
INTRODUCTION
Multiple myeloma (MM), the second most common hematologic malignancy in the United States, is characterized by the accumulation of clonal plasma cells in the bone marrow [1]. Although MM patients initially respond to therapy, they inevitably relapse due to the development of treatment resistance [2]. The introduction of the proteasome inhibitor bortezomib has improved clinical outcome of MM patients [3]. However, MM cells acquire resistance to bortezomib by diverse mechanisms [4-8]. Carfilzomib is a second generation proteasome inhibitor that was approved in 2012 by the U.S. Food and Drug Administration for the treatment of relapsed/refractory MM patients who have received at least two prior therapies including bortezomib and an immunomodulatory drug [9]. While encouraging, the overall response rates to carfilzomib monotherapy in pivotal phase II studies were <25% (17.1% in the PX-171-004 study and 23.7% in the PX-171-003-A1 study, respectively) [10, 11], indicating that the majority of the MM cells that became resistant to bortezomib also exhibited resistance to carfilzomib. Thus, it is important to elucidate the underlying mechanisms of proteasome inhibitor resistance in MM and identify novel agents that will enhance therapeutic efficacy of this class of anti-MM drugs.
MM exhibits considerable genetic heterogeneity, with particular cytogenetic abnormalities such as the t(4;14) chromosomal translocation consistently associated with poor outcome [12]. In previous work, we identified upregulated expression of the ABCB1-encoded P-glycoprotein multidrug resistance efflux pump in t(4;14)-positive KMS-34 MM cells following short-term exposure to carfilzomib [13]. To gain further insight into the various mechanisms of carfilzomib resistance, we subjected another t(4;14)-positive MM cell line, KMS-11, together with KMS-34 to long-term selection in increasingly higher concentrations of the drug, deriving carfilzomib-resistant KMS-11/Cfz and KMS-34/Cfz cells, respectively. Of note, overexpression of ABCB1 was not observed in KMS-11/Cfz cells. Gene set enrichment analysis (GSEA) [14] of microarray gene expression profiling data implicated increased expression of the pluripotency reprogramming factor Kruppel-like factor 4 (KLF4) [15] as contributing to carfilzomib resistance in both cases.
Depending on the type of cancer and genetic context, KLF4 can act as either a tumor suppressor or an oncogene [16]. Notably, high levels of KLF4 expression often occur in MM patients carrying the t(4;14) translocation [17, 18]. Moreover, it was previously reported that exogenous expression of KLF4 partially protected some MM cell lines from cytotoxicity induced by the alkylating agent melphalan, and the partial protection was attributed to a proliferation block [19]. In the current study, we found that acquisition of carfilzomib resistance in both t(4;14)-positive MM cell line models was associated with reduced cell proliferation, decreased plasma cell maturation, and activation of prosurvival autophagy. Specifically, we show that KLF4 plays a role in prosurvival autophagy by binding to the promoter regions and increasing the expression of SQSTM1 encoding the ubiquitin-binding adaptor protein sequestosome (SQSTM1/p62) that links the proteasomal and selective autophagic protein degradation pathways [20, 21]. Furthermore, resensitization of KMS-11/Cfz and KMS-34/Cfz cells to carfilzomib could be achieved by cotreatment with the autophagy inhibitor chloroquine [22].
RESULTS
KLF4 contributes to molecular phenotype of carfilzomib-resistant MM cells
KMS-11 and KMS-34 cells were exposed to stepwise increasing concentrations of carfilzomib over a period of 18 weeks: cells adapted to growth in 4 nM carfilzomib by 4 weeks, in 6 nM in another 6 weeks and in 12 nM after a further 8 weeks, albeit proliferating slower than parental cells not exposed to the drug. The resulting MM cell cultures, denoted KMS-11/Cfz and KMS-34/Cfz, respectively, retained resistance to carfilzomib even when tested after removal of selective pressure for approximately 8 weeks. In the current study, KMS-11/Cfz and KMS-34/Cfz cells were profiled for gene expression after 1 week of growth in the absence of carfilzomib together with parental KMS-11 and KMS-34 cells which had not been selected in the drug.
We employed GSEA to query gene sets in the Molecular Signature Database (MSigDB) to uncover processes or pathways shared between KMS-11/Cfz and KMS-34/Cfz cells that potentially contributed to carfilzomib resistance [14]. We first applied GSEA to examine gene sets from the canonical pathways (C2:CP) collection of MSigDB (1,330 gene sets). The most significantly enriched set of upregulated genes in the carfilzomib-resistant derivatives was the proteasome pathway (Kegg: hsa03050), with PSMB5 encoding the β5 proteasome subunit targeted by carfilzomib as the top-ranked gene (normalized enrichment score, NES = 2.62, false discovery rate, FDR < 0.001; Figure S1A) [23]. The strength of the GSEA method is its utility in identifying modest changes in expression of groups of genes distributed across entire networks or pathways [14]. Real-time reverse transcription polymerase chain reaction (qRT-PCR) analysis validated the microarray expression data that PSMB5 mRNA levels were only slightly increased (Table 1). Likewise, no marked increase was observed in mRNA for the immunoproteasome β5i/LMP7 subunit (encoded by PSMB8) that is also specifically targeted by carfilzomib [23]. Based on prior findings that bortezomib-resistant cell lines with 2- to 4-fold increased PSMB5 mRNA levels retained sensitivity to carfilzomib [24], these results suggested that additional mechanisms may contribute to carfilzomib resistance in KMS-11/Cfz and KMS-34/Cfz cells.
Table 1: Gene expression changes associated with acquisition of carfilzomib resistance (KMS-11/Cfz and KMS-34/Cfz) and KLF4 overexpression (KMS-11/KLF4) in MM cells
Gene |
Fold Change |
||
KMS-11/Cfz |
KMS-34/Cfz |
KMS-11/KLF4 |
|
CCND1 |
0.06 |
0.17 |
1.12 |
CYP1A1 |
3.02 |
2.77 |
2.17 |
GLIPR1 |
1.19 |
1.22 |
0.70 |
HGF |
0.76 |
0.45 |
0.65 |
HOXB7 |
1.33 |
1.69 |
1.38 |
ID1 |
3.61 |
2.29 |
1.63 |
IFIT3 |
1.25 |
1.87 |
0.73 |
IGF1 |
0.69 |
0.93 |
1.07 |
MAPT |
1.21 |
0.94 |
1.13 |
NQO1 |
1.85 |
1.25 |
1.21 |
NQO2 |
1.09 |
1.05 |
0.75 |
P4HA2 |
0.70 |
0.67 |
0.68 |
PSMB5 |
1.09 |
1.11 |
0.83 |
SLAMF7 |
0.12 |
0.53 |
0.59 |
TLR4 |
1.43 |
3.56 |
1.13 |
Shown are fold changes relative to corresponding parental cells (mean values of three qRT-PCR experiments).
It was recently demonstrated that MM cells can acquire resistance to bortezomib via de-commitment to plasma cell differentiation [7]. Notably, among 1,910 gene sets in the immunologic signatures (C7) collection, three of those that were highly scored reflected a partial reversal of plasma cell maturation in the carfilzomib-resistant MM derivatives. The most significantly enriched gene set in KMS-11/Cfz and KMS-34/Cfz cells corresponded to genes with increased expression in IgM-memory B cells versus plasma cells (NES = 1.75, FDR = 0.005; Figure 1A). A set containing genes more highly expressed in naive B cells than in plasma cells (NES = 1.49, FDR = 0.06; Figure 1B) and one containing genes with higher levels of expression in Ig isotype-switched memory B cells relative to plasma cells (NES = 1.46, FDR = 0.06; Figure 1C) were also significantly enriched. We observed that KLF4 was included within the leading edge subset of upregulated genes in all three gene sets, in line with its higher expression in naive and memory B cells than in plasma cells [25-27]. Furthermore, using GeneSpring analysis software, we found overrepresentation of KLF4 target genes previously characterized by genome-wide chromatin immunoprecipitation (ChIP) in embryonic stem cells by Orkin and colleagues [28] among the differentially expressed genes in KMS-11/Cfz (89 out of 887 genes, fold change, FC ≥ 1.4; P = 2.02 x 10-3) and KMS-34/Cfz (92 out of 888 genes, FC ≥ 1.5; P = 6.47 x 10-4) (Table S1), suggesting that upregulation of KLF4 may contribute to carfilzomib resistance.
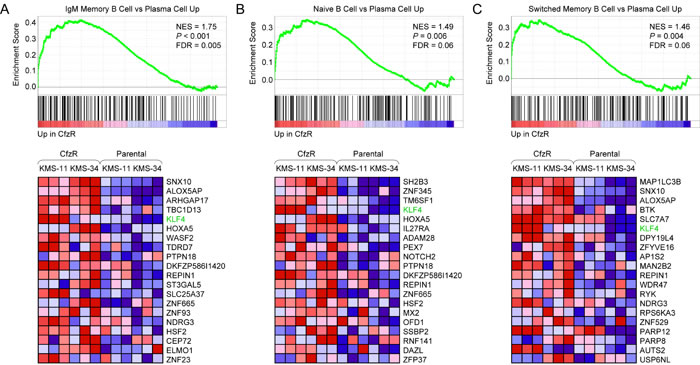
Figure 1: GSEA enrichment plots and heat maps of differentially expressed B lineage-related genes associated with acquisition of carfilzomib resistance in KMS-11 and KMS-34 cells. A. Gene set: GSE13411_IGM_MEMORY_BCELL_VS_PLASMA_CELL_UP (M3249). B. Gene set: GSE13411_NAIVE_BCELL_VS_PLASMA_CELL_UP (M3243). C. Gene set: GSE13411_SWITCHED_MEMORY_BCELL_VS_PLASMA_CELL_UP (M3251). FDR, false discovery rate; NES, normalized enrichment score; CfzR, carfilzomib-resistant derivatives; KLF4 is indicated.
We confirmed increased expression of KLF4 mRNA in carfilzomib-resistant MM cells by qRT-PCR analysis (Figure 2A), which was paralleled by a corresponding increase in KLF4 protein levels (~3.0 ± 0.7-fold, n = 4, P < 0.009 by paired Student’s t test) detected by western blotting (Figure 2B). Consistent with its function as a transcriptional regulator and potential role in the carfilzomib-resistant phenotype [29, 30], immunofluorescence confocal microscopy revealed more prominent nuclear localization of KLF4 in the carfilzomib-resistant MM derivatives (Figure 2C).
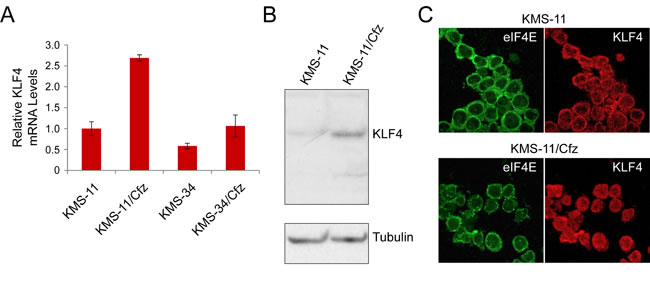
Figure 2: KLF4 expression in KMS-11 and carfilzomib-resistant KMS-11/Cfz cells. A. Relative KLF4 mRNA levels as determined by qRT-PCR. Also shown are KLF4 mRNA levels in KMS-34 and KMS-34/Cfz cells relative to KLF4 mRNA levels in KMS-11 cells. B. KLF4 protein levels were detected by western blotting with rabbit anti-KLF4 monoclonal antibodies against the carboxyl terminus (D1F2; Cell Signaling). Representative of four experiments. KLF4 signal in KMS-11 parental cells is less than in KMS-11/Cfz (P < 0.009). C. Cells were labeled with anti-KLF4 (Alexa Fluor 568, red) or anti-eIF4E (Alexa Fluor 488, green) antibodies, and immunofluorescence staining was analyzed by confocal laser scanning microscopy.
We tested the hypothesis that KLF4 might contribute to the molecular phenotype of carfilzomib-resistant MM cells by short-term expression of a KLF4 cDNA in KMS-11 cells (denoted KMS-11/KLF4). Fourteen genes showing differential expression in KMS-11/Cfz (FC ≥ 1.4) and/or KMS-34/Cfz (FC ≥ 1.5) versus parental MM cells were selected, and their expression levels compared to those in KMS-11/KLF4 cells by qRT-PCR analysis (Table 1). A substantial degree of correlation was found between the expression changes associated with the introduction of exogenous KLF4 and acquisition of carfilzomib resistance in KMS-11/Cfz (r = 0.77) and KMS-34/Cfz cells (r = 0.59) for the selected set of genes (Table 1). Of note, several of the genes that exhibited KLF4-induced changes in expression — CYP1A1, NQO1, HOXB7 and ID1 — were previously identified as direct KLF4 targets by genome-wide ChIP analysis [28]. Another of the genes, SLAMF7 encodes CD319, a cell surface marker specifically upregulated at the plasma cell stage during B cell differentiation [27, 31]. We verified that SLAMF7 mRNA levels were downregulated in the carfilzomib-resistant MM derivatives in accord with the less differentiated plasma cell phenotype revealed by GSEA analysis above. Moreover, SLAMF7 mRNA levels were also reduced following ectopic expression of KLF4 in KMS-11 cells. Together, the accumulated data supported the notion that carfilzomib-resistant MM cells had increased KLF4 transcriptional activity which was associated with the partial reversal of plasma cell maturation during acquisition of drug resistance.
GSEA identifies altered expression of KLF4 target genes associated with autophagy and metabolic pathways in carfilzomib-resistant MM cells
We next used GSEA to examine gene sets from the C2:CGP chemical and genetic perturbations (3,395 gene sets) and C6 oncogenic signatures (189 gene sets) collections of MSigDB. A highly enriched C2:CGP gene set in KMS-11/Cfz and KMS-34/Cfz cells (NES = 2.02, FDR < 0.007; Figure 3A) represented molecular targets that were upregulated during inhibition of MM cell growth following treatment with adaphostin [32]. Bortezomib reportedly triggered similar effects, and a recent study identifying c-Abl as a regulator of proteasome homeostasis provides some insight into the cross-talk between the pathways involved [33]. Interestingly, included in the leading edge subset were several genes associated with autophagy (highlighted in Figure 3). Among them were MAP1LC3B encoding the autophagic effector protein microtubule-associated protein 1 light chain Cβ, and SQSTM1 encoding the selective autophagy receptor sequestosome 1/p62 [34, 35]. During autophagy, a soluble form of LC3B (LC3B-I) is converted to a form (LC3B-II) that specifically associates with autophagosomes and is recognized by SQSTM1/p62 [36]. Also included was GADD45A, a growth arrest and DNA repair gene previously shown to be a KLF4-inducible gene in Hodgkin lymphoma cells and a stimulator of autophagy during skeletal muscle atrophy [37, 38]. Moreover, the most highly enriched gene set in the C6 collection (NES = 1.70, FDR = 0.05) corresponded to genes in MCF-7 breast cancer cells adapted for estrogen-independent growth in culture that were upregulated in the carfilzomib-resistant MM cells; KLF4 and GADD45A were included in the leading edge subset (Figure 3B). A further association with autophagy-related processes was suggested by a recent study reporting that one of the genes in the leading edge subset, LAMP3 encoding lysosome-associated membrane protein 3, is involved in resistance to the anti-estrogen tamoxifen in breast cancer cells by promoting prosurvival autophagy [39]. Another gene, ISG15, encoding an ubiquitin-like protein, was recently shown to interact with SQSTM1/p62, augmenting association with LC3B-II and facilitating autophagic degradation of aggresomes, in response to various types of cellular stress including proteasome inhibition [40]. Considered together, the genes represented in the leading edge subsets of the gene sets presented in Figure 3A, 3B suggested potential upregulation of autophagy pathways in the carfilzomib-resistant MM derivatives.
Conversely, among the downregulated sets of genes that were enriched in KMS-11/Cfz and KMS-34/Cfz cells, one of the most highly scored in the C2:CGP collection corresponded to genes belonging to an IRF4 regulatory network that are expressed at higher levels during B cell differentiation (NES = -1.98, FDR = 0.01; Figure 3C) [41]. SLAMF7 was included in the leading edge subset of downregulated genes. We noted that many of the other downregulated genes in the leading edge subset are involved in lipid metabolism (e.g., INSIG1, HMGCR, SCD, PAM, LDLR, SQLE, CYP51A1, MVK), and complementary analysis of the C2:CP canonical pathways collection (1,330 gene sets) by GSEA indicated downregulation of genes involved in cholesterol biosynthesis (Reactome: REACT_9405.3) in the carfilzomib-resistant MM cells as being statistically significant (NES = -2.41, FDR = 0.001; Figure S1B). Consistent with the inverse correlation observed between KLF4 levels and expression levels of these genes, several were previously demonstrated to be negatively regulated by ectopic KLF4 expression [42]. Examples included three genes involved in cholesterol biosynthesis: HMGCR encoding HMG-CoA reductase and MVK encoding mevalonate kinase, early enzymes in the mevalonate pathway; and CYP51A1 encoding a cytochrome P450 family member involved in the conversion of lanosterol to cholesterol. Additionally, although no significant enrichment of glycolysis-related gene sets was revealed by GSEA, LDHA, which encodes a subunit of lactate dehydrogenase, a key enzyme in the glycolytic phenotype of cancer cells (known as the Warburg effect) [43] previously identified as a KLF4-repressed gene [44] was downregulated in the carfilzomib-resistant MM derivatives (Figure 3C). In view of increasing evidence that enhanced lipid catabolism and low glycolytic activity in slowly proliferating tumor cells is associated with autophagy induction and treatment resistance [45], the above observations raised the possibility that activation of prosurvival autophagy could be a mechanism adopted by the carfilzomib-resistant MM cells to counteract the deficiency of proteasome function and metabolic stress induced by carfilzomib treatment.
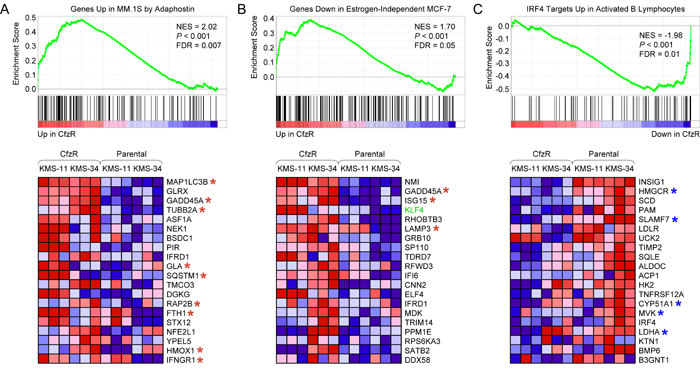
Figure 3: GSEA enrichment plots and heat maps of gene expression changes in sets of genes coregulated in response to chemical and genetic perturbations associated with acquisition of carfilzomib resistance in KMS-11 and KMS-34 cells. A. Gene set: SHAFFER_IRF4_TARGETS_IN_ACTIVATED_B_LYMPHOCYTE (M11189). B. Gene set: PODAR_RESPONSE_TO_ADAPHOSTIN_UP (M16336). C. Gene set: LTE2_UP.V1_DN (M2721). Abbreviations are as in Figure 1. Selected autophagy-related genes are indicated with a red asterisk. KLF4-repressed genes are indicated with a blue asterisk.
Carfilzomib resistance is associated with prosurvival autophagy in MM cells
We first examined whether autophagic activity was increased in carfilzomib-resistant KMS-11/Cfz and KMS-34/Cfz cells by flow cytometry using the fluorescent dye Cyto-ID Green [46, 47]. Fluorescence signals reflect steady state levels of autophagosomes as a result of two offsetting processes: formation of autophagosomes and dissolution of autophagosomes upon lysosomal fusion. We found higher steady state levels of autophagosomes in resistant cells versus their parental counterparts in both MM cell line models (relative increase in autophagic activity based on Cyto-ID mean fluorescence intensity values = 16.6 ± 1.3, P < 0.05, paired Student’s t test; see Materials and Methods for details) (Figure 4A).
Second, we investigated autophagic flux by measuring the processing of LC3B, an ubiquitin-like protein that is converted during autophagy to a lipidated form (LC3B-II) associated with autophagosome membranes [34]. To distinguish between synthesis, modification and degradation rates of LC3B, the ratio of the LC3B-II faster migrating form to the nonlipidated precursor (LC3B-I) is determined in the absence or presence of inhibitors of lysosome-mediated proteolysis [48]. Both carfilzomib-resistant MM derivatives exhibited higher levels of synthesis and modification of LC3B (as measured by LC3B-II/LC3B-I ratio) in comparison to parental cells in the presence of a mixture of the lysosomal protease inhibitors E64d, pepstatin A and leupeptin (Figure 4B). Increased levels of the ubiquitin cargo receptor and autophagy substrate SQSTM1/p62 in KMS-11/Cfz cells in the presence of the lysosomal protease inhibitors was also consistent with more active autophagy (Figure 4C).
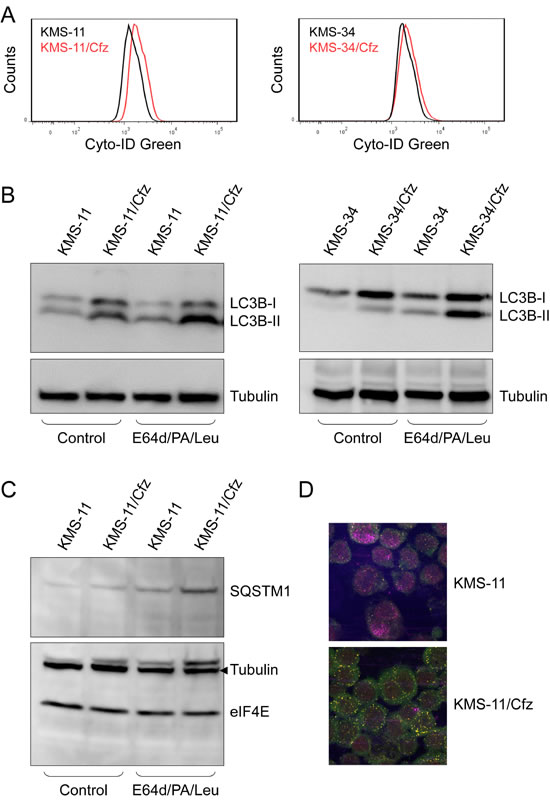
Figure 4: Autophagy is induced in carfilzomib-resistant KMS-11/Cfz and KMS-34/Cfz cells. A. Fluorescence histograms of cells stained with the Cyto-ID Green autophagy detection reagent. Representative examples of three independent experiments are shown (P < 0.05, paired Student’s t test). B., C. To assess complete autophagic flux, cells were treated overnight with or without the lysosomal protease inhibitors E64d (10 µg/ml), pepstatin A (PA; 10 µg/ml), and leupeptin (Leu; 1 µg/ml) [52]. Cell lysates were probed with anti-LC3B B. or anti-SQSTM1 C.. D. Cells were labeled with anti-LC3B (Alexa Fluor 568, red) or anti-SQSTM1 (Alexa Fluor 488, green) antibodies, and immunofluorescence staining was analyzed by confocal laser scanning microscopy. Increased colocalization of the two factors in KMS-11/Cfz versus parental KMS-11 cells (yellow signals) correlated with acquisition of carfilzomib resistance (P < 0.003).
Next, since SQSTM1/p62 is recruited into autophagosomes by lipidated LC3B-II [49], we assessed the subcellular localization of LC3B and SQSTM1/p62 proteins by immunofluorescence confocal microscopy. In both KMS-11 and KMS-34 cell line models increased colocalization of the two factors correlated with acquisition of carfilzomib resistance (P < 0.003; Figure 4D). To further characterize autophagic activity, we used the fluorescent autophagosome-specific reporter construct encoding a GFP-LC3 fusion protein [50], and examined GFP-LC3 signals by fluorescence confocal microscopy in KMS-11 and KMS-11/Cfz cells treated with or without carfilzomib. An increased number of GFP-LC3-II puncta was observed in KMS-11/Cfz cells (Figure 5A). To determine whether this was associated with increased autophagic flux, cells were treated with carfilzomib in the presence of lysosomal protease inhibitors and the levels of endogenous LC3B-I and LC3B-II measured by western blotting. As shown in Figure 5B, the degree of LC3B-II stabilization as reflected by the relative increase in the LC3B-II/LC3B-I ratio was greater in carfilzomib-resistant versus parental MM cells (P < 0.0001). Similar results were obtained when the cells were cotreated with chloroquine which, by increasing lysosomal pH, inhibits autophagic protein degradation and blocks autophagosome-lysosome fusion (Figure 5B) [51].
The above results are consistent with the view that increased autophagic flux was occurring in the carfilzomib-resistant MM cells. Inhibition of autophagy by chloroquine treatment was previously reported to enhance carfilzomib-induced cell death of head and neck squamous cell carcinoma cell lines [52]. To test whether concomitant inhibition of autophagy would sensitize KMS-11/Cfz and KMS-34/Cfz to carfilzomib, the cells were treated with carfilzomib in the presence or absence of chloroquine, and cell growth was measured by alamarBlue assay. Importantly, cotreatment with chloroquine diminished carfilzomib resistance in both KMS-11/Cfz and KMS-34/Cfz cell lines, indicating that prosurvival autophagy contributes to acquired drug resistance in these MM cell line models (Figure 5C).
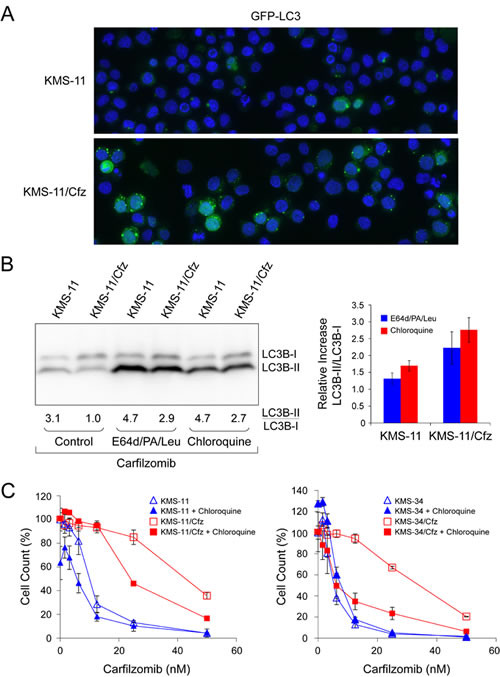
Figure 5: Prosurvival autophagy induction in carfilzomib-resistant MM cells is antagonized by lysosomal inhibitors and chloroquine. A. Increased GFP-LC3 puncta formation in KMS-11/Cfz versus parental KMS-11 cells treated overnight with carfilzomib (50 nM). B. Increased stabilization of LC3B-II in KMS-11/Cfz versus parental KMS-11 cells upon autophagy inhibition. Representative western blot of LC3B levels of four independent experiments is shown (P < 0.0001). Cells were treated overnight with carfilzomib (50 nM) in the presence or absence of the lysosomal protease inhibitors E64d (10 µg/ml), pepstatin A (PA; 10 µg/ml), and leupeptin (Leu; 1 µg/ml) or chloroquine (10 µM). Densitometry was used to calculate the LC3B-II/LC3B-I ratios and the relative increases in the ratios upon autophagy inhibition with respect to carfilzomib only-treated controls are indicated in the graph on the right. C. Cells were treated with the indicated concentrations of carfilzomib for 72 hours in the presence or absence of chloroquine (10 µM). Cell viability was determined by alamarBlue assay. The data are presented as the mean ± S.D of three experiments. IC50 values were calculated by linear regression of the plots in the absence or presence of chloroquine: KMS-11/Cfz, 35 nM versus 22 nM; KMS-11, 12 nM versus 12 nM; KMS-34/Cfz, 35 nM versus 6 nM; KMS-34, 6 nM versus 6 nM.
KLF4 regulates the autophagy receptor gene SQSTM1
The SQSTM1/p62 protein is of particular interest given its role as an adaptor for both proteasomal and autophagic degradation of ubiquitinated proteins [20, 21]. As shown in Figure 6A, we confirmed that SQSTM1 mRNA levels were higher in the carfilzomib-resistant MM derivatives and we demonstrated that the levels increased upon ectopic expression of KLF4 in KMS-11 cells by qRT-PCR analysis. A review of the literature revealed that SQSTM1 was included in a list of direct KLF4 binding targets detected by genome-wide ChIP analysis [53]. We therefore analyzed the genomic regions upstream of the SQSTM1 transcription start sites, and identified evolutionally conserved KLF4-binding motifs (Figure 6B; Figure S2). We performed qPCR-based ChIP analysis in KMS-11 and KMS-11/Cfz cells and confirmed binding to these regions by endogenous KLF4 (Figure 6C). In these experiments, the KLF4 promoter region was included as a positive control [55], whereas GATA6 genomic regions were used as negative controls [28, 53]. These observations support the proposal that KLF4 contributes to carfilzomib resistance by upregulating SQSTM1 expression in these MM cell line models.
To further investigate the potential relevance of a KLF4-associated autophagy component in MM, we identified KLF4 profile neighbors across 304 MM patient samples in the Multiple Myeloma Research Consortium reference collection dataset (GEO accession number GSE26760). In agreement with earlier work [17, 18], the two genes dysregulated by the t(4;14) translocation, WHSC1 and FGFR3 [56], were among the top KLF4 neighbors (1,470 out of 54,675 total on the array). Moreover, the set was enriched for genes associated with the annotation term “autophagy” in the NCBI Gene database (P = 2.34 x 10-4). Importantly, the list included SQSTM1 (Table S2). These results, demonstrating similarity in expression pattern and shared function [57], suggest that contribution to prosurvival autophagy is a clinically relevant aspect of KLF4 expression as regards MM pathobiology.
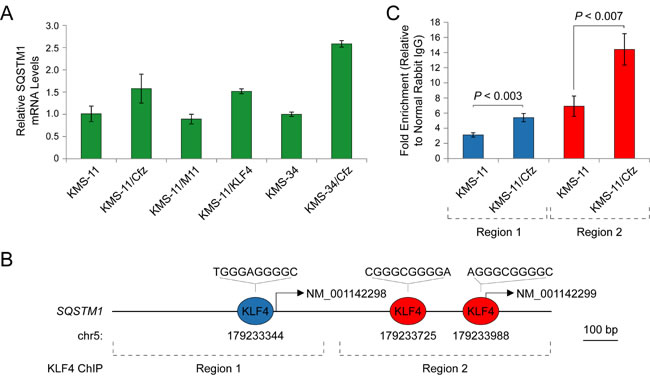
Figure 6: SQSTM1 is a direct target of KLF4 upregulated in carfilzomib-resistant KMS-11/Cfz and KMS-34/Cfz cells. A. Expression of SQSTM1 determined by qRT-PCR. KMS-11/KLF4, KMS-11 cells expressing a KLF4 cDNA. KMS-11/M11, KMS-11 cells transfected with a control vector. B. Evolutionarily conserved KLF4-binding motifs in the SQSTM1 promoter regions upstream of the NM_001142298 and NM_001142299 transcription start sites (Figure S2; see also Table S3 of Zaret and colleagues [54]). C. Increased binding of KLF4 to the SQSTM1 promoter regions indicated in B. in KMS-11/Cfz versus parental KMS-11 cells as determined by ChIP-qPCR.
Elevated expression of KLF4 and SQSTM1 is prognostic of poor survival in a subgroup of WHSC1-positive MM patients
Shaughnessy and colleagues defined a high-risk subgroup of MM patients by gene expression profiling (designated PR) based on expression of certain cell cycle and proliferation-associated genes [58]. The PR subgroup signature was present in approximately 18% of newly diagnosed MM and increased during disease progression to 45% of relapsed cases; strikingly, many of the WHSC1-defined t(4;14)-positive samples were found to cluster in this subgroup [58]. We used the recently published PROGgeneV2 prognostic biomarker identification tool [59] to study the implications of WHSC1, KLF4 and SQSTM1 gene expression on overall survival of 47 MM patients in the PR subgroup (GEO accession number GSE2658). Using median gene expression values as bifurcation points, Cox proportional hazards analyses showed that elevated expression of these genes was associated with inferior 3-year survival outcomes (Figure 7). Kaplan-Meier plots indicated significant segregation in survival outcomes for patients with high versus low WHSC1 expression (hazard ratio, HR = 1.95; 95% confidence interval/CI, 1.16-3.28; P = 0.01), with increasingly worse prognosis when high level coexpression of KLF4 (HR = 4.33; 95% CI, 2.04-9.18; P = 1.0 x 10-4) (Figure 7B), and KLF4 plus SQSTM1 (HR = 8.40; 95% CI, 2.80-25.17; P = 1.0 x 10-4) (Figure 7C) were also considered. When high levels of coexpression of the autophagy-associated genes MAP1LC3B and GADD45A were also taken into account, significantly inferior overall survival outcomes were observed (HR = 51.21; 95% CI, 7.36-356.61; P = 1.0 x 10-4) (Figure 7D). Moreover, reduced tumor cell metabolism (e.g., as exemplified by low HMGCR or LDHA expression) was also associated with adverse outcomes, with high expression ratios of KLF4 to HMGCR (HR = 3.47; 95% CI, 1.61-7.48; P = 0.02) (Figure 7E) and KLF4 to LDHA (HR = 3.74; 95% CI, 1.66-8.43; P = 0.001) (Figure 7F) prognostic of poor 3-year survival rates.
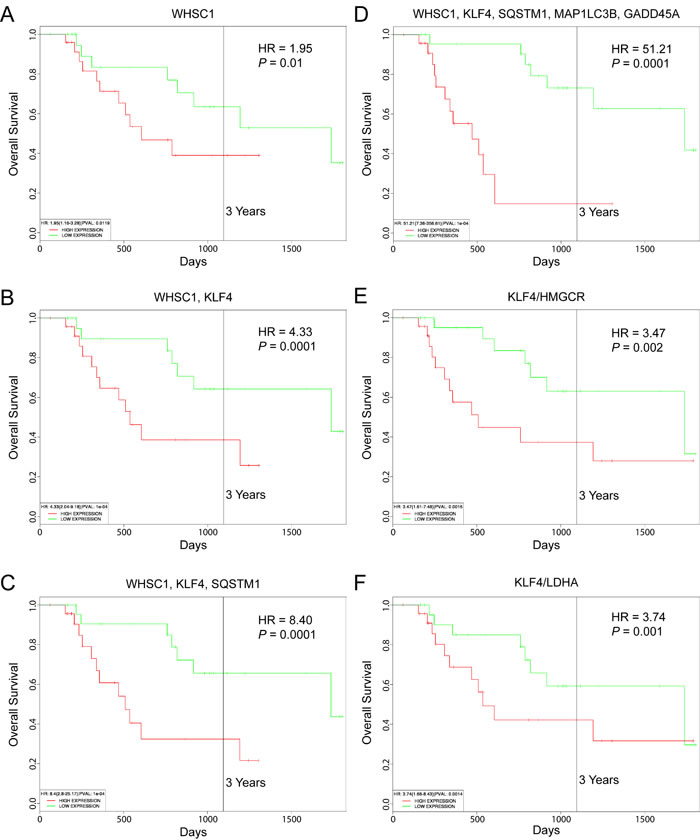
Figure 7: Prognostic value of WHSC1, KLF4 and SQSTM1 expression in MM patient survival outcomes. Kaplan-Meier survival plots of 47 MM patients in a high-risk subgroup associated with refractory/relapsed disease (GEO accession number GSE2658) created using PROGgeneV2. A. WHSC1 expression. B. Coexpression of WHSC1 and KLF4. C. Coexpression of WHSC1, KLF4 and SQSTM1. D. Coexpression of WHSC1, KLF4, SQSTM1, MAP1LC3B, and SQSTM1. E. Ratio of KLF4 to HMGCR expression. F. Ratio of KLF4 to LDHA expression. Median gene expression values were used as bifurcation points. HR, hazard ratio determined by Cox proportional hazards model.
DISCUSSION
The molecular mechanisms associated with the development of clinical resistance to proteasome inhibitors remain to be fully elucidated [4-8]. Toward this end, we have endeavored to establish clinically-relevant carfilzomib-resistant MM cell lines [60]. In a previously published study, we found that upregulation of the ABCB1-encoded multidrug resistance efflux transporter P-glycoprotein contributed to increased carfilzomib resistance in KMS-34 MM cells cultured in low concentrations (6 nM) of the drug [13]. Notably, our analysis of microarray data acquired for a MM patient treated with carfilzomib revealed increased ABCB1 expression during disease progression [61], suggesting that the mechanism could be relevant to carfilzomib resistance observed in the clinic [62]. To further elaborate mechanisms of carfilzomib resistance in MM, we adapted KMS-34/Cfz to growth in 12 nM carfilzomib and likewise established KMS-11/Cfz cells resistant to 12 nM carfilzomib by exposure to stepwise increasing concentrations of the drug. Unlike KMS-34/Cfz, KMS-11/Cfz did not exhibit increased expression of ABCB1. Instead, we found that a common mechanism of carfilzomib resistance was elevated levels of prosurvival autophagy, and that the pluripotency-associated transcription factor KLF4 [15] contributed to the carfilzomib-resistant phenotype. Our findings are in accord with those of two publications that appeared during revision of this manuscript reporting that KLF4 participates in autophagic pathways activated during stress responses in other settings [63, 64].
Substrates destined for selective autophagic destruction are recognized by receptor proteins which anchor the material to the expanding phagophore membrane via co-binding to lipidated LC3B-II [35, 36]. In recent years, the ubiquitin-proteasome and autophagy-lysosome systems, once considered independent processes for protein degradation, are now regarded as being interconnected. Impairment of either pathway has been reported to impact the other. Ubiquitination of proteins targeted for destruction serves as the mechanism of crosstalk and ubiquitin-binding cargo protein SQSTM1/p62 is a critical link [20, 21]. By upregulating SQSTM1/p62 levels in carfilzomib-resistant MM cells, KLF4 is postulated to contribute to prosurvival autophagy by facilitating delivery of aggregated substrates to autophagosomes via LC3B-II for subsequent destruction [65]. The relevance of this process in the MM therapeutic response to proteasome inhibition is supported by the recent finding that short hairpin RNA knockdown of SQSTM1 mRNA resulted in failure of autophagosomes to trap ubiquitinated cargo in MM cells, converting prosurvival autophagy to apoptosis [66].
KLF4 expression is consistently associated with MM patients carrying the t(4;14) translocation [17, 18]. Two genes are aberrantly expressed as a consequence of the translocation: WHSC1, encoding a histone methyltransferase (also referred to as MMSET or NSD2), and FGFR3 encoding a transmembrane tyrosine kinase [56]. Gain- and loss-of function studies in MM cell lines have demonstrated that WHSC1 histone methyltransferase activity is involved in the epigenetic upregulation of KLF4 [67, 68]. Notably, there is highly significant overlap (P < 1 x 10-33) between the WHSC1 target genes identified and the differentially expressed genes in KMS-11/Cfz (86 out of 887; FC ≥ 1.4) and KMS-34/Cfz (69 out of 888 genes, FC ≥ 1.5). Therefore, inhibition of WHSC1 enzymatic function or its ability to interact with chromatin may represent a promising future combination therapy for this subgroup of MM patients [67, 68].
Although the upstream pathways remain undefined, it is worth noting that induction of KLF4 transcription was previously observed in endothelial cells treated with bortezomib or epoxomicin (the natural epoxyketone from which carfilzomib was derived) [69]. Increased levels of KLF4 have also been observed following inhibition of translation by depletion of the translation initiation factor eIF4GI [70]. As eIF4GI depletion was reported to phenocopy mTOR inhibition and promote autophagy [70], and inhibition of mTOR activity by rapamycin induced KLF4 expression in other cells [71], mechanisms linking protein turnover and protein synthesis are implicated in KLF4 upregulation in the context of acquired proteasome inhibitor resistance [72].
Overexpression of KLF4 in MM cells was previously reported to result in cell cycle arrest [19]. We also observed that very high levels of exogenous KLF4 expression reduced proliferation rates in KMS-11 and KMS-34 cells, with diminished transgene expression in cell populations that exhibited a growth advantage after 1 month of culture (Figure S3). In this regard, KLF4 has been reported to exert growth suppressive effects in B-cell non-Hodgkin lymphoma [37], yet a recent study found that high nuclear expression of KLF4 in Burkitt pediatric lymphoma was indicative of inferior overall survival [73]. Thus, as in other systems [16], KLF4 regulation of B cell proliferation is complex and likely dependent on multiple factors. Along these lines, KLF4 was originally characterized as an epithelial oncogene by Rupert’s group imparting a slow growth phenotype to the transformed cells [74]. In follow-up studies of breast cancer cases, these investigators found that small primary tumors having preferential nuclear localization of KLF4 correlated with an increased risk of death [30]. The relative proliferative activity of WHSC1-positive KLF4-expressing MM cells with respect to other MM samples in the PR subgroup defined by Shaughnessy and colleagues [58] is not known. However, the fact that the hazard ratio increased significantly for these MM patients upon consideration of autophagy-associated gene expression levels (Figure 7D), implicates activation of autophagy as a contributing factor to relapsed/refractory disease.
Beyond its function as a direct cell cycle regulator, KLF4 exerts inhibitory effects on metabolic pathways and macromolecular synthesis [75], which include repression of genes encoding key enzymes involved in glycolysis and cholesterol biosynthesis (i.e., HMGCR, HMGCS1, MVK, LDHA) [42, 44]. Recent studies have shown that autophagy plays important roles in glucose homeostasis and lipid metabolism [76, 77], and the inverse relationship between autophagy and cell growth is becoming increasingly appreciated [78]. Although the KLF4 metabolic target genes in Figure 3C, including HMGCR and LDHA were only modestly downregulated upon acquisition of carfilzomib resistance, inhibition of HMGCR or LDHA enzymatic activity has resulted in autophagy induction in cancer cells [79, 80]. Furthermore, high gene expression ratios of KLF4 to HMGCR and KLF4 to LDHA were associated with adverse outcomes of certain MM patients (Figure 7E, 7F). Collectively, these results may raise a cautionary note regarding potential anti-MM therapeutic strategies that target these metabolic enzymes [81, 82].
KLF4 levels decrease during B cell activation and differentiation into plasma cells [25-27]. Increased expression of KLF4 in the carfilzomib-resistant KMS-11/Cfz and KMS-34/Cfz cells was indicative of “dedifferentiation” to an earlier maturation stage (Figure 1). Negative regulation of SLAMF7 encoding plasma cell-specific CD319 [27, 31] by ectopic KLF4 expression in KMS-11 cells (Table 1) suggests a potential role in the process. That this MM cell line phenomenon is biologically relevant is supported by work from Yaccoby who first described the ability of primary MM cells to dedifferentiate into an immature plasmablastic phenotype upon long-term co-culture on osteoclasts [83]. More recently, Karadimitris and colleagues reported epigenetic plasticity in MM patient samples and xenograft assays, where clinical drug resistance was attributed to bidirectional transitions between MM plasma cells and more quiescent pre-plasma cells [84]. Our results also complement the recent findings of Tiedemann and colleagues implicating less mature pre-plasma cells as contributing to therapeutic proteasome inhibitor resistance in MM [7]. Considered from this perspective, it is notable in view of its activity as a reprogramming factor that KLF4 target genes in embryonic stem cells were enriched in the differentially expressed genes in KMS-11/Cfz and KMS-34/Cfz cells [28]. These results suggest potential parallels with putative cancer stem-like cells [13, 85]. In line with this notion, it has been reported that overexpression of KLF4 in breast cancer cells led to an increase of the cancer stem cell-like population [86]. In that work, KLF4-mediated maintenance of stem cell-like characteristics was accompanied by increased cell migration and invasion of the malignant cells. Our future studies will further examine the mechanistic ramifications of KLF4 expression as regards relapse and treatment resistance in MM, and the utility of the KMS-11/Cfz and KMS-34/Cfz MM models for the development of novel autophagy-targeting combination therapies.
MATERIALS AND METHODS
Cell lines, plasmids and transfections
KMS-11 and KMS-34 MM cells were a kind gift from Dr. P. Leif Bergsagel (Mayo Clinic, Scottsdale, AZ) [87]. Cells were cultured in RPMI 1640 with GlutaMAX (Life Technologies) supplemented with 10% fetal bovine serum (Cambrex BioScience), 100 U/ml penicillin and 100 μg/ml streptomycin. The KLF4 expression vector containing a KLF4 cDNA (Accession No. BC030811.1) under the control of a cytomegalovirus promoter in the pReceiver-M11 backbone was from GeneCopoeia (Cat. No. EX-Z0482-M11). The pBABEpuro GFP-LC3 autophagy reporter plasmid was from Addgene (Plasmid No. 22405) [86]. Transfections were performed using the Amaxa nucleofector system with solution V and program X-001 settings (Lonza). Transfected cell lines were selected in 0.5 mg/ml G418 (pReceiver-M11 KLF4) or 0.5 μg/ml puromycin (pBABEpuro GFP-LC3). GFP-LC3-expressing cells were sorted on a FACSAria instrument equipped with FACSDiva software (BD Biosciences).
Antibodies and reagents
The following antibodies were used: anti-KLF4 (D1F2) rabbit mAb (Cell Signaling Technology, Cat. No. 12173); anti-α-tubulin mouse mAb (DM1A) (EMD Millipore Corporation, Cat. No. CP06); anti-p62/SQSTM1 (Clone 3) mouse mAb (BD Transduction Laboratories, Cat. No. 610832); anti-LC3B affinity isolated rabbit polyclonal antibody (Sigma-Aldrich, Cat. No. L7543); and anti-eIF4E (P-2) mouse mAb (Santa Cruz Biotechnology, Cat. No. sc-9976). Carfilzomib was obtained from Active Biochem (Cat. No. A-1098), chloroquine was purchased from Selleck Chemicals (Cat. No. S4157), and the lysosomal inhibitors E-64d (Cat. No. E8640), leupeptin (Cat. No. 103476-89-7) and pepstatin A (Cat. No. 77170) were from Sigma-Aldrich.
Microarray gene expression analysis and quantitative real-time qRT-PCR validation
Total RNA was isolated with the miRNeasy mini kit (Qiagen, Cat. No. 217004). Microarray gene expression analysis of triplicate samples was carried out by Expression Analysis, Inc. (Durham, NC) using Affymetrix GeneChip Human Genome U133 Plus 2.0 arrays. The data have been deposited in GEO (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE69078. Reverse transcription was performed with the SuperScript VILO cDNA synthesis kit (Life Technologies, Catalog No. 11754-250). Real-time qRT-PCR was performed using the Power SYBR Green reagent (Life Technologies, Cat No. 4368708) on an ABI Prism 7000 Sequence Detection System (Life Technologies) as previously described [13]. Primers synthesized by Sigma-Aldrich included: CCND1, forward, GCTCACGCTTACCTCAACCA, reverse, GACAGACAAAGCGTCCCTCA; CYP1A1, forward, GCTGCCTTCTGGCCTTGTAA, reverse, TGCCTGGATATGTGCACTCC; GLIPR1, forward, GACTGCGTTCGAATCCATAACA, reverse, GCTGGGTCCCAAGTCATGTA; HGF, forward, GACGCAGCTACAAGGGAACA, reverse, GGCAAAAAGCTGTGTTCGTG; HOXB7, forward, TGCAGTTTTGTAAGCCCTCT, reverse, GCAACCACAGGGTTAGTCCA; ID1, forward, CCAGCACGTCATCGACTACA, reverse, GGGGGTTCCAACTTCGGATT; IFIT3, forward, CTGGGTGGAAACCTCTTCAGC, reverse, GACCTCACTCATGATGGCTGTTTC; IGF1, forward, TGCAGGAGGGACTCTGAAAC, reverse, GCTGCGTGATATTTGAAAGGT; KLF4, forward, TCCATTACCAAGAGCTCATGCC, reverse, CGCGTAATCACAAGTGTGGG; MAPT, forward, AGCTTGTAGCTGCCAACCTC, reverse, TTTCCAAGGGGGTGTGTTCC; NQO1, forward, TAGCATTGGGCACACTCCAG, reverse, CCAGGCGTTTCTTCCATCCT; P4HA2, forward, GACACTTCCCTCTGTGACCA, reverse, TCATGTGCCCAATAGAGGTG; PSMB5, forward, CAGTACAAAGGCATGGGGCT, reverse, TCAGACACAGGGCCTCTCTTA; SLAMF7, forward, AGTCTGGCACGTAAGATGAACA, reverse, TCAAAAGCAGCCATTCCCCT; SQSTM1, forward, CTCCGCGTTCGCTACAAAAG, reverse, CAGAAGGTAGGCCTTCACGG; and TLR4, forward, TGCCGTTTTATCACGGAGGT, reverse, GGGAGGTTGTCGGGGATTTT.
Chromatin immunoprecipitation (ChIP)
ChIP was performed on the SQSTM1 promoter regions with 20 µg total chromatin and 5 µg of anti-KLF4 (D1F2) rabbit mAb using the SimpleChIP Enzymatic Chromatin IP Kit (Magnetic Beads) (Cell Signaling, Cat. No. 9003), and 4% of the precipitated material was used per qPCR reaction. GATA6 was included as a non-target promoter. Background ChIP levels were obtained using 5 µg of normal rabbit IgG (Cat. No. 2729). Primers used were: SQSTM1 promoter region 1, forward, AGCTTTGTGCCCTGTACTCA, reverse, TGCAGTGAGCCTGATACCTG; SQSTM1 promoter region 2, ACCTCTGTGACCTTGGGTCT, reverse, GCTGTCCCGACGCTGAG; GATA6 promoter region 1, forward, TGTTGAACTGTGCAGCTTTTCT, reverse, TAATGATGCACACACAACCTGA; GATA6 promoter region 2, forward, ATTCCCAGCAGGCTTATTGTAAA, reverse, CAGAAGCAGACAACCACGATAG.
Confocal microscopy
Cells (2.5 x 105) were centrifuged onto a microscope slide at 1,000 rpm for 5 minutes using a Shandon Cytospin 4 instrument. The cells were then immediately fixed in 3.7% formaldehyde for 5 minutes at room temperature and permeabilized with 0.5% Triton X-100 in phosphate-buffered saline (PBS) for 15 minutes at room temperature. Following permeabilization, the cells were rinsed with PBS and blocked in PBS containing 10% goat serum and 0.01% Triton X-100 for 1 hour at room temperature. The cells were then incubated with anti-KLF4 and anti-eIF4E antibodies diluted to final concentrations of 0.4 µg/ml, in PBS containing 1% goat and 0.01% Triton X-100 for 1 hour at room temperature. The cells were rinsed with PBS and then incubated with Alexa Fluor 568-conjugated goat anti-rabbit and Alexa Fluor 488-conjugated goat anti-mouse secondary antibodies (Life Technologies) diluted 1:500 in PBS containing 1% goat serum and 0.01% Triton X-100 for 1 hour at room temperature. The cells were rinsed with PBS and mounted with Fluoromount G (Electron Microscopy Sciences). Cells expressing the GFP-LC3 reporter were fixed in 1% methanol free paraformaldehyde (Electron Microscopy Sciences) in PBS overnight at 4oC then centrifuged onto a microscope slide and mounted with Fluoromount G. Imaging analysis was performed on a Cell Observer SD spinning disk confocal system equipped with Zen software (Carl Zeiss Microscopy).
Cytotoxicity assay
Cells were treated with carfilzomib and chloroquine at the indicated concentrations and cell growth was measured using the alamarBlue cell viability and proliferation reagent (Life Technologies) as previously described [13].
Autophagy detection
Autophagy was measured with the Cyto-ID autophagy detection kit (Enzo, Cat. No. ENZ-51031-K200) using a FACSAria instrument, and data were analyzed with FlowJo Mac v10.0.2 (Tree Star). Autophagy activity factor (AAF) values were calculated using the following equation: AAF = 100 × (MFI carfilzomib-resistant − MFI parental) / MFI carfilzomib resistant. MFI is mean fluorescence intensity, and AAF expresses the level of autophagy in live cells as the difference between the amount of Cyto-ID Green autophagy dye within cells [46]. GFP-LC3 puncta were detected by confocal microscopy as described above.
ACKNOWLEDGMENTS and funding
We thank Leif Bergsagel for providing the KMS-11 and KMS-34 cell lines, and Alex Tzatsos for the GFP-LC3 plasmid. This work was supported by a Grant from the Dr. Cyrus and Myrtle Katzen Cancer Research Center at The George Washington University and a King Fahd Endowment from The George Washington University School of Medicine and Health Sciences. The authors also gratefully acknowledge the generous support of Marc Cohen.
Conflicts of Interest
The authors declare no conflict of interest.
REFERENCES
1. Bianchi G, Anderson KC. Understanding biology to tackle the disease: Multiple myeloma from bench to bedside, and back. CA Cancer J Clin. 2014; 64: 422-444.
2. Abdi J, Chen G, Chang H. Drug resistance in multiple myeloma: latest findings and new concepts on molecular mechanisms. Oncotarget. 2013; 4: 2186-2207.
3. Moreau P, Richardson PG, Cavo M, Orlowski RZ, San Miguel JF, Palumbo A, Harousseau JL. Proteasome inhibitors in multiple myeloma: 10 years later. Blood. 2012; 120: 947-959.
4. Oerlemans R, Franke NE, Assaraf YG, Cloos J, van Zantwijk I, Berkers CR, Scheffer GL, Debipersad K, Vojtekova K, Lemos C, van der Heijden JW, Ylstra B, Peters GJ et al. Molecular basis of bortezomib resistance: proteasome subunit beta5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood. 2008; 112: 2489-2499.
5. Kuhn DJ, Berkova Z, Jones RJ, Woessner R, Bjorklund CC, Ma W, Davis RE, Lin P, Wang H, Madden TL, Wei C, Baladandayuthapani V, Wang M et al. Targeting the insulin-like growth factor-1 receptor to overcome bortezomib resistance in preclinical models of multiple myeloma. Blood. 2012; 120: 3260-3270.
6. Lichter DI, Danaee H, Pickard MD, Tayber O, Sintchak M, Shi H, Richardson PG, Cavenagh J, Blade J, Facon T, Niesvizky R, Alsina M, Dalton W et al. Sequence analysis of beta-subunit genes of the 20S proteasome in patients with relapsed multiple myeloma treated with bortezomib or dexamethasone. Blood. 2012; 120: 4513-4516.
7. Leung-Hagesteijn C, Erdmann N, Cheung G, Keats JJ, Stewart AK, Reece DE, Chung KC, Tiedemann RE. Xbp1s-negative tumor B cells and pre-plasmablasts mediate therapeutic proteasome inhibitor resistance in multiple myeloma. Cancer Cell. 2013; 24: 289-304.
8. Niewerth D, Jansen G, Assaraf YG, Zweegman S, Kaspers GJ, Cloos J. Molecular basis of resistance to proteasome inhibitors in hematological malignancies. Drug Resist Updat. 2015; 18: 18-35.
9. Herndon TM, Deisseroth A, Kaminskas E, Kane RC, Koti KM, Rothmann MD, Habtemariam B, Bullock J, Bray JD, Hawes J, Palmby TR, Jee J, Adams W et al. U.S. Food and Drug Administration approval: carfilzomib for the treatment of multiple myeloma. Clin Cancer Res. 2013; 19: 4559-4563.
10. Vij R, Siegel DS, Jagannath S, Jakubowiak AJ, Stewart AK, McDonagh K, Bahlis N, Belch A, Kunkel LA, Wear S, Wong AF, Wang M. An open-label, single-arm, phase 2 study of single-agent carfilzomib in patients with relapsed and/or refractory multiple myeloma who have been previously treated with bortezomib. Br J Haematol. 2012; 158: 739-748.
11. Siegel DS, Martin T, Wang M, Vij R, Jakubowiak AJ, Lonial S, Trudel S, Kukreti V, Bahlis N, Alsina M, Chanan-Khan A, Buadi F, Reu FJ et al. A phase 2 study of single-agent carfilzomib (PX-171-003-A1) in patients with relapsed and refractory multiple myeloma. Blood. 2012; 120: 2817-2825.
12. Chng WJ, Dispenzieri A, Chim CS, Fonseca R, Goldschmidt H, Lentzsch S, Munshi N, Palumbo A, Miguel JS, Sonneveld P, Cavo M, Usmani S, Durie BG et al. IMWG consensus on risk stratification in multiple myeloma. Leukemia. 2014; 28: 269-277.
13. Hawley TS, Riz I, Yang W, Wakabayashi Y, DePalma L, Chang YT, Peng W, Zhu J, Hawley RG. Identification of an ABCB1 (P-glycoprotein)-positive carfilzomib-resistant myeloma subpopulation by the pluripotent stem cell fluorescent dye CDy1. Am J Hematol. 2013; 88: 265-272.
14. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005; 102: 15545-15550.
15. Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007; 131: 861-872.
16. Rowland BD, Peeper DS. KLF4, p21 and context-dependent opposing forces in cancer. Nat Rev Cancer. 2006; 6: 11-23.
17. Mattioli M, Agnelli L, Fabris S, Baldini L, Morabito F, Bicciato S, Verdelli D, Intini D, Nobili L, Cro L, Pruneri G, Callea V, Stelitano C et al. Gene expression profiling of plasma cell dyscrasias reveals molecular patterns associated with distinct IGH translocations in multiple myeloma. Oncogene. 2005; 24: 2461-2473.
18. Agnelli L, Bicciato S, Mattioli M, Fabris S, Intini D, Verdelli D, Baldini L, Morabito F, Callea V, Lombardi L, Neri A. Molecular classification of multiple myeloma: a distinct transcriptional profile characterizes patients expressing CCND1 and negative for 14q32 translocations. J Clin Oncol. 2005; 23: 7296-7306.
19. Schoenhals M, Kassambara A, Veyrune JL, Moreaux J, Goldschmidt H, Hose D, Klein B. Kruppel-like factor 4 blocks tumor cell proliferation and promotes drug resistance in multiple myeloma. Haematologica. 2013; 98: 1442-1449.
20. Kirkin V, McEwan DG, Novak I, Dikic I. A role for ubiquitin in selective autophagy. Mol Cell. 2009; 34: 259-269.
21. Korolchuk VI, Menzies FM, Rubinsztein DC. Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett. 2010; 584: 1393-1398.
22. White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer. 2012; 12: 401-410.
23. Kuhn DJ, Chen Q, Voorhees PM, Strader JS, Shenk KD, Sun CM, Demo SD, Bennett MK, van Leeuwen FW, Chanan-Khan AA, Orlowski RZ. Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin-proteasome pathway, against preclinical models of multiple myeloma. Blood. 2007; 110: 3281-3290.
24. Verbrugge SE, Al M, Assaraf YG, Niewerth D, van MJ, Cloos J, van d, V, Scheffer GL, Peters GJ, Chan ET, Anderl JL, Kirk CJ, Zweegman S et al. Overcoming bortezomib resistance in human B cells by anti-CD20/rituximab-mediated complement-dependent cytotoxicity and epoxyketone-based irreversible proteasome inhibitors. Exp Hematol Oncol. 2013; 2: 2.
25. Klaewsongkram J, Yang Y, Golech S, Katz J, Kaestner KH, Weng NP. Kruppel-like factor 4 regulates B cell number and activation-induced B cell proliferation. J Immunol. 2007; 179: 4679-4684.
26. Good KL, Tangye SG. Decreased expression of Kruppel-like factors in memory B cells induces the rapid response typical of secondary antibody responses. Proc Natl Acad Sci U S A. 2007; 104: 13420-13425.
27. Good KL, Avery DT, Tangye SG. Resting human memory B cells are intrinsically programmed for enhanced survival and responsiveness to diverse stimuli compared to naive B cells. J Immunol. 2009; 182: 890-901.
28. Kim J, Chu J, Shen X, Wang J, Orkin SH. An extended transcriptional network for pluripotency of embryonic stem cells. Cell. 2008; 132: 1049-1061.
29. Shields JM, Yang VW. Two potent nuclear localization signals in the gut-enriched Kruppel-like factor define a subfamily of closely related Kruppel proteins. J Biol Chem. 1997; 272: 18504-18507.
30. Pandya AY, Talley LI, Frost AR, FitzGerald TJ, Trivedi V, Chakravarthy M, Chhieng DC, Grizzle WE, Engler JA, Krontiras H, Bland KI, LoBuglio AF, Lobo-Ruppert SM et al. Nuclear localization of KLF4 is associated with an aggressive phenotype in early-stage breast cancer. Clin Cancer Res. 2004; 10: 2709-2719.
31. Hsi ED, Steinle R, Balasa B, Szmania S, Draksharapu A, Shum BP, Huseni M, Powers D, Nanisetti A, Zhang Y, Rice AG, van Abbema A, Wong M et al. CS1, a potential new therapeutic antibody target for the treatment of multiple myeloma. Clin Cancer Res. 2008; 14: 2775-2784.
32. Podar K, Raab MS, Tonon G, Sattler M, Barila D, Zhang J, Tai YT, Yasui H, Raje N, DePinho RA, Hideshima T, Chauhan D, Anderson KC. Up-regulation of c-Jun inhibits proliferation and induces apoptosis via caspase-triggered c-Abl cleavage in human multiple myeloma. Cancer Res. 2007; 67: 1680-1688.
33. Li D, Dong Q, Tao Q, Gu J, Cui Y, Jiang X, Yuan J, Li W, Xu R, Jin Y, Li P, Weaver DT, Ma Q et al. c-Abl regulates proteasome abundance by controlling the ubiquitin-proteasomal degradation of PSMA7 subunit. Cell Rep. 2015; 10: 484-496.
34. Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000; 19: 5720-5728.
35. Rogov V, Dotsch V, Johansen T, Kirkin V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol Cell. 2014; 53: 167-178.
36. Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007; 282: 24131-24145.
37. Guan H, Xie L, Leithauser F, Flossbach L, Moller P, Wirth T, Ushmorov A. KLF4 is a tumor suppressor in B-cell non-Hodgkin lymphoma and in classic Hodgkin lymphoma. Blood. 2010; 116: 1469-1478.
38. Ebert SM, Dyle MC, Kunkel SD, Bullard SA, Bongers KS, Fox DK, Dierdorff JM, Foster ED, Adams CM. Stress-induced skeletal muscle Gadd45a expression reprograms myonuclei and causes muscle atrophy. J Biol Chem. 2012; 287: 27290-27301.
39. Nagelkerke A, Sieuwerts AM, Bussink J, Sweep FC, Look MP, Foekens JA, Martens JW, Span PN. LAMP3 is involved in tamoxifen resistance in breast cancer cells through the modulation of autophagy. Endocr Relat Cancer. 2014; 21: 101-112.
40. Nakashima H, Nguyen T, Goins WF, Chiocca EA. Interferon-stimulated gene 15 (ISG15) and ISG15-linked proteins can associate with members of the selective autophagic process, histone deacetylase 6 (HDAC6) and SQSTM1/p62. J Biol Chem. 2015; 290: 1485-1495.
41. Shaffer AL, Emre NC, Lamy L, Ngo VN, Wright G, Xiao W, Powell J, Dave S, Yu X, Zhao H, Zeng Y, Chen B, Epstein J et al. IRF4 addiction in multiple myeloma. Nature. 2008; 454: 226-231.
42. Whitney EM, Ghaleb AM, Chen X, Yang VW. Transcriptional profiling of the cell cycle checkpoint gene Kruppel-like factor 4 reveals a global inhibitory function in macromolecular biosynthesis. Gene Expr. 2006; 13: 85-96.
43. Doherty JR, Cleveland JL. Targeting lactate metabolism for cancer therapeutics. J Clin Invest. 2013; 123: 3685-3692.
44. Shi M, Cui J, Du J, Wei D, Jia Z, Zhang J, Zhu Z, Gao Y, Xie K. A novel KLF4/LDHA signaling pathway regulates aerobic glycolysis in and progression of pancreatic cancer. Clin Cancer Res. 2014; 20: 4370-4380.
45. Ye F, Zhang Y, Liu Y, Yamada K, Tso JL, Menjivar JC, Tian JY, Yong WH, Schaue D, Mischel PS, Cloughesy TF, Nelson SF, Liau LM et al. Protective properties of radio-chemoresistant glioblastoma stem cell clones are associated with metabolic adaptation to reduced glucose dependence. PLoS One. 2013; 8: e80397.
46. Chan LL, Shen D, Wilkinson AR, Patton W, Lai N, Chan E, Kuksin D, Lin B, Qiu J. A novel image-based cytometry method for autophagy detection in living cells. Autophagy. 2012; 8: 1371-1382.
47. Oeste CL, Seco E, Patton WF, Boya P, Perez-Sala D. Interactions between autophagic and endo-lysosomal markers in endothelial cells. Histochem Cell Biol. 2013; 139: 659-670.
48. Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, Ahn HJ, Ait-Mohamed O, Ait-Si-Ali S et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012; 8: 445-544.
49. Shvets E, Fass E, Scherz-Shouval R, Elazar Z. The N-terminus and Phe52 residue of LC3 recruit p62/SQSTM1 into autophagosomes. J Cell Sci. 2008; 121: 2685-2695.
50. Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004; 15: 1101-1111.
51. Glaumann H, Ahlberg J. Comparison of different autophagic vacuoles with regard to ultrastructure, enzymatic composition, and degradation capacity—formation of crinosomes. Exp Mol Pathol. 1987; 47: 346-362.
52. Zang Y, Thomas SM, Chan ET, Kirk CJ, Freilino ML, DeLancey HM, Grandis JR, Li C, Johnson DE. Carfilzomib and ONX 0912 inhibit cell survival and tumor growth of head and neck cancer and their activities are enhanced by suppression of Mcl-1 or autophagy. Clin Cancer Res. 2012; 18: 5639-5649.
53. Sen GL, Boxer LD, Webster DE, Bussat RT, Qu K, Zarnegar BJ, Johnston D, Siprashvili Z, Khavari PA. ZNF750 is a p63 target gene that induces KLF4 to drive terminal epidermal differentiation. Dev Cell. 2012; 22: 669-677.
54. Soufi A, Donahue G, Zaret KS. Facilitators and impediments of the pluripotency reprogramming factors’ initial engagement with the genome. Cell. 2012; 151: 994-1004.
55. Mahatan CS, Kaestner KH, Geiman DE, Yang VW. Characterization of the structure and regulation of the murine gene encoding gut-enriched Kruppel-like factor (Kruppel-like factor 4). Nucleic Acids Res. 1999; 27: 4562-4569.
56. Chesi M, Nardini E, Lim RS, Smith KD, Kuehl WM, Bergsagel PL. The t(4;14) translocation in myeloma dysregulates both FGFR3 and a novel gene, MMSET, resulting in IgH/MMSET hybrid transcripts. Blood. 1998; 92: 3025-3034.
57. Thompson HG, Harris JW, Wold BJ, Quake SR, Brody JP. Identification and confirmation of a module of coexpressed genes. Genome Res. 2002; 12: 1517-1522.
58. Zhan F, Huang Y, Colla S, Stewart JP, Hanamura I, Gupta S, Epstein J, Yaccoby S, Sawyer J, Burington B, Anaissie E, Hollmig K, Pineda-Roman M et al. The molecular classification of multiple myeloma. Blood. 2006; 108: 2020-2028.
59. Goswami CP, Nakshatri H. PROGgeneV2: enhancements on the existing database. BMC Cancer. 2014; 14: 970.
60. McDermott M, Eustace AJ, Busschots S, Breen L, Crown J, Clynes M, O’Donovan N, Stordal B. In vitro development of chemotherapy and targeted therapy drug-resistant cancer cell lines: a practical guide with case studies. Front Oncol. 2014; 4: 40.
61. Keats JJ, Chesi M, Egan JB, Garbitt VM, Palmer SE, Braggio E, Van Wier S, Blackburn PR, Baker AS, Dispenzieri A, Kumar S, Rajkumar SV, Carpten JD et al. Clonal competition with alternating dominance in multiple myeloma. Blood. 2012; 120: 1067-1076.
62. Abraham J, Salama NN, Azab AK. The role of P-glycoprotein in drug resistance in multiple myeloma. Leuk Lymphoma. 2015; 56: 26-33.
63. Liu C, DeRoo EP, Stecyk C, Wolsey M, Szuchnicki M, Hagos EG. Impaired autophagy in mouse embryonic fibroblasts null for Kruppel-like Factor 4 promotes DNA damage and increases apoptosis upon serum starvation. Mol Cancer. 2015; 14: 101.
64. Wu Y, Li Y, Zhang H, Huang Y, Zhao P, Tang Y, Qiu X, Ying Y, Li W, Ni S, Zhang M, Liu L, Xu Y et al. Autophagy and mTORC1 regulate the stochastic phase of somatic cell reprogramming. Nat Cell Biol. 2015; May 18. doi: 10.1038/ncb3172. [Epub ahead of print]
65. Viiri J, Hyttinen JM, Ryhanen T, Rilla K, Paimela T, Kuusisto E, Siitonen A, Urtti A, Salminen A, Kaarniranta K. p62/sequestosome 1 as a regulator of proteasome inhibitor-induced autophagy in human retinal pigment epithelial cells. Mol Vis. 2010; 16: 1399-1414.
66. Chen S, Zhou L, Zhang Y, Leng Y, Pei XY, Lin H, Jones R, Orlowski RZ, Dai Y, Grant S. Targeting SQSTM1/p62 induces cargo loading failure and converts autophagy to apoptosis via NBK/Bik. Mol Cell Biol. 2014; 34: 3435-3449.
67. Huang Z, Wu H, Chuai S, Xu F, Yan F, Englund N, Wang Z, Zhang H, Fang M, Wang Y, Gu J, Zhang M, Yang T et al. NSD2 is recruited through its PHD domain to oncogenic gene loci to drive multiple myeloma. Cancer Res. 2013; 73: 6277-6288.
68. Popovic R, Martinez-Garcia E, Giannopoulou EG, Zhang Q, Zhang Q, Ezponda T, Shah MY, Zheng Y, Will CM, Small EC, Hua Y, Bulic M, Jiang Y et al. Histone methyltransferase MMSET/NSD2 alters EZH2 binding and reprograms the myeloma epigenome through global and focal changes in H3K36 and H3K27 methylation. PLoS Genet. 2014; 10: e1004566.
69. Hiroi T, Deming CB, Zhao H, Hansen BS, Arkenbout EK, Myers TJ, McDevitt MA, Rade JJ. Proteasome inhibitors enhance endothelial thrombomodulin expression via induction of Kruppel-like transcription factors. Arterioscler Thromb Vasc Biol. 2009; 29: 1587-1593.
70. Ramirez-Valle F, Braunstein S, Zavadil J, Formenti SC, Schneider RJ. eIF4GI links nutrient sensing by mTOR to cell proliferation and inhibition of autophagy. J Cell Biol. 2008; 181: 293-307.
71. Wang Y, Zhao B, Zhang Y, Tang Z, Shen Q, Zhang Y, Zhang W, Du J, Chien S, Wang N. Kruppel-like factor 4 is induced by rapamycin and mediates the anti-proliferative effect of rapamycin in rat carotid arteries after balloon injury. Br J Pharmacol. 2012; 165: 2378-2388.
72. Zhang Y, Nicholatos J, Dreier JR, Ricoult SJ, Widenmaier SB, Hotamisligil GS, Kwiatkowski DJ, Manning BD. Coordinated regulation of protein synthesis and degradation by mTORC1. Nature. 2014; 513: 440-443.
73. Valencia-Hipolito A, Hernandez-Atenogenes M, Vega GG, Maldonado-Valenzuela A, Ramon G, Mayani H, Pena Alonso Y, Martinez-Maza O, Mendez-Tenorio A, Huerta-Yepez S, Bonavida B, Vega MI. Expression of KLF4 is a predictive marker for survival in pediatric Burkitt lymphoma. Leuk Lymphoma. 2014; 55: 1806-1814.
74. Foster KW, Ren S, Louro ID, Lobo-Ruppert SM, Kie-Bell P, Grizzle W, Hayes MR, Broker TR, Chow LT, Ruppert JM. Oncogene expression cloning by retroviral transduction of adenovirus E1A-immortalized rat kidney RK3E cells: transformation of a host with epithelial features by c-MYC and the zinc finger protein GKLF. Cell Growth Differ. 1999; 10: 423-434.
75. Bellance N, Pabst L, Allen G, Rossignol R, Nagrath D. Oncosecretomics coupled to bioenergetics identifies alpha-amino adipic acid, isoleucine and GABA as potential biomarkers of cancer: Differential expression of c-Myc, Oct1 and KLF4 coordinates metabolic changes. Biochim Biophys Acta. 2012; 1817: 2060-2071.
76. Karsli-Uzunbas G, Guo JY, Price S, Teng X, Laddha SV, Khor S, Kalaany NY, Jacks T, Chan CS, Rabinowitz JD, White E. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov. 2014; 4: 914-927.
77. Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ. Autophagy regulates lipid metabolism. Nature. 2009; 458: 1131-1135.
78. Neufeld TP. Autophagy and cell growth—the yin and yang of nutrient responses. J Cell Sci. 2012; 125: 2359-2368.
79. Parikh A, Childress C, Deitrick K, Lin Q, Rukstalis D, Yang W. Statin-induced autophagy by inhibition of geranylgeranyl biosynthesis in prostate cancer PC3 cells. Prostate. 2010; 70: 971-981.
80. Yang Y, Su D, Zhao L, Zhang D, Xu J, Wan J, Fan S, Chen M. Different effects of LDH-A inhibition by oxamate in non-small cell lung cancer cells. Oncotarget. 2014; 5: 11886-11896.
81. Pandyra A, Mullen PJ, Kalkat M, Yu R, Pong JT, Li Z, Trudel S, Lang KS, Minden MD, Schimmer AD, Penn LZ. Immediate utility of two approved agents to target both the metabolic mevalonate pathway and its restorative feedback loop. Cancer Res. 2014; 74: 4772-4782.
82. Maiso P, Huynh D, Moschetta M, Sacco A, Aljawai Y, Mishima Y, Asara JM, Roccaro AM, Kimmelman AC, Ghobrial IM. Metabolic signature identifies novel targets for drug resistance in multiple myeloma. Cancer Res. 2015; 75: 2071-2082.
83. Yaccoby S. The phenotypic plasticity of myeloma plasma cells as expressed by dedifferentiation into an immature, resilient, and apoptosis-resistant phenotype. Clin Cancer Res. 2005; 11: 7599-7606.
84. Chaidos A, Barnes CP, Cowan G, May PC, Melo V, Hatjiharissi E, Papaioannou M, Harrington H, Doolittle H, Terpos E, Dimopoulos M, Abdalla S, Yarranton H et al. Clinical drug resistance linked to interconvertible phenotypic and functional states of tumor-propagating cells in multiple myeloma. Blood. 2013; 121: 318-328.
85. Hawley RG. The cancer stem cell conundrum in multiple myeloma. J Stem Cell Res Ther. 2012; 2: 1000e110.
86. Yu F, Li J, Chen H, Fu J, Ray S, Huang S, Zheng H, Ai W. Kruppel-like factor 4 (KLF4) is required for maintenance of breast cancer stem cells and for cell migration and invasion. Oncogene. 2011; 30: 2161-2172.
87. Keats JJ, Fonseca R, Chesi M, Schop R, Baker A, Chng WJ, Van WS, Tiedemann R, Shi CX, Sebag M, Braggio E, Henry T, Zhu YX et al. Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell. 2007; 12: 131-144.