and Schultz N. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012; 2:401-404.
43. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C and Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013; 6:pl1.
44. Geback T, Schulz MM, Koumoutsakos P and Detmar M. TScratch: a novel and simple software tool for automated analysis of monolayer wound healing assays. BioTechniques. 2009; 46:265-274.
45. McFarland BC, Ma JY, Langford CP, Gillespie GY, Yu H, Zheng Y, Nozell SE, Huszar D and Benveniste EN. Therapeutic potential of AZD1480 for the treatment of human glioblastoma. Mol Cancer Ther. 2011; 10:2384-2393.
46. Bredel M, Bredel C, Juric D, Duran GE, Yu RX, Harsh GR, Vogel H, Recht LD, Scheck AC and Sikic BI. Tumor necrosis factor-alpha-induced protein 3 as a putative regulator of nuclear factor-kappaB-mediated resistance to O6-alkylating agents in human glioblastomas. J Clin Oncol. 2006; 24:274-287.
47. Fleiss JL, Levin, B., Paik, M. Cho, Fleiss, J. (1981). Statistical Methods for Rates and Proportions (2nd Ed.). (New York: John Wiley & Sons).