
INTRODUCTION
MicroRNAs (miRNAs) represent a class of small non-coding single-stranded RNA molecules of 17–27-nucleotides in length that act as master regulators of gene expression post-transcriptionally by inhibiting the translation or inducing the degradation of target messenger RNAs (mRNAs) with partially complementary sites in the 3′-untranslated regions [1, 2]. Given these multiple roles, miRNAs aberrant expression and dysregulation often results in human diseases and cancer [3–9]. The first miRNAs related with cancer were miR-15a/miR-16-1 whose cluster is located in the chromosome region 13q, commonly deleted in chronic lymphocytic leukemia (CLL) [10]. The deletion was proposed as one of the primary genetic event in CLL, due to a decreased expression of the miR-15a/miR-16-1 targeting the BCL2 gene [10]. Other evidence of the association between miRNA dysregulation and cancer came from studies revealing that many miRNAs are effectively located in genomic regions frequently involved in chromosomal alterations, including breakpoint deletions or amplifications related to cancer [11, 12]. Overall considered, these studies indicate that the contribute of miRNAs to cancer pathogenesis is dependent of two opposite functions: either they can act as tumor suppressors, as in the case of the miR-15a/miR-16-1 cluster in CLL, or they can act as oncogenes, as it has been proposed for the members of the miR-17-92 cluster that is the topic of the present review [13].
In this review we first describe the miR-17-92 cluster locus along with the main mechanism(s) of expression regulation and the major molecular interactions of the miR-17-92 cluster in normal and neoplastic B cells. Then, we review the main clinical and pathogenetic implications of miR-17-92 cluster expression in lymphoproliferative disorders and the potential applications of anti-miRNAs based therapies targeting miRNAs belonging to the cluster.
The miR-17-92 chromosomal locus
The miR-17-92 polycistronic miRNA cluster is located in a region of 800 bp in the non-protein-coding gene C13orf25 at 13q31.3 (Figure 1A) [14]. The precursor transcript derived from the miR-17-92 gene (a.k.a. MIR17HG) contains six tandem-loop hairpin structures that ultimately yield the six mature miRNAs miR-17, miR-18a, miR-19a, miR-20a, miR-19b-1 and miR-92a-1 [14]. Moreover, complementary miRNAs derived from the opposite strands of each miR-17-92 pre-miRNA have been identified. The biological importance of the miR-17-92 cluster is also underlined by the presence of paralogs on chromosome X and chromosome 7, the miR-106a-363 cluster and the miR-106b-25 cluster, respectively, that both contains homologous miRNAs to a subset of miR-17-92 components (Figure 1B). Three separate miRNA families according to miRNA seed sequences have been defined: the miR-17 (that includes miR-17, miR-20a and miR-18a), miR-19 (that includes miR-19a and miR-19b-1), and miR-92 families (Figure 1C). Both miR-17 and miR-19 families are composed also by miRNAs belonging to the paralogs (Figure 1C). All these miRNAs derive from an unique gene that, during the early evolution of vertebrates, underwent a series of different dysregulations, such as duplications, mutations and losses [15].

Figure 1: The miR-17-92 cluster. A. Genomic localization of the miR-17-92 cluster (MIR17HG). The two transcripts of the gene are reported in blue. The miRNAs belonging to the cluster are reported in red. The panel of the figure was created modifying the output of the UCSC genome browser. B. Structure of the miR-17-92 cluster (MIR17HG, located on chromosome 13) and of the two paralogs (located on chromosomes X and 7). Both paralogs contains homologous miRNAs to a subset of miR-17-92 components. C. miRNA families of the miR-17-92 cluster. Four separate miRNA families according to miRNA seed sequences have been defined: the miR-17, miR-18, miR-19 and miR-92 families. miR-17, miR-18 and miR-19 families are composed also by miRNAs belonging to the paralogs. Seed sequences are reported in bold.
The six members of the miR-17-92 cluster can act independently and/or coordinately to target various mRNA, according to the degree of binding affinity and the seed sequences of the various members of the cluster (Figure 1C). As a strong oncogene, miR-17-92 regulates multiple cellular processes that favor malignant transformation promoting cell survival, rapid cell proliferation, and increased angiogenesis [16–19]. Given the oncogenic role of the miR-17-92 cluster, the primary transcript for these miRNAs was named ‘OncomiR-1’ [17].
The 13q31.3 human genomic locus undergoes amplification in several types of lymphoproliferative disorders and solid tumors [20, 21], and, consistently, aberrant overexpression of the miR-17-92 cluster in the absence of amplification is also frequently observed in some tumors. The miR-17-92 cluster has been reported to be involved in hematopoietic malignancies including diffuse large B-cell lymphoma (DLBCL), mantle cell lymphoma (MCL), Burkitt’s lymphoma (BL), and CLL (Table 1) [14, 21–26]. In general, a significant over expression of pri-miR-17-92 has been observed in 65% of B-cell lymphoma patients [17].
Table 1: miR-17-92 cluster overexpression in lymphoproliferative disorders
Disease |
miR-17-92 dysregulation |
Genomic aberrations |
References |
DLBCL |
overexpression |
13q31.3 amplification; MYC aberration in complex karyotypes |
|
MCL |
overexpression |
13q31.3 amplification |
[24] |
BL |
overexpression |
13q31.3 amplification; MYC translocation |
|
CLL |
overexpression upon microenvironmental stimuli compared to the unstimulated counterpart |
absence of associated genomic aberrations |
Abbreviations: DLBCL, diffuse large B cell lymphoma; MCL, mantle cell lymphoma; BL, Burkitt’s lymphoma; CLL, chronic lymphocytic leukemia.
Expression regulation of the miR-17-92 cluster
A number of oncogenic transcription factors (TFs) regulate the expression of miR-17-92 cluster thus influencing its oncogenic activity (Figure 2) [27, 28]. In particular, MYC, the first identified transcription regulator of miR-17-92, activates miR-17-92 expression by directly binding to its genomic locus [17, 18, 29]. Similarly, miR-17-92 can be also upregulated by MYCN, as demonstrated in neuroblastoma cells [30, 31]. In addition to MYC, TFs belonging to the E2F family, e.g. E2F1, E2F2, E2F3, are other potent inducers of the miR-17-92 cluster [32]. The E2F TFs are essential for cell cycle progression; in fact, they activate a large number of S phase genes, including thymidine kinase, DNA polymerase, Cyclin A and Cyclin E. Thus, cycling cells are likely to have elevated levels of miR-17-92 due to periodic burst of E2F activity during S phase, while quiescent cells may have reduced miR-17-92 levels. Other TFs that directly activate the transcription of miR-17-92 are two TFs belonging to the ETS family, SPI-1 and FLI-1, as demonstrated in the murine Friend’s leukemia model [33]. Finally, STAT3 can also regulate the expression of the miR-17-92 cluster at transcriptional level [34]. Notably, miR-17-92 can be also transcriptionally repressed by tumor suppressors, e.g. TP53 in hypoxia-treated cells, or via histone modification [35–37].
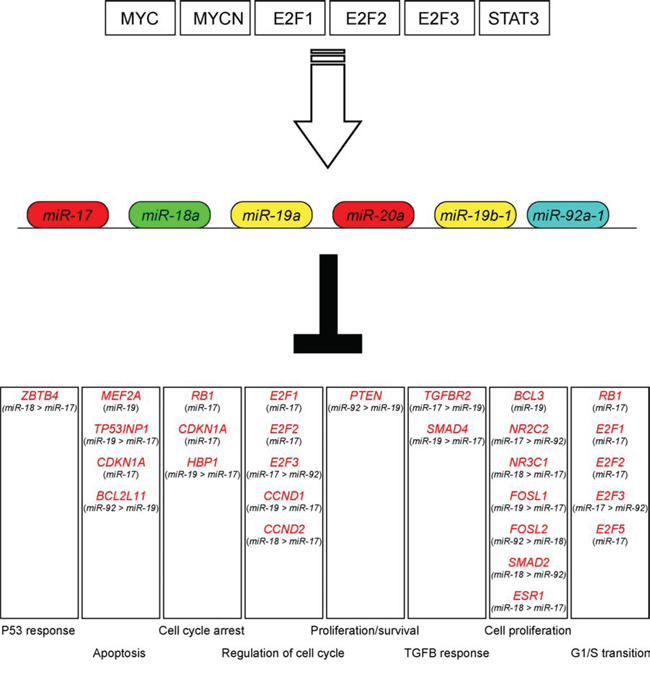
Figure 2: Major interactions of the miR-17-92 cluster. A number of oncogenic transcription factors positively (white arrow) regulate the expression of miRNAs from the miR-17-92 cluster (upper part of the figure). miRNA belonging to the miR-17-92 cluster target specific genes involved in different biological processes. The miRNA families (as indicated in Figure 1C) with the highest scores for each specific target gene are reported according to the algorithms Target Scan Human (http://www.targetscan.org) and http://Microrna.org; Targets and Expression (http://www.microrna.org).
Major molecular interactions of the miR-17-92 cluster
The major functional evidence of the oncogenic activity of miR-17-92 comes from several studies employing B-cell lymphoma in-vivo mouse models. In a first model, in which MYC is driven by the immunoglobulin heavy chain enhancer (Eμ) as a transgene, the enforced expression of a truncated miR-17-92 lacking the miR-92a-1, e.g. miR-17-19b, collaborated with the MYC to accelerate lymphomagenesis. Of note, the overexpression of miR-17-19b not only promotes the oncogenesis of MYC expressing B cells, but also alters the cell fate of transformed B cells. In fact, while the majority of B cell lymphomas derived from the Eμ-MYC transgene have a mature B-cell phenotype, B-cell lymphomas resulting from the collaboration between miR-17-19b and MYC are mostly derived from precursor cells [17]. In this context, a key oncogenic role is conducted by miR-19a and miR-19b-1 as indicated by the fact that mutations of both miR-19 miRNAs nearly abolish the oncogenic cooperation between MYC and miR-17-92 [38–40]. On the other hand, in Eμ-MYC chimeric mice, it was demonstrated that this oncogenic cooperation is significant stronger when miR-92a-1 is deleted within the oncomir, as well as when the seed sequence of the miR-92a-1 is mutated [41]. Moreover, mutations of miR-20a or of miR-17 did not affect oncogenesis in the Eμ-MYC model [41].
Regarding the oncogenic cooperation between MYC and miR-17-92 [42], it has been demonstrated that MYC directly suppresses through miR-17-92 the expression of the chromatin regulatory genes SIN3B, HBP1, SUV420 and BTG1 and the proapoptotic gene BCL2L11 (a.k.a. BIM), this suppression contributing to maintain survival, autonomous proliferation, and self-renewal.
Moreover, according to another study [43], mice with exogenously induced miR-17-92 overexpression in lymphocytes developed an aggressive lymphoproliferative disorder associated with autoimmunity followed by a premature death. This study also reported that, in the transgenic lymphocytes, the miR-17-92 cluster down-regulated PTEN and BCL2L11, thus enhancing proliferation and survival [43]. This observation was confirmed by other studies employing B cell specific transgenic mouse models, in which miR-17-92 overexpression induced B cell malignancies such as splenic B cell lymphomas or B cell leukemia/lymphomas [44, 45]. Seventeen genes, predicted to be targets of the miR-17-92 cluster, have been found consistently downregulated in Eμ miR-17-92 cells [45]. In transgenic mice specifically overexpressing miR-17-92 in B cells [29], the miR-17-92 cluster drives lymphomagenesis by suppressing the expression of multiple inhibitors of the PI3K and NFkB pathways and by inhibiting the mitochondrial apoptosis pathway [29]. In the same study, miR-17-92-driven lymphoma cells showed constitutive activation of the PI3K and NFkB pathways, and chemical inhibition of these pathways was useful to treat these lymphomas [29]. Moreover, as described in mice overexpressing miR-17-92 in hematopoietic lineages [46], the expression of the miR-17-92 cluster in a limited number of hematopoietic cells is sufficient to develop B cell malignancies, further highlighting the ability of miR-17-92 to act as driver of tumorigenesis. On the other hand, the role of the miR-17-92 cluster in the B cell development is also defined by the fact that a deficiency of miR-17-92 impairs this process, particularly at the pro-B to pre-B transition stage, due to enhanced apoptosis occurring in the pro-B cells during both fetal and adult B cell development [19].
Among the miRNAs belonging to the miR-17-92 cluster, miR-17 and miR-20a are those that target the largest number of genes. In particular, these miRNAs are able to control the expression of genes with antagonizing functions, e.g. promoting or suppressing cell cycle progression [47], thus reflecting the complex and partially conflicting effects of miRNAs in tumor growth. In this context, the E2F TFs, direct targets of MYC [48], are among the genes down-regulated by miR-17 and miR-20a. Thus, a tightly controlled proliferative loop is represented by MYC that simultaneously activates the E2F TFs and limits their translation by a miRNA-based mechanism [18]. In this process, a role can be exerted by RB1 and the other hypophosphorylated retinoblastoma proteins that inhibit E2F TFs and, in turn, are repressed by miR-17 and miR-20a [49, 50]. CCDN1 (a.k.a. Cyclin D1) is also regulated by miR-17 and miR-20a. On the other hand, both E2Fs and CCDN1 are able to bind the miR-17-92 promoter establishing a negative feedback loop [32, 51, 52].
The miR-17 can also act as regulator of cell cycle by directly targeting more than 20 genes involved in the G1/S phase cell cycle transition. In this context, miR-17, by inhibiting the mitogen-activated kinase JNK2, a cell cycle promoting protein [47], may have also a tumor suppressive role that has been demonstrated by evidence showing that loss of heterozygosity at 13q31.3 is associated with tumor progression and poor prognosis in several cancers [12, 53].
Interaction of the miR-17-92 cluster with the B cell receptor (BCR)
The role of the miR-17-92 cluster has also been investigated in the regulation of the BCR pathway. In this context, information has been obtained by taking advantage of the P493-6 cell line, an Epstein-Barr virus-immortalized lymphoblastoid cell line with a tet-repressible MYC gene and a significant enrichment of MYC repressed genes, all with the predicted binding sites for miRNAs of the miR-17-92 cluster [54]. In particular, genes for the ITIM-containing proteins CD22 and CD32b were identified as direct targets of the miR-17-92 cluster. Moreover, either MYC or miR-17-92 expression have been found necessary to sustain phosphorylation of the BCR pathway proteins SYK and BLNK upon BCR ligation. Furthermore, stimulation of the BCR response in miR-17-92 overexpressing cells results in enhanced calcium influx and elevated levels of MYC. Consistently, inhibition of the miR-17-92 cluster was demonstrated to diminish the BCR response as measured by SYK and BLNK phosphorylation [54].
Dysregulation of the miR-17-92 cluster in DLBCL
DLBCL is a heterogeneous lymphoproliferative disease in which different molecular subtypes have been identified involving the deregulation of distinct signalling pathways [55]. Among the three molecular DLBCL subtypes recognized by gene-expression profiling, those classified as germinal centre B cell-like (GCB) DLBCL harbour frequent 13q31.3 amplification. Overexpression of the miR-17-92 cluster members was a consequence of such an amplification, as found in leukemia/lymphoma cell lines bearing 13q amplification [56], or due to an overexpression of MYC [17, 26, 57–59]. In DLBCL, MYC gene aberrations were found in less than 10% of the cases at diagnosis [60] and in almost 20% at first relapse [61, 62]. MYC aberration in DLBCL usually associates with complex karyotypes and BCL2 and BCL6 rearrangements [63]. MYC protein overexpression was recently identified in about 30% of cases, without significant difference between GCB-DLBCL and non-GCB-DLBCL subtypes [62, 64, 65].
Another genetic aberration related to miR-17-92 cluster overexpression in GCB DLBCL is a complex rearrangement t(3;13)(q27;q31)t(12;13)(p11;q31), which targets both BCL6 and the miR-17-92 cluster. In particular, this rearrangement was shown to deregulate miR-17-92 by increasing histone acetylation at its upstream breakpoint near the MIR17HG promoter [56].
DLBCL heterogeneity is also related to differences in anatomical presentation in extranodal sites. An expression profiling study in DLBCL cases with different extranodal locations showed no significant difference in the expression of miR-17-92 cluster members in comparison with the nodal DLBCLs [66]. Nevertheless, others studies, focused on DLBCL primarily originating from the central nervous system showed higher levels of miR-20a and/or miR-17 compared to nodal DLBCL [66, 67]. Consistently, high levels of miR-19b-1 and miR-92a-1 were detected in the cerebrospinal fluid of these lymphomas [68].
In keeping with the capability of miRNAs from the miR-17-92 cluster to regulate specific BCR pathway genes, DLBCLs characterized by higher levels of many components of the BCR signalling cascade (e.g. CD19, IG, CD79a, BLK, SYK, PLCγ2, and MAP4K) has been found to express higher MYC and miR-17-92 transcript levels than other DLBCL subtypes [54, 69]. In this context, studies investigating copy number variations identified the MYC and MIR17HG loci as frequently amplified, consistently with the idea that DLBCLs require high levels of MYC or of miRNAs from the miR-17-92 cluster to sustain BCR response [54, 69].
DLBCL could arise as transformation of follicular lymphoma (FL) or CLL, leading to the so-called Richter’s syndrome (RS). In these cases, amplification of the 13q31.3 region containing the miR-17-92 cluster, described to be acquired at the time of transformation, was coupled with the gain of MYC and loss of TP53 suggesting a late involvement of the miR-17-92 cluster in the acquisition of a more biologically aggressive phenotype [70, 71].
Finally, individuals infected by HIV have an increased risk for developing non-Hodgkin’s lymphomas (AIDS-NHL) including DLBCL, and overexpression of miR-17-92 and its paralogs have been found in these and other AIDS-NHL subtypes [72].
Dysregulation of the miR-17-92 cluster in MCL
Overexpression of the miR-17-92 cluster in MCL is usually associated with gains and high number amplifications of its locus [24]. The same study evidenced that at least two miRNAs of this cluster, miR-17 and miR-20a, when associated with high MYC mRNA levels, identified a MCL subset with a more aggressive clinical behaviour [24]. A correlation of high miR-17-92 expression with poor prognosis was also confirmed in a subsequent independent study [73]. It has been proposed that this effect is related with the targeting by miRNAs of the miR-17-92 cluster on several negative regulators of the PI3K/AKT pathway, e.g. PHLPP2 and PTEN, as well as in the blocking of chemotherapy-induced apoptosis through the targeting of BCL2L11 [19]. Consistently, the knockdown of miRNAs from the miR-17-92 cluster inhibits tumor growth in xenograft MCL mouse models [19] and the enforced expression of the same miRNAs facilitates cell proliferation and apoptosis suppression of lymphocytes [19, 43]. This cell proliferative effect of miRNAs belonging to this cluster can be also detected in MCL primary samples in studies either involving peripheral blood or lymph node samples [24, 74]. Targeting on additional cell cycle regulators are surely involved in this effect as, for example, it has been shown for CDKN1A in MCL cells [75].
Dysregulation of the miR-17-92 cluster in BL
In a recent study [76], high miR-17-92 overexpression has been found in BL, thus confirming that the activation of the MYC/miR-17-92 axis is a general feature of this disease. In addition, gain of 13q31.3 has been found to occur in about 10–20% of BLs and to associate with miR-17-92 overexpression [77, 78]. Of note, the MYC gene is translocated to one of the immunoglobulin loci in virtually all BLs. In particular, the typical translocation of MYC into the immunoglobulin heavy chain locus is observed in about 80% of BLs whereas the variant translocation into either the κ or λ light chain loci occurs at a frequency of about 10% [79]. The juxtaposition of the immunoglobulin heavy chain or of the κ/λ light chain loci with MYC are required for the induction of translocated MYC transcription [80–82]. The transcription of the translocated MYC is greater than that seen in resting B-cells and similar to that of actively dividing non-malignant B cells such those infected by EBV [79]. This overexpression can induce higher miR-17-92 levels. Moreover, the deletion of miR-17-92 in established MYC-driven lymphoma cell lines decreased the capability to growth in tissue culture and immunodeficient hosts, in keeping with the concept that miR-17-92 levels influences proliferation of these cells [39].
Dysregulation of the miR-17-92 cluster in CLL
In a study in which miRNA expression was compared in CLL with or without TP53 abnormalities [83], a significant down regulation of miR-17 in cases harbouring TP53 deletion/mutation was shown. These data were subsequently confirmed in an another study in which CLL patients lacking TP53 expression and displaying aggressive disease exhibit reduced miR-17 and miR-20a level of expression and increased miR-19a, miR-19b-1 and miR-92a-1, whereas miR-18a levels were unchanged, compared to healthy normal controls [44]. In contrast, patients expressing wild type TP53, exhibit increased levels of miR-17 and miR-20a, unchanged levels of miR-18a, miR-19a, miR-19b-1 and lower levels of miR-92a-1 [44]. The loss of TP53 may selectively effect the processing of miR-17-92 miRNA, resulting in the imbalanced expression of its encoded units [84].
Concerning the ability of miRNA belonging to the miR-17-92 cluster to predict disease outcome, the debate is still open. In this context, miR-20a expression has been found to correlate with time to treatment in CLL [85].
Regarding the capacity of microenvironmental stimuli to induce miRNA expression, in a study of some of us [22], expression of miRNAs from the miR-17-92 cluster were up-regulated upon TLR9 triggering by CpG, compared to unstimulated cells, in CLL cells bearing an unmutated BCR, whereas no difference was detected in the context of a mutated configuration. In this study, the miR-17-92 overexpression due to TLR9 stimulation was correlated with the concomitant gene expression signature. In particular, it has been shown that the enforced expression of miR-17 in primary unmutated IGHV CLL cells reduces the expression of the tumor suppressor genes E2F5, TP53INP1, TRIM8 and ZBTB4. Among these genes, ZBTB4 and TP53INP1 are involved in apoptosis regulation through CDKN1A and TP53 [86, 87], E2F5 is involved in the G1 arrest [88], and TRIM8, is involved in the degradation of SOCS1, a well-known regulator of the response to CpG [89]. All these genes were identified as direct targets of miR-17, as evidenced by their significant downregulation upon ectopic miR-17 overexpression. In the same study [22], we also demonstrated that the stable and sustained up-regulation of miRNAs belonging to the miR-17-92 cluster in unmutated IGHV CLL by CpG is preceded by induction of MYC, thus providing evidence of an associative interaction between MYC and miR-17-92 cluster also in CLL cells. In agreement with these results, miR-17 transfection was also demonstrated to be sufficient to reduce apoptosis induced by serum deprivation in a series of primary unmutated IGHV CLL cells. Consistently, as indicated by experiments in primary CLL cells, transfection with miR-17-92 cluster antagomiRs reduces bromo-deoxy-uridine incorporation in CpG-stimulated unmutated IGHV CLL cells [22]. Moreover, the expression of miR-17 was demonstrated to be significant higher in unmutated IGHV CLL cells expressing high ZAP70 compared with the IGHV-mutated/ZAP70-low counterpart [22]. This evidence could suggest that IGHV-unmutated/ZAP70-high CLL cells are more frequently subjected to signals capable of activating the TLR9 pathway, or, alternatively, that the former cells have a greater capacity to respond to microenvironmental signals, including those delivered through TLR9 stimulation [90–93].
Further evidence regarding the induction of miR-17-92 expression by microenvironmental stimuli has been provided in another study in which miRNAs belonging to the miR-17-92 cluster have been found to be differentially expressed and up regulated by the co-culture with stromal cells with or without the co-stimulation with the CD154 molecule [94]. Specifically, four out of the six members of the miR-17-92 cluster, i.e. miR-17, miR-20a, miR-18a and miR-19b-1, resulted significantly regulated by CD154. Other members of the family were also induced but with lower, not significant extent, i.e. miR-19a and miR-92a-1. In the same study, it has been also shown that, for all the members of the miR-17-92 cluster except miR-92a-1, stromal cell culture produced an increase in miRNA expression, which was further increased by CD154. In this context, the most highly expressed members of the cluster following CD154 culture were miR-17 and miR-19b-1 [94]. Of note, also in this case, miR-17-92 up-regulation was associated with MYC, in particular, MYC expression was induced by stromal cell culture with a further increase due to CD154 culture [94].
These data indicate that miR-17-92 cluster over expression may be not only due to genomic abnormalities but also to microenvironmental stimuli capable to influence miRNA expression [22, 94].
Future perspectives
The discovery of miRNAs acting as oncogenes or oncosuppressors have introduced novel treatment approaches. The more frequently proposed modality of therapy targeting miRNAs is to silence oncomiRs through anti-miRNAs oligonucleotides or to overexpress oncosuppressors miRNAs using miRNA mimics [95–97]. In general, artificial miRNAs or anti-miRNAs are small, stable, and, theoretically, easy to deliver to cells, possibly by packaging them into lipid based and/or antibody-conjugated nanoparticles [95–97]. However, these oligonucleotides still remain to be fully tested for efficacy and safety for a therapeutic use, although very recently a few phase II clinical trials have been performed [98, 99].
Given the multiple and potent effects on cellular proliferation and apoptosis exerted by the miR-17-92 cluster, miRNAs belonging to the cluster surely represent attractive targets for cancer therapy. In this context, in a study by us [100], it has been recently demonstrated that in-vitro administration of a specific oligonucleotide targeting endogenous miR-17 effectively reduces miR-17 expression and the proliferation of CLL-like MEC-1 cells. Consistently, it has been also demonstrated that, when injected in-vivo in tumor generated by subcutaneously injected MEC-1 cells in SCID mice, this specific oligonucleotide dramatically reduces tumor growth and significantly increase mouse survival. This evidence could represent a proof of principle for the use of specific anti-miRNAs oligonucleotides targeting miRNAs of the miR-17-92 cluster as a therapeutic tool in CLL and in lymphoproliferative disorders where miR-17-92 amplification and/or overexpression have a proved pathogenetic role, as discussed in the present review.
ACKNOWLEDGMENTS
The authors are indebted with Professor Elias Campo, Department of Pathology, Hospital Clinic, Institut d’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS), University of Barcelona, for his continuous support.
This work was supported by: Progetto Giovani Ricercatori n. GR-2011-02347441, n. GR-2010-2317594, n. GR-2009-1475467, n. GR-2008-1138053, Ministero della Salute, Rome, Italy; Progetto Ricerca Finalizzata RF-2010-2307262, Ministero della Salute, Rome, Italy; Associazione Italiana contro le Leucemie, linfomi e mielomi (AIL), Venezia Section, Pramaggiore Group; the Associazione Italiana Ricerca Cancro (AIRC), Investigator Grant IG-13227, MFAG-10327, IG-12939, “5 × 1000 Intramural Program”, Centro di Riferimento Oncologico, Aviano, Italy. This work was partially developed at the Centro Esther Koplowitz, Barcelona, Spain. This work was also supported by the Instituto de Salud Carlos III (ISCIII), Fondo Investigaciones Sanitarias & European Regional Development Fund (Union Europea. “Una manera de hacer Europa”; PI12/01302; to L.H.)
CONFLICTS OF INTEREST
There are no conflict of interest to disclose.
REFERENCES
1. Visone R, Croce CM. MiRNAs and cancer. Am J Pathol. 2009; 174:1131–1138.
2. Kim VN, Nam JW. Genomics of microRNA. Trends Genet. 2006; 22:165–173.
3. Ambros V. The functions of animal microRNAs. Nature. 2004; 431:350–355.
4. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004; 116:281–297.
5. Cho WC. OncomiRs: the discovery and progress of microRNAs in cancers. Mol Cancer. 2007; 6:60.
6. Poy MN, Spranger M, Stoffel M. microRNAs and the regulation of glucose and lipid metabolism. Diabetes Obes Metab. 2007; 9:67–73.
7. Sullivan CS, Ganem D. MicroRNAs and viral infection. Mol Cell. 2005; 20:3–7.
8. Lovis P, Gattesco S, Regazzi R. Regulation of the expression of components of the exocytotic machinery of insulin-secreting cells by microRNAs. Biol Chem. 2008; 389:305–312.
9. Xiao C, Rajewsky K. MicroRNA control in the immune system: basic principles. Cell. 2009; 136:26–36.
10. Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, Aldler H, Rattan S, Keating M, Rai K, Rassenti L, Kipps T, Negrini M, et al. Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 2002; 99:15524–15529.
11. Calin GA, Sevignani C, Dumitru CD, Hyslop T, Noch E, Yendamuri S, Shimizu M, Rattan S, Bullrich F, Negrini M, Croce CM. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci U S A. 2004; 101:2999–3004.
12. Zhang L, Huang J, Yang N, Greshock J, Megraw MS, Giannakakis A, Liang S, Naylor TL, Barchetti A, Ward MR, Yao G, Medina A, O’brien-Jenkins A, et al. microRNAs exhibit high frequency genomic alterations in human cancer. Proc Natl Acad Sci U S A. 2006; 103:9136–9141.
13. Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006; 6:857–866.
14. Ota A, Tagawa H, Karnan S, Tsuzuki S, Karpas A, Kira S, Yoshida Y, Seto M. Identification and characterization of a novel gene, C13orf25, as a target for 13q31-q32 amplification in malignant lymphoma. Cancer Res. 2004; 64:3087–3095.
15. Petrocca F, Vecchione A, Croce CM. Emerging role of miR-106b-25/miR-17-92 clusters in the control of transforming growth factor beta signaling. Cancer Res. 2008; 68:8191–8194.
16. Dews M, Fox JL, Hultine S, Sundaram P, Wang W, Liu YY, Furth E, Enders GH, El-Deiry W, Schelter JM, Cleary MA, Thomas-Tikhonenko A. The myc-miR-17~92 axis blunts TGF{beta} signaling and production of multiple TGF{beta}-dependent antiangiogenic factors. Cancer Res. 2010; 70:8233–8246.
17. He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, Powers S, Cordon-Cardo C, Lowe SW, Hannon GJ, Hammond SM. A microRNA polycistron as a potential human oncogene. Nature. 2005; 435:828–833.
18. O’Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005; 435:839–843.
19. Ventura A, Young AG, Winslow MM, Lintault L, Meissner A, Erkeland SJ, Newman J, Bronson RT, Crowley D, Stone JR, Jaenisch R, Sharp PA, Jacks T. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell. 2008; 132:875–886.
20. Mendell JT. miRiad roles for the miR-17-92 cluster in development and disease. Cell. 2008; 133:217–222.
21. Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M, Prueitt RL, Yanaihara N, Lanza G, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A. 2006; 103:2257–2261.
22. Bomben R, Gobessi S, Dal BM, Volinia S, Marconi D, Tissino E, Benedetti D, Zucchetto A, Rossi D, Gaidano G, Del PG, Laurenti L, Efremov DG, et al. The miR-17 approximately 92 family regulates the response to Toll-like receptor 9 triggering of CLL cells with unmutated IGHV genes. Leukemia. 2012; 26:1584–1593.
23. Lu J, Getz G, Miska EA, varez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA, Downing JR, Jacks T, Horvitz HR, et al. MicroRNA expression profiles classify human cancers. Nature. 2005; 435:834–838.
24. Navarro A, Bea S, Fernandez V, Prieto M, Salaverria I, Jares P, Hartmann E, Mozos A, Lopez-Guillermo A, Villamor N, Colomer D, Puig X, Ott G, et al. MicroRNA expression, chromosomal alterations, and immunoglobulin variable heavy chain hypermutations in Mantle cell lymphomas. Cancer Res. 2009; 69:7071–7078.
25. Schmidt H, Bartel F, Kappler M, Wurl P, Lange H, Bache M, Holzhausen HJ, Taubert H. Gains of 13q are correlated with a poor prognosis in liposarcoma. Mod Pathol. 2005; 18:638–644.
26. Tagawa H, Seto M. A microRNA cluster as a target of genomic amplification in malignant lymphoma. Leukemia. 2005; 19:2013–2016.
27. Mogilyansky E, Rigoutsos I. The miR-17/92 cluster: a comprehensive update on its genomics, genetics, functions and increasingly important and numerous roles in health and disease. Cell Death Differ. 2013; 20:1603–1614.
28. Olive V, Jiang I, He L. miR-17-92, a cluster of miRNAs in the midst of the cancer network. Int J Biochem Cell Biol. 2010; 42:1348–1354.
29. Jin HY, Oda H, Lai M, Skalsky RL, Bethel K, Shepherd J, Kang SG, Liu WH, Sabouri-Ghomi M, Cullen BR, Rajewsky K, Xiao C. MicroRNA-17~92 plays a causative role in lymphomagenesis by coordinating multiple oncogenic pathways. EMBO J. 2013; 32:2377–2391.
30. Schulte JH, Horn S, Otto T, Samans B, Heukamp LC, Eilers UC, Krause M, Astrahantseff K, Klein-Hitpass L, Buettner R, Schramm A, Christiansen H, Eilers M, et al. MYCN regulates oncogenic MicroRNAs in neuroblastoma. Int J Cancer. 2008; 122:699–704.
31. Mestdagh P, Bostrom AK, Impens F, Fredlund E, Van PG, De AP, von SK, Ghesquiere B, Schulte S, Dews M, Thomas-Tikhonenko A, Schulte JH, Zollo M, et al. The miR-17-92 microRNA cluster regulates multiple components of the TGF-beta pathway in neuroblastoma. Mol Cell. 2010; 40:762–773.
32. Woods K, Thomson JM, Hammond SM. Direct regulation of an oncogenic micro-RNA cluster by E2F transcription factors. J Biol Chem. 2007; 282:2130–2134.
33. Kayali S, Giraud G, Morle F, Guyot B. Spi-1, Fli-1 and Fli-3 (miR-17-92) oncogenes contribute to a single oncogenic network controlling cell proliferation in friend erythroleukemia. PLoS One. 2012; 7:e46799.
34. Brock M, Trenkmann M, Gay RE, Michel BA, Gay S, Fischler M, Ulrich S, Speich R, Huber LC. Interleukin-6 modulates the expression of the bone morphogenic protein receptor type II through a novel STAT3-microRNA cluster 17/92 pathway. Circ Res. 2009; 104:1184–1191.
35. Yan HL, Xue G, Mei Q, Wang YZ, Ding FX, Liu MF, Lu MH, Tang Y, Yu HY, Sun SH. Repression of the miR-17-92 cluster by p53 has an important function in hypoxia-induced apoptosis. EMBO J. 2009; 28:2719–2732.
36. Mi S, Li Z, Chen P, He C, Cao D, Elkahloun A, Lu J, Pelloso LA, Wunderlich M, Huang H, Luo RT, Sun M, He M, et al. Aberrant overexpression and function of the miR-17-92 cluster in MLL-rearranged acute leukemia. Proc Natl Acad Sci U S A. 2010; 107:3710–3715.
37. Pospisil V, Vargova K, Kokavec J, Rybarova J, Savvulidi F, Jonasova A, Necas E, Zavadil J, Laslo P, Stopka T. Epigenetic silencing of the oncogenic miR-17-92 cluster during PU.1-directed macrophage differentiation. EMBO J. 2011; 30:4450–4464.
38. Mavrakis KJ, Van Der MJ, Wolfe AL, Liu X, Mets E, Taghon T, Khan AA, Setty M, Rondou P, Vandenberghe P, Delabesse E, Benoit Y, Socci NB, et al. A cooperative microRNA-tumor suppressor gene network in acute T-cell lymphoblastic leukemia (T-ALL). Nat Genet. 2011; 43:673–678.
39. Mu P, Han YC, Betel D, Yao E, Squatrito M, Ogrodowski P, de SE, D’Andrea A, Sander C, Ventura A. Genetic dissection of the miR-17~92 cluster of microRNAs in Myc-induced B-cell lymphomas. Genes Dev. 2009; 23:2806–2811.
40. Olive V, Bennett MJ, Walker JC, Ma C, Jiang I, Cordon-Cardo C, Li QJ, Lowe SW, Hannon GJ, He L. miR-19 is a key oncogenic component of miR-17-92. Genes Dev. 2009; 23:2839–2849.
41. Olive V, Sabio E, Bennett MJ, De Jong CS, Biton A, McGann JC, Greaney SK, Sodir NM, Zhou AY, Balakrishnan A, Foth M, Luftig MA, Goga A, et al. A component of the miR-17-92 polycistronic oncomir promotes oncogene-dependent apoptosis. Elife. 2013; 2:e00822.
42. Li Y, Choi PS, Casey SC, Dill DL, Felsher DW. MYC through miR-17-92 suppresses specific target genes to maintain survival, autonomous proliferation, and a neoplastic state. Cancer Cell. 2014; 26:262–272.
43. Xiao C, Srinivasan L, Calado DP, Patterson HC, Zhang B, Wang J, Henderson JM, Kutok JL, Rajewsky K. Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat Immunol. 2008; 9:405–414.
44. Li Y, Vecchiarelli-Federico LM, Li YJ, Egan SE, Spaner D, Hough MR, Ben-David Y. The miR-17-92 cluster expands multipotent hematopoietic progenitors whereas imbalanced expression of its individual oncogenic miRNAs promotes leukemia in mice. Blood. 2012; 119:4486–4498.
45. Sandhu SK, Fassan M, Volinia S, Lovat F, Balatti V, Pekarsky Y, Croce CM. B-cell malignancies in microRNA Emu-miR-17~92 transgenic mice. Proc Natl Acad Sci U S A. 2013; 110:18208–18213.
46. Danielson LS, Reavie L, Coussens M, Davalos V, Castillo-Martin M, Guijarro MV, Coffre M, Cordon-Cardo C, Aifantis I, Ibrahim S, Liu C, Koralov SB, Hernando E. Limited miR-17-92 overexpression drives hematologic malignancies. Leuk Res. 2015; 39:335–341.
47. Cloonan N, Brown MK, Steptoe AL, Wani S, Chan WL, Forrest AR, Kolle G, Gabrielli B, Grimmond SM. The miR-17-5p microRNA is a key regulator of the G1/S phase cell cycle transition. Genome Biol. 2008; 9:R127.
48. Leone G, DeGregori J, Sears R, Jakoi L, Nevins JR. Myc and Ras collaborate in inducing accumulation of active cyclin E/Cdk2 and E2F. Nature. 1997; 387:422–426.
49. Tamrakar S, Rubin E, Ludlow JW. Role of pRB dephosphorylation in cell cycle regulation. Front Biosci. 2000; 5:D121–D137.
50. Trompeter HI, Abbad H, Iwaniuk KM, Hafner M, Renwick N, Tuschl T, Schira J, Muller HW, Wernet P. MicroRNAs MiR-17, MiR-20a, and MiR-106b act in concert to modulate E2F activity on cell cycle arrest during neuronal lineage differentiation of USSC. PLoS One. 2011; 6:e16138.
51. Sylvestre Y, De G V, Querido E, Mukhopadhyay UK, Bourdeau V, Major F, Ferbeyre G, Chartrand P. An E2F/miR-20a autoregulatory feedback loop. J Biol Chem. 2007; 282:2135–2143.
52. Yu Z, Wang C, Wang M, Li Z, Casimiro MC, Liu M, Wu K, Whittle J, Ju X, Hyslop T, McCue P, Pestell RG. A cyclin D1/microRNA 17/20 regulatory feedback loop in control of breast cancer cell proliferation. J Cell Biol. 2008; 182:509–517.
53. Coller HA, Forman JJ, Legesse-Miller A. “Myc’ed messages”: myc induces transcription of E2F1 while inhibiting its translation via a microRNA polycistron. PLoS Genet. 2007; 3:e146.
54. Psathas JN, Doonan PJ, Raman P, Freedman BD, Minn AJ, Thomas-Tikhonenko A. The Myc-miR-17-92 axis amplifies B-cell receptor signaling via inhibition of ITIM proteins: a novel lymphomagenic feed-forward loop. Blood. 2013; 122:4220–4229.
55. Pasqualucci L. The genetic basis of diffuse large B-cell lymphoma. Curr Opin Hematol. 2013.
56. Schneider B, Nagel S, Ehrentraut S, Kaufmann M, Meyer C, Geffers R, Drexler HG, Macleod RA. Neoplastic MiR-17~92 deregulation at a DNA fragility motif (SIDD). Genes Chromosomes Cancer. 2012; 51:219–228.
57. Lawrie CH, Soneji S, Marafioti T, Cooper CD, Palazzo S, Paterson JC, Cattan H, Enver T, Mager R, Boultwood J, Wainscoat JS, Hatton CS. MicroRNA expression distinguishes between germinal center B cell-like and activated B cell-like subtypes of diffuse large B cell lymphoma. Int J Cancer. 2007; 121:1156–1161.
58. Lenz G, Wright GW, Emre NC, Kohlhammer H, Dave SS, Davis RE, Carty S, Lam LT, Shaffer AL, Xiao W, Powell J, Rosenwald A, Ott G, et al. Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc Natl Acad Sci U S A. 2008; 105:13520–13525.
59. Tagawa H, Karube K, Tsuzuki S, Ohshima K, Seto M. Synergistic action of the microRNA-17 polycistron and Myc in aggressive cancer development. Cancer Sci. 2007; 98:1482–1490.
60. Savage KJ, Johnson NA, Ben-Neriah S, Connors JM, Sehn LH, Farinha P, Horsman DE, Gascoyne RD. MYC gene rearrangements are associated with a poor prognosis in diffuse large B-cell lymphoma patients treated with R-CHOP chemotherapy. Blood. 2009; 114:3533–3537.
61. Cuccuini W, Briere J, Mounier N, Voelker HU, Rosenwald A, Sundstrom C, Cogliatti S, Hirchaud E, Ysebaert L, Bron D, Soulier J, Gaulard P, Houlgatte R, et al. MYC+ diffuse large B-cell lymphoma is not salvaged by classical R-ICE or R-DHAP followed by BEAM plus autologous stem cell transplantation. Blood. 2012; 119:4619–4624.
62. Horn H, Ziepert M, Becher C, Barth TF, Bernd HW, Feller AC, Klapper W, Hummel M, Stein H, Hansmann ML, Schmelter C, Moller P, Cogliatti S, et al. MYC status in concert with BCL2 and BCL6 expression predicts outcome in diffuse large B-cell lymphoma. Blood. 2013; 121:2253–2263.
63. Petrich AM, Nabhan C, Smith SM. MYC-associated and double-hit lymphomas: a review of pathobiology, prognosis, and therapeutic approaches. Cancer. 2014; 120:3884–3895.
64. Johnson NA, Slack GW, Savage KJ, Connors JM, Ben-Neriah S, Rogic S, Scott DW, Tan KL, Steidl C, Sehn LH, Chan WC, Iqbal J, Meyer PN, et al. Concurrent expression of MYC and BCL2 in diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J Clin Oncol. 2012; 30:3452–3459.
65. Green TM, Nielsen O, de SK, Xu-Monette ZY, Young KH, Moller MB. High levels of nuclear MYC protein predict the presence of MYC rearrangement in diffuse large B-cell lymphoma. Am J Surg Pathol. 2012; 36:612–619.
66. Robertus JL, Harms G, Blokzijl T, Booman M, de Jong D, van Imhoff G, Rosati S, Schuuring E, Kluin P. Specific expression of miR-17–5p and miR-127 in testicular and central nervous system diffuse large B-cell lymphoma. Mod Pathol. 2009; 22:547–555.
67. Fischer L, Hummel M, Korfel A, Lenze D, Joehrens K, Thiel E. Differential micro-RNA expression in primary CNS and nodal diffuse large B-cell lymphomas. Neuro Oncol. 2011; 13:1090–1098.
68. Baraniskin A, Kuhnhenn J, Schlegel U, Schmiegel W, Hahn S, Schroers R. MicroRNAs in cerebrospinal fluid as biomarker for disease course monitoring in primary central nervous system lymphoma. J Neurooncol. 2012; 109:239–244.
69. Monti S, Savage KJ, Kutok JL, Feuerhake F, Kurtin P, Mihm M, Wu B, Pasqualucci L, Neuberg D, Aguiar RC, Dal CP, Ladd C, Pinkus GS, et al. Molecular profiling of diffuse large B-cell lymphoma identifies robust subtypes including one characterized by host inflammatory response. Blood. 2005; 105:1851–1861.
70. Lawrie CH, Chi J, Taylor S, Tramonti D, Ballabio E, Palazzo S, Saunders NJ, Pezzella F, Boultwood J, Wainscoat JS, Hatton CS. Expression of microRNAs in diffuse large B cell lymphoma is associated with immunophenotype, survival and transformation from follicular lymphoma. J Cell Mol Med. 2009; 13:1248–1260.
71. Scandurra M, Rossi D, Deambrogi C, Rancoita PM, Chigrinova E, Mian M, Cerri M, Rasi S, Sozzi E, Forconi F, Ponzoni M, Moreno SM, Piris MA, et al. Genomic profiling of Richter’s syndrome: recurrent lesions and differences with de novo diffuse large B-cell lymphomas. Hematol Oncol. 2009.
72. Thapa DR, Li X, Jamieson BD, Martinez-Maza O. Overexpression of microRNAs from the miR-17-92 paralog clusters in AIDS-related non-Hodgkin's lymphomas. PLoS One. 2011; 6:e20781.
73. Rao E, Jiang C, Ji M, Huang X, Iqbal J, Lenz G, Wright G, Staudt LM, Zhao Y, McKeithan TW, Chan WC, Fu K. The miRNA-17 approximately 92 cluster mediates chemoresistance and enhances tumor growth in mantle cell lymphoma via PI3K/AKT pathway activation. Leukemia. 2012; 26:1064–1072.
74. Iqbal J, Shen Y, Liu Y, Fu K, Jaffe ES, Liu C, Liu Z, Lachel CM, Deffenbacher K, Greiner TC, Vose JM, Bhagavathi S, Staudt LM, et al. Genome-wide miRNA profiling of mantle cell lymphoma reveals a distinct subgroup with poor prognosis. Blood. 2012; 119:4939–4948.
75. Tagawa H, Karnan S, Suzuki R, Matsuo K, Zhang X, Ota A, Morishima Y, Nakamura S, Seto M. Genome-wide array-based CGH for mantle cell lymphoma: identification of homozygous deletions of the proapoptotic gene BIM. Oncogene. 2005; 24:1348–1358.
76. Schmitz R, Young RM, Ceribelli M, Jhavar S, Xiao W, Zhang M, Wright G, Shaffer AL, Hodson DJ, Buras E, Liu X, Powell J, Yang Y, et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature. 2012; 490:116–120.
77. Schiffman JD, Lorimer PD, Rodic V, Jahromi MS, Downie JM, Bayerl MG, Sanmann JN, Althof PA, Sanger WG, Barnette P, Perkins SL, Miles RR. Genome wide copy number analysis of paediatric Burkitt lymphoma using formalin-fixed tissues reveals a subset with gain of chromosome 13q and corresponding miRNA over expression. Br J Haematol. 2011; 155:477–486.
78. Scholtysik R, Kreuz M, Klapper W, Burkhardt B, Feller AC, Hummel M, Loeffler M, Rosolowski M, Schwaenen C, Spang R, Stein H, Thorns C, Trumper L, et al. Detection of genomic aberrations in molecularly defined Burkitt’s lymphoma by array-based, high resolution, single nucleotide polymorphism analysis. Haematologica. 2010; 95:2047–2055.
79. Boxer LM, Dang CV. Translocations involving c-myc and c-myc function. Oncogene. 2001; 20:5595–5610.
80. Erikson J, ar-Rushdi A, Drwinga HL, Nowell PC, Croce CM. Transcriptional activation of the translocated c-myc oncogene in burkitt lymphoma. Proc Natl Acad Sci U S A. 1983; 80:820–824.
81. Strobl LJ, Eick D. Hold back of RNA polymerase II at the transcription start site mediates down-regulation of c-myc in vivo. EMBO J. 1992; 11:3307–3314.
82. Taub R, Moulding C, Battey J, Murphy W, Vasicek T, Lenoir GM, Leder P. Activation and somatic mutation of the translocated c-myc gene in burkitt lymphoma cells. Cell. 1984; 36:339–348.
83. Mraz M, Malinova K, Kotaskova J, Pavlova S, Tichy B, Malcikova J, Stano KK, Smardova J, Brychtova Y, Doubek M, Trbusek M, Mayer J, Pospisilova S. miR-34a, miR-29c and miR-17–5p are downregulated in CLL patients with TP53 abnormalities. Leukemia. 2009; 23:1159–1163.
84. Suzuki HI, Yamagata K, Sugimoto K, Iwamoto T, Kato S, Miyazono K. Modulation of microRNA processing by p53. Nature. 2009; 460:529–533.
85. Moussay E, Wang K, Cho JH, van MK, Pierson S, Paggetti J, Nazarov PV, Palissot V, Hood LE, Berchem G, Galas DJ. MicroRNA as biomarkers and regulators in B-cell chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 2011; 108:6573–6578.
86. Okamura S, Arakawa H, Tanaka T, Nakanishi H, Ng CC, Taya Y, Monden M, Nakamura Y. p53DINP1, a p53-inducible gene, regulates p53-dependent apoptosis. Mol Cell. 2001; 8:85–94.
87. Weber A, Marquardt J, Elzi D, Forster N, Starke S, Glaum A, Yamada D, Defossez PA, Delrow J, Eisenman RN, Christiansen H, Eilers M. Zbtb4 represses transcription of P21CIP1 and controls the cellular response to p53 activation. EMBO J. 2008; 27:1563–1574.
88. Gaubatz S, Lindeman GJ, Ishida S, Jakoi L, Nevins JR, Livingston DM, Rempel RE. E2F4 and E2F5 play an essential role in pocket protein-mediated G1 control. Mol Cell. 2000; 6:729–735.
89. Fujimoto M, Naka T. SOCS1, a Negative Regulator of Cytokine Signals and TLR Responses, in Human Liver Diseases. Gastroenterol Res Pract. 2010; 2010.
90. Allsup DJ, Kamiguti AS, Lin K, Sherrington PD, Matrai Z, Slupsky JR, Cawley JC, Zuzel M. B-cell receptor translocation to lipid rafts and associated signaling differ between prognostically important subgroups of chronic lymphocytic leukemia. Cancer Res. 2005; 65:7328–7337.
91. Chen L, Apgar J, Huynh L, Dicker F, Giago-McGahan T, Rassenti L, Weiss A, Kipps TJ. ZAP-70 directly enhances IgM signaling in chronic lymphocytic leukemia. Blood. 2005; 105:2036–2041.
92. Gobessi S, Laurenti L, Longo PG, Sica S, Leone G, Efremov DG. ZAP-70 enhances B-cell-receptor signaling despite absent or inefficient tyrosine kinase activation in chronic lymphocytic leukemia and lymphoma B cells. Blood. 2007; 109:2032–2039.
93. Mockridge CI, Potter KN, Wheatley I, Neville LA, Packham G, Stevenson FK. Reversible anergy of sIgM-mediated signaling in the two subsets of CLL defined by VH-gene mutational status. Blood. 2007; 109:4424–4431.
94. Willimott S, Wagner SD. Stromal cells and CD40 ligand (CD154) alter the miRNome and induce miRNA clusters including, miR-125b/miR-99a/let-7c and miR-17-92 in chronic lymphocytic leukaemia. Leukemia. 2012; 26:1113–1116.
95. Croce CM. Causes and consequences of microRNA dysregulation in cancer. Nat Rev Genet. 2009; 10:704–714.
96. Krutzfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. 2005; 438:685–689.
97. Stenvang J, Petri A, Lindow M, Obad S, Kauppinen S. Inhibition of microRNA function by antimiR oligonucleotides. Silence. 2012; 3:1.
98. Janssen HL, Reesink HW, Lawitz EJ, Zeuzem S, Rodriguez-Torres M, Patel K, van der Meer AJ, Patick AK, Chen A, Zhou Y, Persson R, King BD, Kauppinen S, et al. Treatment of HCV infection by targeting microRNA. N Engl J Med. 2013; 368:1685–1694.
99. van der Ree MH, van der Meer AJ, de BJ, Maan R, van VA, Welzel TM, Zeuzem S, Lawitz EJ, Rodriguez-Torres M, Kupcova V, Wiercinska-Drapalo A, Hodges MR, Janssen HL, et al. Long-term safety and efficacy of microRNA-targeted therapy in chronic hepatitis C patients. Antiviral Res. 2014; 111:53–59.
100. Dereani S, Macor P, D’Agaro T, Mezzaroba N, Dal-Bo M, Capolla S, Zucchetto A, Tissino E, Del PG, Zorzet S, Gattei V, Bomben R. Potential therapeutic role of antagomiR17 for the treatment of chronic lymphocytic leukemia. J Hematol Oncol. 2014; 7:79.