
INTRODUCTION
The peroxisome proliferator-activated receptor (PPAR) nuclear receptor subfamily regulates a number of metabolic processes, including fatty acid β-oxidation, glucose utilization, cholesterol transport, energy balance and adipocyte differentiation [1-4]. PPARs also play important roles in modulating inflammation, proliferation, angiogenesis and neoplasia [5-8]. PPARs function as heterodimeric partners with RXR, and require high-affinity binding of PPAR isotype-specific ligands to engage transcription. Of the three subtypes, PPARγ is the major species expressed in the mammary gland and in primary and metastatic breast cancer and breast cancer cell lines [5].
PPARγ and PPARδ modulate cell fate in the mammary gland [6, 9, 10], suggesting that PPAR agonists or antagonists may have the potential to regulate differentiation and hence tumor progression. PPARγ agonists are potent chemopreventive agents in mammary carcinogenesis [11], which is consistent with the enhancement of mammary tumorigenesis by PPARγ heterozygosity [12]. In a large percentage of follicular thyroid cancers, PPARγ exists as the dominant-negative fusion protein, Pax8-PPARγ, associated with the t(2;3)(q13;p25) translocation [13]. Pax8PPARγ potently blocks PPARγ function [13, 14], rather than merely serving as a low affinity receptor that can be activated at high ligand concentrations [15]. Importantly, the irreversible PPARγ ‘suicide’ inhibitor, GW9662 [16], mimics the growth promoting effects of Pax8PPARγ in thyroid cells [17], suggesting that selective pharmacological manipulation of PPARγ is feasible.
Although many studies have addressed the interactions between different nuclear receptor subfamilies, an area of relevance to breast cancer is the inhibitory effect of PPARγ on ERα (ER) promoter activation through its interaction with ER response elements [18]. Conversely, ER may bind to PPARγ response elements (PPREs) to inhibit PPAR-dependent transcription [19]. The ER and PPARγ pathways produce opposite effects on PI3K/AKT signaling, accounting in part, for the divergent responses produced by their cognate ligands in estrogen-dependent human breast cancer cells [19]. These findings suggest that suppression of PPARγ may upregulate ER expression in tumors to allow the implementation of anti-estrogen therapy. As a proof of principle, this was demonstrated by the effectiveness of the ER antagonist, fulvestrant, in preventing mammary tumorigenesis in MMTV-Pax8PPARγ mice, in which tumors normally present with a more aggressive progenitor cell phenotype [10]. Therefore, from a chemoprevention perspective, it would be important to be able to mimic the MMTV-Pax8PPARγ transgene pharmacologically by administering a PPARγ antagonist to increase the percentage of ER+ tumors and render them amenable to anti-estrogen therapy. This approach would be dependent on whether a PPAR antagonist could be developed with favorable specificity and pharmacokinetic properties to achieve selective and sustained inhibition of PPARγ. Examples of PPARγ antagonists are the suicide inhibitors, GW9662 (2-chloro-5-nitro-N-phenylbenzamide) [16], 2-bromo-5-nitro-N-phenylbenzamide [20] and the structurally similar T0070907 [21], as well as the partial PPARγ agonists, GW0072 [22] and L-764406 [23]. Although, GW9662 and T0070907 have also been reported to produce off-target effects in vitro [24-26], their in vivo selectivity has yet to be demonstrated. In this report, we show that GW9662 when administered continuously in the diet beginning at the onset of mammary carcinogenesis induces ER-responsive tumors susceptible to fulvestrant therapy. Furthermore, GW9662 inhibited a PPARγ-dependent metabolic gene expression signature, including PPARγ itself. These results are the first to demonstrate that GW9662 is at least in part PPARγ-selective, and can induce sensitivity to anti-estrogen therapy.
RESULTS
GW9662 induces sensitivity to antiestrogen therapy
To evaluate the chemopreventive effect of GW9662 on mammary tumor development, carcinogenesis was induced in FVB mice by progestin and DMBA treatment. Animals were maintained on either a control diet or a diet supplemented with 0.1% GW9662 beginning one day after the last dose of DMBA, and both groups were administered either vehicle or 250 mg/kg fulvestrant by subcutaneous injection every other week (Figure 1). Animals maintained on GW9662 alone exhibited a modest reduction in survival (Figure 1A) similar to what was observed previously in MMTV-Pax8PPARγ transgenic mice [10], but not a reduction in the total number of tumors (Figure 1B). While no significant difference in survival was noted for fulvestrant-treated control mice, a marked increase in survival (Figure 1A) and a reduction in tumor number (Figure 1B) were observed in animals maintained on GW9662 and treated with fulvestrant. Consistent with these findings was an increase in ER expression in tumors from GW9662-treated mice in comparison to animals maintained on the control diet as determined by immunohistochemical (Figure 2A) and western analyses (Figure 2B). Increased ER, as well as PR expression, was accompanied by an increase in Esr1 and Pgr mRNA levels (Figure 3A). GW9662 treatment also resulted in a reduction of PPARγ protein (Figure 2B) and mRNA (Figure 3A). Histological evaluation of the tumors indicated that GW9662, but not fulvestrant, produced a significant increase in the percentage of adenocarcinomas (P=0.0333) (Table S1).
Table 1: Metabolic genes downregulated by GW9662. Shown are genes whose signal was >300 in either group and were changed [3] 2.5-fold in GW9662-treated animals vs. control. The full list of changes in gene expression are presented in Table S2. Gene symbols in bold contain PPREs.
|
Raw Value |
|||
Gene symbol |
Gene Title |
Fold Change |
WT |
GW9662 |
Ces3 |
carboxylesterase 3 |
-105.7 |
2733 |
25 |
Gys2 |
glycogen synthase 2 |
-74.7 |
558 |
7 |
Lep |
leptin |
-74.7 |
1231 |
16 |
Aqp7 |
aquaporin 7 |
-55.6 |
3523 |
63 |
Pnpla3 |
patatin-like phospholipase domain containing 3 |
-53.9 |
1301 |
24 |
Cox8b |
cytochrome c oxidase, subunit VIIIb |
-51.0 |
1324 |
26 |
Cyp2e1 |
cytochrome P450 family 2, subfamily e, polypeptide 1 |
-44.3 |
5209 |
118 |
Pck1 |
phosphoenolpyruvate carboxykinase 1, cytosolic |
-43.9 |
3071 |
70 |
Retn |
resistin |
-35.7 |
10637 |
298 |
Rbp4 |
retinol binding protein 4, plasma |
-33.9 |
3187 |
94 |
Lao1 |
L-amino acid oxidase 1 |
-30.2 |
3092 |
103 |
Fabp3 |
fatty acid binding protein 3, muscle and heart |
-27.0 |
886 |
33 |
Cd36 |
CD36 antigen |
-22.5 |
6726 |
306 |
Car4 |
carbonic anhydrase 4 |
-22.3 |
982 |
44 |
Fabp4 |
fatty acid binding protein 4, adipocyte |
-21.7 |
7777 |
3543 |
Adipoq |
adiponectin, C1Q and collagen domain containing |
-21.6 |
10299 |
522 |
Adig |
adipogenin |
-20.8 |
1676 |
85 |
Acsl1 |
acyl-CoA synthetase long-chain family member 1 |
-18.6 |
3172 |
374 |
Lipe |
lipase, hormone sensitive |
-16.2 |
1329 |
82 |
Hsd11b1 |
hydroxysteroid-11-beta dehydrogenase 1 |
-15.7 |
28.15 |
1.79 |
Pparg |
peroxisome proliferator activated receptor gamma |
-13.9 |
967 |
69 |
Pc |
pyruvate carboxylase |
-13.1 |
1588 |
95 |
Dgat2 |
diacylglycerol O-acyltransferase 2 |
-12.4 |
4000 |
521 |
Cel |
carboxyl esterase lipase |
-12.1 |
955 |
79 |
Acacb |
acetyl-Coenzyme A carboxylase beta |
-11.5 |
530 |
46 |
Acaa1b |
acetyl-Coenzyme A acyltransferase 1B |
-10.6 |
696 |
66 |
Ephx2 |
epoxide hydrolase 2, cytoplasmic |
-10.0 |
1402 |
140 |
Lpl |
lipoprotein lipase |
-9.6 |
6823 |
713 |
Pgam2 |
phosphoglycerate mutase 2 |
-8.9 |
628 |
70 |
Cox6a2 |
cytochrome c oxidase, subunit VI a, polypeptide 2 |
-8.1 |
405 |
50 |
Fasn |
fatty acid synthase |
-7.3 |
11558 |
1579 |
Ptger3 |
prostaglandin E receptor 3 (subtype EP3) |
-7.1 |
1106 |
157 |
Sorbs1 |
sorbin and SH3 domain containing 1 |
-6.5 |
2532 |
581 |
Pygl |
liver glycogen phosphorylase |
-6.4 |
1600 |
250 |
Scd1 |
stearoyl-Coenzyme A desaturase 1 |
-6.4 |
7943 |
2026 |
Chpt1 |
choline phosphotransferase 1 |
-5.8 |
1658 |
327 |
Slc1a5 |
solute carrier family 1 (neutral amino acid transporter), member 5 |
-5.6 |
2664 |
476 |
Acss2 |
acyl-CoA synthetase short-chain family member 2 |
-5.5 |
969 |
160 |
Mgll |
monoglyceride lipase |
-5.5 |
3443 |
632 |
Pnpla2 |
patatin-like phospholipase domain containing 2 |
-5.1 |
4552 |
890 |
Eno3 |
enolase 3, beta muscle |
-4.9 |
672 |
136 |
Cyp2f2 |
cytochrome P450 family 2, subfamily f, polypeptide 2 |
-4.9 |
550 |
112 |
Lpin1 |
lipin 1 |
-4.8 |
1167 |
268 |
Ido1 |
indoleamine 2,3-dioxygenase 1 |
-4.8 |
406 |
85 |
Sod3 |
superoxide dismutase 3, extracellular |
-4.7 |
678 |
145 |
Cyp4b1 |
cytochrome P450 family 4, subfamily b, polypeptide 1 |
-4.6 |
1286 |
283 |
Igf1 |
insulin-like growth factor 1 |
-4.3 |
558 |
153 |
Aacs |
acetoacetyl-CoA synthetase |
-4.1 |
1176 |
320 |
Acox1 |
acyl-Coenzyme A oxidase 1, palmitoyl |
-4.1 |
920 |
225 |
Xdh |
xanthine dehydrogenase |
-3.9 |
1400 |
362 |
Gpd1 |
glycerol-3-phosphate dehydrogenase 1 (soluble) |
-3.6 |
1710 |
283 |
Gpt2 |
glutamic pyruvate transaminase (alanine aminotransferase) 2 |
-3.6 |
1338 |
438 |
Gpt |
glutamic pyruvic transaminase, soluble |
-3.6 |
577 |
159 |
Abca8a |
ATP-binding cassette, sub-family A (ABC1), member 8a |
-3.5 |
1675 |
478 |
Me1 |
malic enzyme 1, NADP(+)-dependent, cytosolic |
-3.4 |
2810 |
900 |
Aqp1 |
aquaporin 1 |
-3.4 |
2841 |
848 |
Retsat |
retinol saturase (all trans retinol 13,14 reductase) |
-3.3 |
488 |
146 |
Slc27a1 |
solute carrier family 27 (fatty acid transporter), member 1 |
-3.2 |
522 |
163 |
Lipa |
lysosomal acid lipase A |
-3.2 |
374 |
117 |
Fads3 |
fatty acid desaturase 3 |
-3.2 |
1545 |
485 |
Alox12e |
arachidonate lipoxygenase, epidermal |
-3.1 |
818 |
262 |
Elovl6 |
ELOVL family member 6, elongation of long chain fatty acids (yeast) |
-3.1 |
1088 |
320 |
Gpam |
glycerol-3-phosphate acyltransferase, mitochondrial |
-3.0 |
2818 |
947 |
Nr1h3 |
nuclear receptor subfamily 1, group H, member 3 (LXR) |
-3.0 |
1137 |
379 |
Acly |
ATP citrate lyase |
-2.9 |
993 |
343 |
Pik3r1 |
phosphatidylinositol 3-kinase, regulatory subunit, polypeptide 1 (p85 alpha) |
-2.9 |
558 |
192 |
Rbp7 |
retinol binding protein 7, cellular |
-2.9 |
1212 |
418 |
Slc2a4 |
solute carrier family 2 (faciltated glucose transporter), member 4 |
-3.2 |
522 |
163 |
Crat |
carnitine acetyltransferase |
-2.8 |
537 |
191 |
Slc2a4 |
solute carrier family 2 (facilitated glucose transporter), member 4 |
-2.8 |
1021 |
364 |
Sord |
sorbitol dehydrogenase |
-2.8 |
700 |
250 |
Ehhadh |
enoyl-Coenzyme A, hydratase/3-hydroxyacyl Coenzyme A dehydrogenase |
-2.7 |
341 |
126 |
Hk2 |
hexokinase 2 |
-2.7 |
1447 |
534 |
Lpgat1 |
lysophosphatidylglycerol acyltransferase 1 |
-2.7 |
399 |
150 |
Gbe1 |
glucan (1,4-alpha-)branching enzyme |
-2.7 |
723 |
267 |
Apod |
apolipoprotein D |
-2.6 |
4011 |
1526 |
Gatm |
glycine amidinotransferase (L-arginine:glycine amidinotransferase |
-2.6 |
400 |
152 |
Ltc4s |
leukotriene C4 synthase |
-2.6 |
467 |
179 |
Pfkfb1 |
6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 1 |
-2.6 |
320 |
124 |
Plin2 |
perilipin 2 |
-2.5 |
6773 |
2671 |
Cebpa |
CCAAT/enhancer binding protein (C/EBP), alpha |
-2.5 |
1819 |
734 |
Dgat1 |
diacylglycerol O-acyltransferase 1 |
-2.5 |
1065 |
425 |
Ptgs1 |
prostaglandin-endoperoxide synthase 1 |
-2.5 |
362 |
145 |
Shown are genes whose signal was >300 in either group and were changed ≥2.5-fold in GW9662-treated animals vs. control. The full list of changes in gene expression are presented in Table S2. Gene symbols in bold contain PPREs |
||||
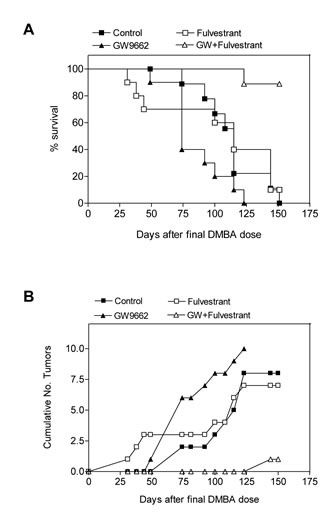
Figure 1: GW9662 enhances the sensitivity of mammary tumors to fulvestrant. (A) Survival curves of mice administered a control diet, a diet supplemented with 0.1% (w/w) GW9662, 250 mg/kg fulvestrant administered s.c. every other week or the combination of the GW9662 diet and fulvestrant. GW9662 treatment alone produced a significant reduction in survival vs. control mice (P=0.0382), but not vs. fulvestrant treatment (P=0.0759); fulvestrant treatment alone did not significantly affect survival (P=0.7223). GW9662 and fulvestrant treatment produced a significant increase in survival vs. fulvestrant (P=0.0008) or GW9662 (P=0.0001) treatment alone. Each group contained 10 mice. Statistical significance was determined by the log rank test. (B) Tumor formation in the experimental groups indicated in (A). Neither GW9662 (P=0.3942) nor fulvestrant (P=0.3339) treatment alone significantly affected tumor number vs. control mice. GW9662 and fulvestrant treatment produced a significant reduction in tumor number vs. either fulvestrant (P=0.0001) or GW9662 (P=0.0004) treatment alone. Each group contained 10 mice. Statistical significance was determined by the two-tailed Student’s t test.
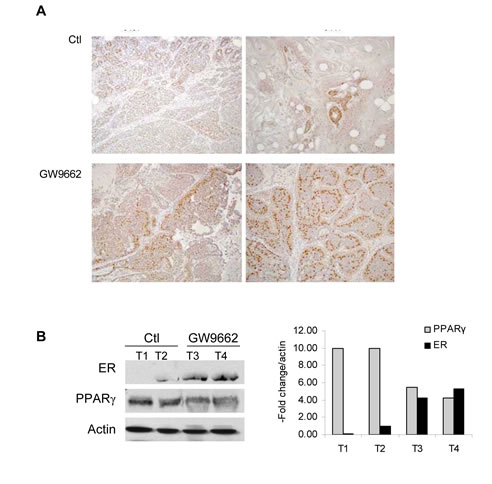
Figure 2: ER expression in adenocarcinomas from control and GW9662 mice. (A) Immunohistochemical detection of ER expression. Two representative tumors from control and GW9662-treated mice are shown. ER expression was increased following GW9662 treatment. Magnification 200X. (B) Western analysis of ER and PPARγ expression. Two representative tumors from control and GW9662-treated mice are shown. ER expression was increased, and PPARγ expression reduced following GW9662 treatment. The bar graph represents quantitation of the western blot normalized to actin expression.
Gene expression analysis
Gene microarray analysis of tumors from control and GW9662-treated animals indicated that 356 genes were differentially affected by GW9662 treatment (Figure 3B). Of the 303 genes downregulated by GW9662, 24% were metabolic genes, and 55% of which contain PPREs (Table 1). In addition, there were 10 genes regulated by transcription factors Cebpa and Pouf1, which are PPAR-regulated. Overall, 67% of the metabolic genes were directly or indirectly regulated by GW9662. Gene ontology of the differentially expressed genes (Table S1) indicated that the largest percentage were associated with transport, glucose and lipid metabolism, and developmental processes (Table 2). Pathway linkage analysis revealed that most of the genes whose expression was downregulated by GW9662 were linked directly or indirectly to PPARγ (Figure 4), whereas, those genes whose expression was increased by GW9662 were connected to Mapk3, Mapk8 and Akt signaling (Figure S1). Interestingly, the majority of the genes upregulated by GW9662 were associated with transcription, splicing, processing and translation of RNA (Table S2). In particular, RBM39, whose expression was increased 6.6-fold by GW9662, was recently reported to be increased in ER-dependent mammary tumors developing in caveolin-1 knockout mice [27].
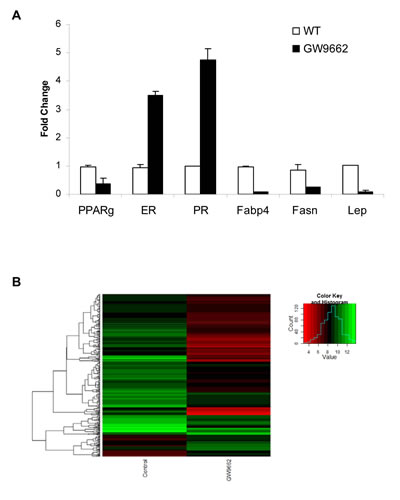
Figure 3: (A) qRT-PCR analysis of gene expression in adenocarcinomas from control and GW9662-treated mice. Gene selection was based on the data in Table 1. (B) Heat map of changes in gene expression based on the data in Table S2.
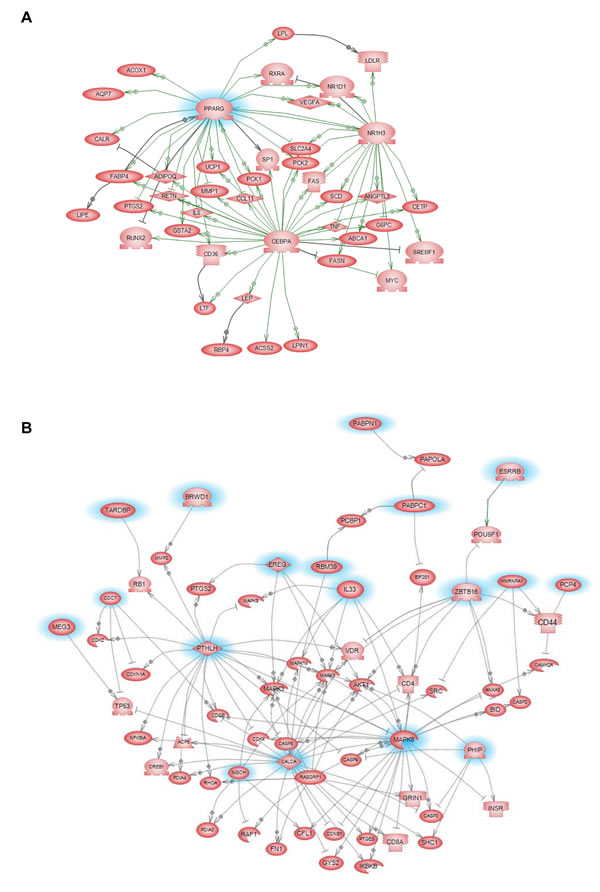
Figure 4: GW9662 signaling pathways in tumors from control and GW9662-treated animals. Pathways are based on the expression of genes that were reduced ≥2.5-fold by GW9662 in Table S1. Metabolic signaling pathways associated with genes that were downregulated by GW9662.
Table 2: Gene ontology of differentially expressed genes affected by GW9662. Shown are enrichment data with P<0.05 by Fisher’s Exact test.
Name |
Total Entities |
Overlap |
Overlapping Entities |
p-value |
DOWNREGULATED: |
|
|||
lipid metabolism |
342 |
23 |
CHPT1,CD36,LEP,LPL,LIPE,APOD,NR1H3,SLC27A1,LIPA,ACSL1, HSD11B1,DGAT2,CRAT,ACLY,LPIN1,ACOX1,EHHADH,PNPLA2, PCK1,PNPLA3,MGLL,AACS,FADS3 |
1.92E-26 |
metabolism |
858 |
21 |
LPGAT1,FASN,LIPE,ACACB,SLC27A1,EPHX2,ACSL1,GPAM, HSD11B1,ME1,PC,ACLY,PFKFB1,ACOX1,EHHADH,PNPLA2, GPD1,PNPLA3,ACSS2,PGAM2,AACS |
3.71E-15 |
transport |
1807 |
15 |
AQP1,CD36,APOD,SLC2A4,SLC27A1,FABP4,FABP3,CRAT,SORBS1, SLC1A5,AQP7,RBP4,CRABP1,RBP7,FADS3 |
6.01E-05 |
oxidation reduction |
702 |
14 |
CYP2E1,PTGS1,SOD3,XDH,FASN,HSD11B1,ME1,ACOX1,EHHADH,GPD1, CYP4B1,SORD,RETSAT,FADS3 |
3.58E-09 |
fatty acid metabolism |
110 |
12 |
CD36,PPARG,SLC27A1,LIPA,FABP4,ACSL1,GPAM,FABP3,CRAT, ACOX1,EHHADH,AACS |
1.21E-16 |
response to drug |
295 |
10 |
ADIPOQ,PPARG,LIPE,ACACB,FABP4,ACSL1,AQP7,SORD,ENO3,AACS |
5.40E-09 |
response to insulin |
37 |
10 |
LEP,RETN,PIK3R1,PFKFB1,PCK1,RBP4,PPARG,SORBS1,LPIN1,NRIH3 |
5.81E-10 |
fat cell differentiation |
29 |
9 |
ADIG,CEBPA,ADIPOQ,PPARG,SLC2A4,FABP4,IGF1,AACS,RETN |
1.21E-10 |
lipid biosynthesis |
115 |
8 |
PTGS1,FASN,DGAT2,PC,ACLY,ELOVL6,ACSS2,FADS3 |
7.24E-10 |
gluconeogenesis |
33 |
7 |
GPT,PC,PFKFB1,GPD1,PCK1,RBP4,PGAM2 |
2.60E-12 |
generation of precursor metabolites & energy |
63 |
7 |
CEBPA,ADIPOQ,GYS2,ACOX1,AQP7,GBE1,COX6A2 |
3.17E-10 |
response to glucocorticoids |
95 |
6 |
CEBPA,ADIPOQ,IGF1,FABP4,PIK3R1,PFKFB1 |
1.93E-07 |
response to nutrients |
117 |
6 |
CEBPA,ADIPOQ,PPARG,ACSL1,GATM,AACS |
6.62E-07 |
lipid catabolism |
113 |
6 |
CEL,LPL,LIPE,LIPA,PNPLA2,PNPLA3 |
5.40E-07 |
glucose homeostasis |
50 |
6 |
ADIPOQ,PPARG,SLC2A4,PCK1,RBP4,PYGL |
3.87E-09 |
spermatogenesis |
353 |
5 |
ADIG,ACOX1,AQP7,RBP4,PGAM2 |
2.38E-03 |
carbohydrate metabolism |
296 |
5 |
SLC2A4,ME1,GPD1,PYGL,GBE1 |
1.10E-03 |
fatty acid biosynthesis |
78 |
5 |
PTGS1,FASN,ACACB,ELOVL6,FADS3 |
1.98E-06 |
triglyceride biosynthesis |
11 |
5 |
LPL,GPAM,DGAT1,DGAT2,PNPLA3 |
4.97E-11 |
glucose metabolism |
115 |
5 |
ADIPOQ,LEP,HK2,PIK3R1,SORD |
1.33E-05 |
inflammatory response |
293 |
4 |
PPARG,LIPA,EPHX2,MGLL |
7.45E-03 |
lung development |
106 |
4 |
CEBPA,LIPA,HSD11B1,RBP4 |
1.78E-04 |
organ regeneration |
49 |
4 |
CEBPA,PPARG,LPIN1,PFKFB1 |
8.49E-06 |
triglyceride catabolism |
13 |
4 |
LPL,LIPE,PNPLA2,PNPLA3 |
3.08E-08 |
fatty acid beta-oxidation |
32 |
4 |
ADIPOQ,FABP3,ACOX1,EHHADH |
1.49E-06 |
glycolysis |
68 |
4 |
HK2,PFKFB1,ENO3,PGAM2 |
3.14E-05 |
regulation of transcription |
159 |
4 |
CEBPA,NR1H3,FABP4,PPARG |
8.53E-03 |
response to ethanol |
83 |
3 |
ADIPOQ,RBP4,AACS |
1.37E-03 |
long-chain fatty acid transport |
12 |
3 |
CD36,PPARG,FABP3 |
3.76E-06 |
aging |
101 |
3 |
PTGS1,PIK3R1,ENO3 |
2.41E-03 |
fatty acid oxidation |
18 |
3 |
CD36,ADIPOQ,PPARG |
1.38E-05 |
glycogen metabolism |
41 |
3 |
GYS2,PYGL,GBE1 |
1.72E-04 |
phospholipid biosynthesis |
49 |
3 |
LPGAT1,CHPT1,GPAM |
2.94E-04 |
regulation of cell proliferation |
135 |
3 |
PTGS1,CEBPA,IGF1 |
5.44E-03 |
negative regulation of foam cell differentiation |
10 |
3 |
ADIPOQ,PPARG,NR1H3 |
2.06E-06 |
UPREGULATED: |
|
|||
regulation of transcription |
2501 |
9 |
ZBTB16,MAPK8,RHOX5,BRWD1,ESRRB,RBM39,TARDBP,NFIB,THRAP3 |
2.24E-02 |
RNA splicing |
238 |
5 |
HNRNPA1,PABPC1,RBM39,TARDBP,RBMX |
2.13E-05 |
mRNA processing |
277 |
5 |
PABPN1,HNRNPA1,PABPC1,RBM39,TARDBP |
4.40E-05 |
cell proliferation |
324 |
4 |
PTHLH,EREG,ZBTB16,NFIB |
1.13E-03 |
central nervous system development |
140 |
3 |
ZBTB16,PCP4,NPTX1 |
1.04E-03 |
translational elongation |
161 |
3 |
RPS25,RPS24,RPL41 |
1.55E-03 |
cell-cell signaling |
275 |
3 |
CALCA,PTHLH,EREG |
6.96E-03 |
apoptosis |
550 |
3 |
ZBTB16,SLC5A8,NISCH |
4.29E-02 |
Shown are enrichment data with P<0.05 by Fisher's Exact test. |
||||
DISCUSSION
The present study was designed to determine if pharmacological inhibition of PPARγ could sensitize mammary tumor growth to antiestrogen therapy. This concept was based on our previous finding that induction of mammary carcinogenesis in transgenic mice expressing the dominant-negative Pax8PPARγ fusion protein resulted in increased ER expression and responsiveness to the ER antagonist, fulvestrant [10]. MMTV-Pax8PPARγ transgenic mice represent a rare mouse model in which the mammary gland exhibits a progenitor cell phenotype that results in the preferential development of ER+ rather than ER- tumors of mixed lineage following progestin/DMBA treatment [10, 28]. A similar mammary tumor phenotype developed in caveolin-1 knockout mice that was also associated with the induction of several stem/progenitor cell markers, including RBM39 [27], as found in the present study. RBM39 functions primarily in RNA splicing and may also be a putative partner of the co-activator Ncoa6/PRIP [29]. Thus, one unexpected finding was that GW9662 upregulated a number of genes associated with transcription, processing, splicing and translation that likely contribute to the diversity of the proteome [30].
GW9662 is an irreversible PPARγ antagonist [16], although in vitro cell studies have also reported off-target effects [24-26]. However, there are no in vivo studies that have established whether GW9662 is PPARγ-selective. In one instance, GW9662 was shown to reduce high fat diet-induced obesity in rats when administered in the diet at a concentration of 0.1% [31], which was identical to the GW9662 diet used in our study. GW9662 was also shown to block the anti-inflammatory effects of the PPARγ agonist, rosiglitazone, in endotoxin-induced acute lung injury after intravenous administration [32]. Based on gene array profiling, we found that GW9662 elicited PPARγ specificity based on its direct and indirect inhibitory effects on the expression of metabolic genes known to be under the control of PPARs.
An important caveat to the use of GW9662 is its ability to induce a modest acceleration of tumorigenesis when administered orally at the onset of carcinogenesis. We also observed a similar effect in MMTV-Pax8PPARγ mice following progestin/DMBA mammary carcinogenesis [10]. While this has not been reported previously, the ability of GW9662 to inhibit cell growth in vitro similarly to PPARγ agonists [24, 33, 34] suggests the presence of “off-target” effects. The increase in tumorigenesis observed with GW9662 and the dominant-negative Pax8PPARγ transgene suggests that partial antagonists rather than full antagonists or drugs with greater specificity may be a useful approach for further studies. Clearly, additional pharmacokinetic and pharmacodynamic studies in vivo are needed to establish the bioavailability and metabolic effects of GW9662. Overall, the positive aspect of inhibiting PPARγ was its ability to sensitize tumors to the ER antagonist fulvestrant, suggesting the potential for such an approach for hormone-insensitive malignancies.
MATERIALS AND METHODS
Animal model
FVB wild-type (WT) mice were obtained from Taconic Farms, Germantown, N.Y. All animal studies were conducted under protocols approved by the Georgetown University Animal Care and Use Committee.
Mammary carcinogenesis
Five week-old WT mice were treated with medroxyprogesterone acetate and DMBA as previously described [9, 28]. Briefly, mice were injected s.c. with 15 mg medroxyprogesterone acetate suspension (Depo-Provera?), and after seven days were administered four weekly doses of 1 mg DMBA/0.1 ml cottonseed oil by gavage. One day after the last dose of DMBA, mice were divided into four groups of 10 mice each: 1) one group was maintained on standard Purina Rodent Chow 5001, 2) one group was maintained on chow supplemented with 0.1% (w/w) GW9962, 2) one group was maintained on chow supplemented with GW9662 and injected s.c. every other week with 250 mg/kg fulvestrant (Faslodex®) and 4) one group was injected with 250 mg/kg fulvestrant every other week. GW9662 was provided by the Chemoprevention Branch, NCI. The histopathology of the resulting tumors is presented in Table S1.
Antibodies
The source of antibodies, their dilution and use were the following: rabbit anti-ERα (sc-542, Santa Cruz Biotechnology, 1:200 for IHC, 1:1,000 for western); rabbit anti-PgR (sc-538, Santa Cruz Biotechnology, 1:200 for IHC, 1:1,000 for western).
Immunohistochemistry
IHC analysis was carried out as previously described [9, 10, 28].
Western Blotting
Western blotting was carried out as previously described [10]. Briefly, tissue was frozen in liquid nitrogen and pulverized in a mortar and pestle, and mixed with lysis buffer containing: 0.1% SDS, 0.5% NP-40, phenylmethylsulfonyl fluoride, 1 mM sodium vanadate, 50 mM sodium fluoride, 10 mM β-glycerophosphate, 5 mM sodium pyrophosphate, and protease inhibitor cocktail (Roche Diagnostics). Following incubation on ice for 30 min, lysates were cleared by centrifugation for 15 min at 13,000 x g at 4oC. Protein concentrations were determined by the Coomassie Plus Protein Assay (Pierce), and 50 μg of lysate was separated in a 4-12% NuPAGE Bis-Tris gel (Invitrogen). After wet transfer, membranes were blocked for 1 hr at room temperature in TBS (pH 7.4) containing 5% non-fat dry milk and 0.1% Tween 20. Primary antibody was incubated overnight at 4°C, and secondary antibody was incubated for 1 hr at room temperature. Proteins were visualized with either SuperSignal West Pico or SuperSignal West Dura (Pierce).
Gene Microarray Analysis
Total RNA was extracted using an RNeasy Mini Kit (Qiagen) following the manufacturer’s protocol as previously described [10, 35]. cRNA was synthesized using the Affymetrix (Santa Clara, CA) protocol with minor modifications as described [28]. Biotin-labeled cRNA was fragmented for 35 min at 94°C and hybridized overnight to an Affymetrix mouse 430A 2.0 GeneChip® representing approximately 22,000 annotated mouse genes by the Genomics and Epigenomics Shared Resource, Lombardi Comprehensive Cancer Center, Georgetown University. Hybridization signals were detected with an Agilent Gene Array scanner, and grid alignment and raw data generation performed with Affymetrix GeneChip® Operating software 1.1. Changes in gene expression with a signal ≥300 (log2 ≥8.1) and ≥3-fold change [9, 35, 36] were clustered hierarchically with CIMiner software (National Cancer Institute, NIH). Array data are presented in Table S2, and complete data files were deposited in the GEO database under accession no. GSE33762.
Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
Total RNA was extracted using the RNAeasy Mini Kit (Qiagen, Valencia, CA) according to the manufacturer’s protocol as previously described [10, 35]. One µg of RNA was reverse transcribed in a total volume of 20 µl using the Cloned AMV First-Strand cDNA Synthesis kit (Invitrogen). PCR was performed in triplicate in an ABI 7900 instrument (Applied Biosystems, Foster City, CA) using SYBRGreen detection (Applied Biosystems, Foster City, CA) according to the manufacturer’s protocol. qRT-PCR primers were designed using the primer design tool at http://www.idtdna.com/Scitools/Applications/RealTimePCR/. Efficiencies of all primer sets (Table S1) were validated using a standard curve of five serial cDNA dilutions in water in duplicate. Primers were acceptable if the deviation from the slope of the standard curve was <0.3, and if the melting curve showed only one product. The expression of each target gene was normalized to the expression of GAPDH, and the relative quantification method was applied using SDS2.3 software (Applied Biosystems, Foster City, CA). Primers are listed in Table S3.
Statistical Analysis
Survival curves were analyzed by Pearson’s log rank test and cumulative tumor formation by Student’s two-tailed t test at a significance level of P≤0.05.
ACKNOWLEDGEMENTS
This study was supported by contract 1NO1 CN43302-WA19 from the National Cancer Institute, NIH, and award P30CA051008 from the National Cancer Institute, NIH to the Lombardi Comprehensive Cancer Center (LCCC). This investigation was conducted using the Animal Research, Flow Cytometry, Genomics and Epigenomics, and Microscopy and Imaging Shared Resources of the LCCC, and by an animal facilities construction grant from the NIH. .
REFERENCES
1. Berger JP, Akiyama TE and Meinke PT. PPARs: therapeutic targets for metabolic disease. Trends Pharmacol Sci. 2005; 26(5):244-251.
2. Barish GD, Narkar VA and Evans RM. PPAR delta: a dagger in the heart of the metabolic syndrome. The Journal of clinical investigation. 2006; 116(3):590-597.
3. Evans RM, Barish GD and Wang YX. PPARs and the complex journey to obesity. Nat Med. 2004; 10(4):355-361.
4. Lehrke M and Lazar MA. The many faces of PPARgamma. Cell. 2005; 123(6):993-999.
5. Michalik L, Desvergne B and Wahli W. Peroxisome-proliferator-activated receptors and cancers: complex stories. Nat Rev Cancer. 2004; 4(1):61-70.
6. Glazer RI, Yuan H, Xie Z and Yin Y. PPARgamma and PPARdelta as Modulators of Neoplasia and Cell Fate. PPAR Res. 2008; 2008:247379.
7. Kopelovich L, Fay JR, Glazer RI and Crowell JA. Peroxisome proliferator-activated receptor modulators as potential chemopreventive agents. Mol Cancer Ther. 2002; 1(5):357-363.
8. Koeffler HP. Peroxisome proliferator-activated receptor gamma and cancers. Clin Cancer Res. 2003; 9(1):1-9.
9. Yin Y, Russell RG, Dettin LE, Bai R, Wei ZL, Kozikowski AP, Kopelovich L and Glazer RI. Peroxisome proliferator-activated receptor delta and gamma agonists differentially alter tumor differentiation and progression during mammary carcinogenesis. Cancer Res. 2005; 65(9):3950-3957.
10. Yin Y, Yuan H, Zeng X, Kopelovich L and Glazer RI. Inhibition of peroxisome proliferator-activated receptor gamma increases estrogen receptor-dependent tumor specification. Cancer Res. 2009; 69(2):687-694.
11. Suh N, Wang Y, Williams CR, Risingsong R, Gilmer T, Willson TM and Sporn MB. A new ligand for the peroxisome proliferator-activated receptor-gamma (PPAR-gamma), GW7845, inhibits rat mammary carcinogenesis. Cancer Res. 1999; 59(22):5671-5673.
12. Nicol CJ, Yoon M, Ward JM, Yamashita M, Fukamachi K, Peters JM and Gonzalez FJ. PPARgamma influences susceptibility to DMBA-induced mammary, ovarian and skin carcinogenesis. Carcinogenesis. 2004; 25(9):1747-1755.
13. Kroll TG, Sarraf P, Pecciarini L, Chen CJ, Mueller E, Spiegelman BM and Fletcher JA. PAX8-PPARgamma1 fusion oncogene in human thyroid carcinoma Science. 2000; 289(5483):1357-1360.
14. Yin Y, Yuan H, Wang C, Pattabiraman N, Rao M, Pestell RG and Glazer RI. 3-Phosphoinositide-Dependent Protein Kinase-1 Activates the Peroxisome Proliferator-Activated Receptor-{gamma} and Promotes Adipocyte Differentiation. Mol Endocrinol. 2006; 20:268-278.
15. Agostini M, Schoenmakers E, Mitchell C, Szatmari I, Savage D, Smith A, Rajanayagam O, Semple R, Luan J, Bath L, Zalin A, Labib M, Kumar S, Simpson H, Blom D, Marais D, et al. Non-DNA binding, dominant-negative, human PPARgamma mutations cause lipodystrophic insulin resistance. Cell Metab. 2006; 4(4):303-311.
16. Leesnitzer LM, Parks DJ, Bledsoe RK, Cobb JE, Collins JL, Consler TG, Davis RG, Hull-Ryde EA, Lenhard JM, Patel L, Plunket KD, Shenk JL, Stimmel JB, Therapontos C, Willson TM and Blanchard SG. Functional consequences of cysteine modification in the ligand binding sites of peroxisome proliferator activated receptors by GW9662. Biochemistry. 2002; 41(21):6640-6650.
17. Gregory Powell J, Wang X, Allard BL, Sahin M, Wang XL, Hay ID, Hiddinga HJ, Deshpande SS, Kroll TG, Grebe SK, Eberhardt NL and McIver B. The PAX8/PPARgamma fusion oncoprotein transforms immortalized human thyrocytes through a mechanism probably involving wild-type PPARgamma inhibition. Oncogene. 2004; 23(20):3634-3641.
18. Keller H, Givel F, Perroud M and Wahli W. Signaling cross-talk between peroxisome proliferator-activated receptor/retinoid X receptor and estrogen receptor through estrogen response elements. Mol Endocrinol. 1995; 9(7):794-804.
19. Bonofiglio D, Gabriele S, Aquila S, Catalano S, Gentile M, Middea E, Giordano F and Ando S. Estrogen receptor alpha binds to peroxisome proliferator-activated receptor response element and negatively interferes with peroxisome proliferator-activated receptor gamma signaling in breast cancer cells. Clin Cancer Res. 2005; 11(17):6139-6147.
20. Lee H, Finck BN, Jones LA, Welch MJ and Mach RH. Synthesis and evaluation of a bromine-76-labeled PPARgamma antagonist 2-bromo-5-nitro-N-phenylbenzamide. Nucl Med Biol. 2006; 33(7):847-854.
21. Lee G, Elwood F, McNally J, Weiszmann J, Lindstrom M, Amaral K, Nakamura M, Miao S, Cao P, Learned RM, Chen JL and Li Y. T0070907, a selective ligand for peroxisome proliferator-activated receptor gamma, functions as an antagonist of biochemical and cellular activities. J Biol Chem. 2002; 277(22):19649-19657.
22. Oberfield JL, Collins JL, Holmes CP, Goreham DM, Cooper JP, Cobb JE, Lenhard JM, Hull-Ryde EA, Mohr CP, Blanchard SG, Parks DJ, Moore LB, Lehmann JM, Plunket K, Miller AB, Milburn MV, et al. A peroxisome proliferator-activated receptor gamma ligand inhibits adipocyte differentiation. Proc Natl Acad Sci U S A. 1999; 96(11):6102-6106.
23. Elbrecht A, Chen Y, Adams A, Berger J, Griffin P, Klatt T, Zhang B, Menke J, Zhou G, Smith RG and Moller DE. L-764406 is a partial agonist of human peroxisome proliferator-activated receptor gamma. The role of Cys313 in ligand binding. J Biol Chem. 1999; 274(12):7913-7922.
24. Seargent JM, Yates EA and Gill JH. GW9662, a potent antagonist of PPARgamma, inhibits growth of breast tumour cells and promotes the anticancer effects of the PPARgamma agonist rosiglitazone, independently of PPARgamma activation. Br J Pharmacol. 2004; 143(8):933-937.
25. Lecomte J, Flament S, Salamone S, Boisbrun M, Mazerbourg S, Chapleur Y and Grillier-Vuissoz I. Disruption of ERalpha signalling pathway by PPARgamma agonists: evidences of PPARgamma-independent events in two hormone-dependent breast cancer cell lines. Breast Cancer Res Treat. 2008.
26. Schaefer KL, Takahashi H, Morales VM, Harris G, Barton S, Osawa E, Nakajima A and Saubermann LJ. PPARgamma inhibitors reduce tubulin protein levels by a PPARgamma, PPARdelta and proteasome-independent mechanism, resulting in cell cycle arrest, apoptosis and reduced metastasis of colorectal carcinoma cells. Int J Cancer. 2007; 120(3):702-713.
27. Cavallo F, Astolfi A, Iezzi M, Cordero F, Lollini PL, Forni G and Calogero R. An integrated approach of immunogenomics and bioinformatics to identify new Tumor Associated Antigens (TAA) for mammary cancer immunological prevention. BMC Bioinformatics. 2005; 6 Suppl 4:S7.
28. Yin Y, Bai R, Russell RG, Beildeck ME, Xie Z, Kopelovich L and Glazer RI. Characterization of medroxyprogesterone and DMBA-induced multilineage mammary tumors by gene expression profiling. Mol Carcinog. 2005; 44(1):42-50.
29. Astolfi A, Landuzzi L, Nicoletti G, De Giovanni C, Croci S, Palladini A, Ferrini S, Iezzi M, Musiani P, Cavallo F, Forni G, Nanni P and Lollini PL. Gene expression analysis of immune-mediated arrest of tumorigenesis in a transgenic mouse model of HER-2/neu-positive basal-like mammary carcinoma. Am J Pathol. 2005; 166(4):1205-1216.
30. David CJ and Manley JL. Alternative pre-mRNA splicing regulation in cancer: pathways and programs unhinged. Genes Dev. 2010; 24(21):2343-2364.
31. Nakano R, Kurosaki E, Yoshida S, Yokono M, Shimaya A, Maruyama T and Shibasaki M. Antagonism of peroxisome proliferator-activated receptor gamma prevents high-fat diet-induced obesity in vivo. Biochem Pharmacol. 2006; 72(1):42-52.
32. Liu D, Zeng BX, Zhang SH, Wang YL, Zeng L, Geng ZL and Zhang SF. Rosiglitazone, a peroxisome proliferator-activated receptor-gamma agonist, reduces acute lung injury in endotoxemic rats. Crit Care Med. 2005; 33(10):2309-2316.
33. Burton JD, Castillo ME, Goldenberg DM and Blumenthal RD. Peroxisome proliferator-activated receptor-gamma antagonists exhibit potent antiproliferative effects versus many hematopoietic and epithelial cancer cell lines. Anticancer Drugs. 2007; 18(5):525-534.
34. Kim KR, Choi HN, Lee HJ, Baek HA, Park HS, Jang KY, Chung MJ and Moon WS. A peroxisome proliferator-activated receptor gamma antagonist induces vimentin cleavage and inhibits invasion in high-grade hepatocellular carcinoma. Oncol Rep. 2007; 18(4):825-832.
35. Pollock C, Yin Y, Yuan H, Zeng X, King S, Li X, Kopelovich L, Albanese C and Glazer RI. PPARδ activation acts cooperatively with 3-phosphoinositide-dependent protein kinase-1 to enhance mammary tumorigenesis. PLoSONE. 2011; 6(1):e16215.
36. Yuan H, Upadhyay G, Yin Y, Kopelovich L and Glazer RI. Stem Cell Antigen-1 Deficiency Enhances the Chemopreventive Effect of Peroxisome Proliferator-Activated Receptor{gamma} Activation. Cancer Prev Res (Phila). 2011; 4(12):1-10.