
INTRODUCTION
Human malignant pleural mesothelioma (MPM) is a highly aggressive neoplasm that leads to patient death within a few months or years [1–3]. Presently, no standard curative therapy exists for MPM, and there are no recognized specific markers for the diagnosis of MPM. In addition, little is known regarding the apparent lack of apoptosis in mesothelioma cells [3–5].
Apoptosis is a highly tuned mechanism that is regulated by distinct and complex pathways. This process can be induced by a variety of physiological and pharmacological stimuli [6], including changes in the Ca2+ distribution within intracellular compartments [7]. Specifically, evidence indicates that both Ca2+ release from intracellular pools and capacitative Ca2+ influx through Ca2+ release-activated Ca2+ channels are sufficient to trigger apoptosis [8–12]. Additionally, mitochondria play a critical role in Ca2+ homeostasis and apoptosis [13, 14]. In conjunction with incremental changes in the cytosolic Ca2+ levels, similar or even larger levels of mitochondrial Ca2+ ([Ca2+]m) uptake have been observed [15]. [Ca2+]m overload is a pro-apoptotic mechanism that induces mitochondrial swelling, which perturbs or ruptures the outer mitochondrial membrane, inducing the release of mitochondrial apoptotic factors into the cytosol [16, 17].
A large body of evidence has indicated the potential role of Ca2+ in the onset or progression of cancer, highlighting Ca2+ handling as a potential target for the pharmacological treatment of tumors [18]. In particular, perturbations in Ca2+ homeostasis and alterations in the principal pumps and channels responsible for appropriate Ca2+ handling have been found in cancers of different histotypes [19–23]. It has been reported that the mineral asbestos, which is considered the main cause of MPM onset/progression, induces an endoplasmic reticulum (ER)-stress response, consequently altering ER-Ca2+ ([Ca2+]ER) release [24]. Furthermore, several compounds considered to be promising molecules for the treatment of MPM are capable of inducing oxidative stress, leading to ER-stress and Ca2+ release [24]. Moreover, mutations and alterations in the expression of many oncogenes and tumor suppressor genes, which are involved in the control of both apoptosis and Ca2+ handling, have been identified in mesothelioma specimens and are considered responsible for MPM onset/progression [25–28].
Collectively, these data suggest the importance of crosstalk between the dysregulation of Ca2+ homeostasis, apoptosis and cancer progression. It is noteworthy that the relevance of these relationships to human MPM could be highly significant for the development of new therapeutic approaches for this cancer, which at present is neglected.
In this study, MPM-specimens exhibited a global reduction in Ca2+ physiology. This characteristic involved the inhibition of Ca2+ transfer from the ER to the mitochondria, preserved mitochondrial integrity and protected against Ca2+-mediated apoptosis. Furthermore, for the first time, we demonstrated that various approaches promoting intracellular Ca2+ accumulation restored the typically deficient levels of apoptosis in MPM cells back to physiological levels.
RESULTS
MPM specimens exhibit dysregulated cellular Ca2+ signaling
Our study aimed to investigate whether intracellular Ca2+ plays a key role in the progression of MPM. To measure intracellular Ca2+ homeostasis, we used aequorin probes targeted to different subcellular compartments. These experiments employed the inositol triphosphate agonist bradykinin (BK), which is known to act via distinct surface membrane receptors to promote ER-Ca2+ mobilization [29]. The data shown in Figure 1A demonstrate that short-term cell cultures derived from biopsies of MPM-affected patients (MPM) exhibited a marked reduction in [Ca2+]m uptake in response to stimulation of the cells with BK when compared with cell cultures derived from biopsies of healthy individuals (HM). In parallel, we found the same features in MPM commercial cell lines (Figure 1B and Supplementary Figure S1A–1B).
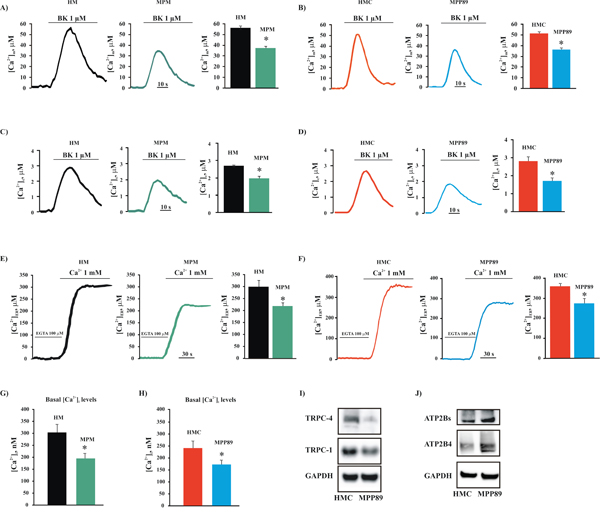
Figure 1: Intracellular and intraorganellar Ca2+ homeostasis in MPM cells. Representative mitochondrial A. and cytosolic B. Ca2+ measurements in primary cell cultures obtained from healthy (HM) and MPM-affected patients (MPM) (peak amplitude [Ca2+]m: 56.83 ± 1.32 μM [HM], 37.89 ± 1.44 μM [MPM], n = 16; peak amplitude [Ca2+]c: 2.87 ± 0.43 μM [HM] vs. 2.02 ± 0.34 μM [MPM]; n = 18). Likewise, the alteration of mitochondrial C. and cytosolic D. Ca2+ handling was assessed in normal mesothelial (HMC) and malignant mesothelioma (MPP89) cell lines (peak amplitude [Ca2+]m: 51.36 ± 1.87 μM [HMC], 36.81 ± 1.98 μM [MPP89], n = 32; peak amplitude [Ca2+]c: 2.83 ± 0.34 μM [HMC], 1.75 ± 0.23 μM [MPP89], n = 37). Additionally, the steady-state [Ca2+]ER was analyzed in primary cell cultures obtained from healthy (HM) and MPM-affected patients (MPM) E. and in normal (HMC) and MPM (MPP89) F. commercial cell lines (steady state [Ca2+]ER: 217.86 ± 14.34 μM [MPM], 298.45 ± 22.21 μM [HM], n = 12; 283.67 ± 18.11 μM [MPP89], 364.49 ± 11.81 μM [HMC], n = 14). Representative traces are shown. Next, primary cell cultures G. and commercial cell lines H. were loaded with the Ca2+-indicator FURA-2/AM to analyze the basal [Ca2+]i (basal [Ca2+]i in commercial cell lines: 238.73 ± 18.24 nM [HMC], 174.78 ± 11.53 nM [MPP89], n = 16; basal [Ca2+]i in primary cell cultures: 304.48 ± 31.65 nM [HM], 193.98 ± 22.72 nM [MPM], n = 14). Finally, the protein expression of C-type TRPCs I. and ATP2Bs J. in normal and mesothelioma cell lines was assessed by immunoblotting. Membrane protein samples (15 μg/lane) were loaded and probed using specific antibodies. GAPDH was used as a loading control. All graphs display the means ± SEM. *p < 0.01. Abbreviations: BK, bradykinin; KRB: Krebs ringer buffer.
To investigate the possibility that this reduced Ca2+ signaling was not restricted to the mitochondrial compartment, we monitored the Ca2+ concentrations in the cytosol ([Ca2+]c). In MPM cells, the [Ca2+]c increases were significantly smaller than those in control cells (Figure 1C–1D). Given that the concentrations of Ca2+ in the mitochondria and cytosol are highly dependent on the amount of Ca2+ in the ER, we investigated the Ca2+ concentrations in the ER compartment [Ca2+]ER. We found that the steady state [Ca2+]ER in the mesothelioma cell was markedly lower than in HMC controls (Figure 1E–1F).
The ER constitutes the principal Ca2+ store and participates in the initial rapid increase in [Ca2+]c by supplying Ca2+ via the inositol 1,4,5-trisphosphate receptors (ITPRs). The ER also participates in the subsequent decrease in [Ca2+]c by removing Ca2+ from the cytoplasm and recovering the internal Ca2+ stores through the action of sarco- and endoplasmic reticulum Ca2+-ATPases (ATP2A2). It is clear that ATP2A2 pumps are the principal regulator for the maintenance of [Ca2+]ER. One of the most common compounds used to induce intracellular Ca2+ accumulation, the sesquiterpene thapsigargin (TG), is a specific and potent inhibitor of ATP2A2. Taking advantage of this feature, we decided to evaluate the native store filling of the ER compartment in normal and mesothelioma cells. Cells were loaded in Ca2+-free medium with the Ca2+-indicator Fura-2-acetoxymethylester (FURA-2/AM) for 30 min, and the levels of the thapsigargin-releasable Ca2+ were assessed. We found that in MPM cells, the thapsigargin-dependent intracellular Ca2+ elevation was significantly lower when compared with those observed in HMC cells (Supplementary Figure S1C–1D). These results suggest that the native store filling of the ER may be seriously affected. As yet reported, the amplitude of the ER-Ca2+ transient is regulated by different ER-resident protein, such as ATP2A2 and ITPR3. For these reasons, we investigate whether the alteration in Ca2+ handling could be due to a change in the expression of an ER-Ca2+ binding proteins.
Interestingly, we found that in MPM cells, the levels of the ATP2A2 and ITPR3 proteins were seriously affected. By contrast, we did not observe alterations in the expression of other ER-Ca2+ binding proteins, such as CALNX (Supplementary Figure S1E). Overall, these data may suggest that MPM cells display a critical perturbation in [Ca2+]ER, most likely due to a lack in the activity of two of the main proteins involved in Ca2+ release and Ca2+ reuptake at ER level.
It is well established that the triggering of apoptosis involves Ca2+ influx via mitochondrial, cytoplasmic and ER-mediated mechanisms [30]. Similarly, it is well known that transformed cells lacking apoptosis exhibit a reduced capacity to maintain an excessive intracellular Ca2+ concentration ([Ca2+]i); this is often due to store-operated Ca2+ entry (SOCE) that is no longer functional and/or suppressed by Ca2+ efflux across the plasma membrane (PM) [8]. To investigate whether this remodeling of Ca2+ signaling is also a characteristic of MPM, a cytosolic aequorin Ca2+ probe and the Ca2+ indicator FURA-2/AM were used to measure capacitative and basal Ca2+ influx, respectively. The data reported in Supplementary Figure S1F show that the MPM cell lines exhibited reduced Ca2+ influx activity. Indeed, MPP89 cells displayed a significant reduction in their ability to take up Ca2+ into the cytosolic compartment compared with HMC. In accordance with these data, the basal [Ca2+]i was markedly lower in the MPM cells compared with the control cells (Figure 1G–1H).
Many studies have established that multiple cancer types are associated with alterations in the expression of proteins involved in the movement of Ca2+ across the PM, specifically Ca2+ channels and pumps such as TRP-channels (TRPCs) and PM Ca2+-ATPase pumps (ATP2Bs) [8].
Thus, we assessed the expression of TRPCs and ATP2Bs in MPM and HMC cells. We found that the protein levels of the TRPC isoforms TRPC1 and TRP4C3 were substantially reduced in mesothelioma cells compared with HMC cells (Figure 1I). Furthermore, consistent with the basal intracellular Ca2+ measurements reported in Figure 1G–1H, we found that expression of the ATP2Bs, including isoform ATP2B4, was increased in MPM cells (Figure 1J). Importantly, it has been demonstrated that this ATP2B4 isoform is strongly involved in tumorigenesis [31].
MPM specimens exhibited alterations of the apoptotic machinery
As described above, Ca2+ plays various key physiological roles in normal human cells. Importantly, Ca2+ overload has been considered to be a final common pathway of apoptosis. Because we found significant differences in Ca2+ homeostasis between MPM and HMC cells, we sought to examine the rate of apoptosis under our experimental conditions. We found that HMC cells display significantly higher apoptotic levels than those observed in MPM cells (Figure 2A–2C and Supplementary Figure S2). Furthermore, these findings were clearly confirmed by analyzing the levels of apoptosis in other mesothelioma specimens (Supplementary Figure S2).
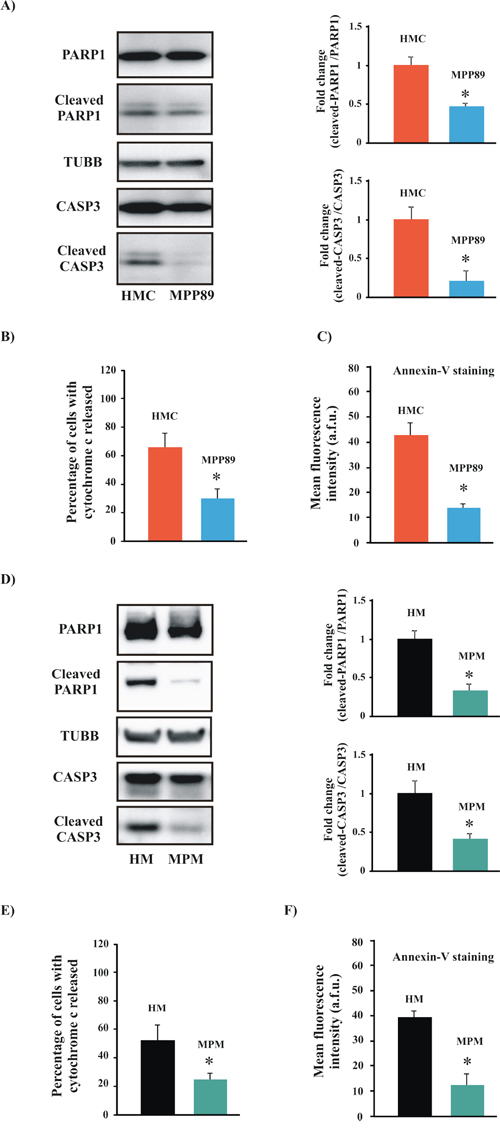
Figure 2: Measurements of apoptotic levels in MPM samples. A. Representative immunoblot showing cleavage of the apoptotic markers PARP1 and CASP3 in normal mesothelial (HMC) and mesothelioma (MPP89) cell lines. The upper and lower graphs are expressed as the means ± SEM and depict the fold-changes in the cleaved-CASP3/CASP3 ratio and the cleaved-PARP1/PARP1 ratio, respectively. B–C. Analysis of cytochrome c release and Annexin-V staining. The subcellular distribution of cytochrome C (B) was visualized using an anti-cytochrome c antibody (percentage of cells with cytochrome c released: 62.76 ± 15.42% [HMC] vs. 29.32 ± 7.56% [MPP89]; n = 7). For detection of PS on the extracellular surface of the PM, cells were stained with Annexin-V (Annexin-V fluorescence intensity: (41.21 ± 7.32 a.f.u. [HMC] vs. 13.45 ± 2.98 a.f.u. [MPP89]; n = 8) (C) The same experiments were replicated in short-term cell cultures obtained from healthy (HM) and MPM-affected patients (MPM). As reported in the text, the apoptotic machinery in the primary MPM cells was affected D–F. (percentage of cells with cytochrome c released: 56.87 ± 11.23% [HM] vs. 22.91 ± 3.65% [MPM]; n = 5; (Annexin-V fluorescence intensity: 38.89 ± 5.01 a.f.u. [HM] vs. 11.77 ± 4.17 a.f.u. [MPP89]; n = 5). All graphs display the means ± SEM. *p < 0.01.
Notably, a major criticism of cell lines is that they grow indefinitely in the laboratory and could differ genetically from primary tissue, accumulating new mutations as they adapt to their artificial environment. To address this issue, we performed the same apoptosis assays on primary cell cultures obtained from biopsies of healthy individuals (HM) or MPM-affected patients. The results of these studies confirmed our hypothesis. Indeed, mesothelioma specimens exhibited a sustained downregulation of the apoptosis pathway compared with normal mesothelial cells (Figure 2D–2F).
Dysregulated cellular Ca2+ signaling exerts adverse effects on the execution of the mitochondrial apoptotic process and on the sensitivity to apoptotic stimuli
Due to the large differences in the modulation of Ca2+ homeostasis and the activity of the apoptotic machinery between normal mesothelial and mesothelioma cells, we sought to examine whether these two aspects of cell death were interrelated. Ca2+ transfer from the ER to the mitochondria has been shown to be involved in several models of apoptosis. Importantly, it has been well demonstrated that this event is directly responsible for mitochondrial Ca2+ overload, mitochondrial permeability transition (MPT) pore opening and caspase-mediated cell death [6]. Various apoptotic stimuli, such as menadione, H2O2 and C2-ceramide (C2), induce the movement of Ca2+ from the ER to the mitochondrial compartment [32, 33]. Based on these findings, we investigated whether treatment with these stimuli promoted the apoptotic process via the use of Ca2+ as a cofactor that sensitizes mesothelioma cells for apoptotic cell death.
First, we analyzed the biochemical characteristics of the apoptotic process in cells subjected to oxidative stress (menadione). As expected, we found that apoptotic activity was strongly induced in HMCs treated with menadione, but not in MPM cells, where the ability of the oxidative agent to induce apoptosis was abrogated (Figure 3A–3B and Supplementary Figure S3A–3B). Then, we measured apoptosis after the administration of other apoptotic agents. We found that treatment with H2O2 and C2 did not activate the apoptotic program in mesothelioma cells (Supplementary Figure S3C).
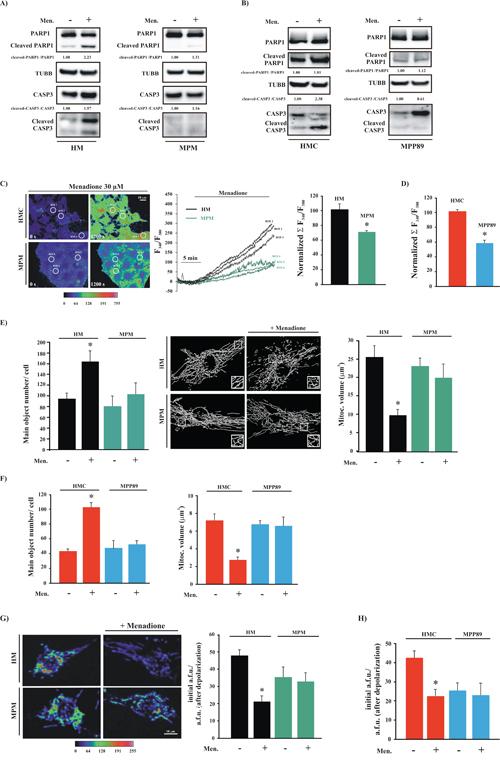
Figure 3: MPM cells are insensitive to pro-apoptotic stimuli. A. Primary cell cultures (HM and MPM) and B. cell lines (HMC and MPP89) cells were treated with 30 μM menadione (Men) or left untreated. After 30 min, the cells were subjected to immunoblotting for the indicated proteins. Normalized fold-change values in the cleaved-CASP3/CASP3 ratio and cleaved-PARP1/PARP1 ratio are shown. C. Cells were loaded with FURA-2/AM and stimulated with 30 μM Menadione (Men) for 30 min. The kinetic properties of the Ca2+ response are presented as the ratio of fluorescence at 340 nm/380 nm. Representative images captured at different time points (0 s and 1200 s) are shown in the left panel and are displayed in pseudo-color. The middle panel depicts the representative kinetic properties of specific ROIs (regions of interest) in the left panel (black for HM and green for MPM). D. Normalized fluorescence ratio 340/380 nm of the Ca2+ response after stimulation with 30 μM menadione in HMC and MPP89 cells. Representative images and kinetics are shown in Supplementary Figure S3D E. Analysis of mitochondrial structures in HM and MPM cells transfected with a green fluorescent protein targeted to the mitochondria (mtGFP). The acquired images were deconvoluted, reconstructed and quantitatively analyzed. Representative reconstructed 3D images are shown. The data represent the means ± SEM of the mitochondrial number/cell (left) and the volume of an individual mitochondrion (right) (number of mitochondria: 94.67 ± 8.73 [HM], 164.43 ± 16.35 [HM+Men], n = 12; 81.03 ± 17.77 [MPM], 102.32 ± 21.37 [MPM+Men], n = 11); (volume of individual mitochondria: 25.98 ± 3.79 μm3 [HM], 9.31 ± 2.19 μm3 [HM+Men], n = 12; 23.71 ± 1.98 μm3 [MPM] vs. 19.89 ± 4.45 μm3 [MPM+Men]; n = 11). F. Quantitative analysis of mitochondrial parameters in normal (HMC) and mesothelioma (MPP89) cell lines: number of mitochondria per cells (41.23 ± 2.31 [HMC], 101.29 ± 5.87 [HMC+Men], n = 21; 43.11 ± 8.38 [MPP89], 49.67 ± 4.33 [MPP89+Men], n = 19) and volume of individual mitochondria: 7.43 ± 1.11 μm3 [HMC], 2.67 ± 0.63 μm3 [HMC+Men], n = 21; 6.88 ± 0.45 μm3 [MPP89] vs. 6.08 ± 1.54 μm3 [MPP89+Men]; n = 19). Representative images are shown in Supplementary Figure S3E. G. Cultured cells were loaded with TMRM and imaged via confocal microscopy to determine the Ψm. Representative images and quantitative results (means ± SEM) are shown (TMRM intensity a.f.u.: 48.22 ± 3.52 [HM], 21.76 ± 2.88 [HM+Men]; n = 13; p < 0.01; 35.11 ± 7.31 [MPM] vs. 31.22 ± 5.99 [MPM+Men]; n = 12). H. Bar graph showing quantification of the TMRM signals measured by fluorescence microscopy in HMC and MPP89 cells. (TMRM intensity a.f.u.: 41.86 ± 3.10 [HMC], 20.99 ± 4.77 [HMC+Men]; n = 12; p < 0.01; 24.09 ± 5.11 [MPP89] vs. 21.11 ± 6.54 [MPP89+Men]; n = 19). To establish the mitochondrial Ψm levels, the ratio between the initial TMRM fluorescence (initial a.f.u.) and the residual TMRM fluorescence (obtained after FCCP addition, a.f.u. after depolarization) was calculated. The values obtained represent the amount of the potential-indicating probe incorporated in the different experimental conditions. Representative images are shown in Supplementary Figure S3F. *p < 0.01.
As mentioned above, exposure to different apoptotic stimuli may trigger or enhance the amount of Ca2+ released from the ER. Thus, we investigated whether the apoptotic characteristics of menadione are associated with Ca2+ signaling via the ER. MPM and HMC cells were loaded with the Ca2+ indicator FURA-2/AM and monitored during stimulation with menadione. The addition of menadione to HMCs caused a rapid elevation of FURA-2-associated intracellular fluorescence during the following 30 min. In contrast, this increase was significantly reduced in MPM cells (Figure 3C–3D and Supplementary Figure S3D).
During apoptosis, mitochondria undergo massive fragmentation with concomitant permeabilization of the outer mitochondrial membrane and cytochrome c release. Furthermore, the mitochondrial membrane potential (Ψm) collapses, and the amount of ROS produced is augmented [34]. As demonstrated, we found remarkable morphological injury to the mitochondria, prominent dissipation of the Ψm and a notable augmentation of mitochondrial ROS production in HMC cells. No difference was detected in MPM cells treated with menadione (Figure 3E–3H and Supplementary Figure S3E–3H).
In recent years, important studies have indicated a key role for Ca2+ in apoptotic cell death based on the finding that specific oncogenes and tumor suppressor genes regulate cell death by perturbing intracellular Ca2+ homeostasis [35]. We therefore examined whether mesothelioma cells displayed some of these characteristics. We detected a prominent increase in the protein expression of activated AKT, BCL2 and BCL2L1 in MPM cells compared with normal mesothelial cells (Figure 4A). Importantly, we also found a significant reduction in the protein levels of the oncosuppressor PML (Figure 4A). Altogether, these findings may explain the observed dysregulated cellular Ca2+ signaling. Several works highlight the presence of a direct interaction between PML and AKT to modulate the Ca2+ handling. Furthermore, it has been widely reported that IP3 receptors (ITPRs) are substrates for AKT [36] and, in particular, that AKT controls the Ca2+-dependent apoptotic process by regulating ITPR3 activity [37]. Thus, we investigated the possible relationship between ITPR3 and AKT in mesothelioma cells. MPM cells (primary and commercial cell lines) were collected, lysed and immunoprecipitated with ITPR3 antibody.
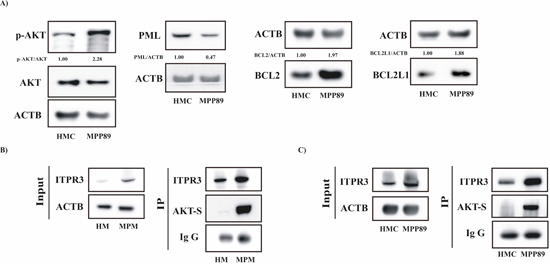
Figure 4: Apoptosis-related proteins regulating the core mechanism of the Ca2+-dependent apoptotic pathway are involved in MPM. A. Protein expression levels of the primary cofactors involved in the regulation of Ca2+ handling. Lysates prepared from HMC and MPP89 cells were loaded on a precast gel and transferred to nitrocellulose membranes. These membranes were probed for the desired proteins using specific primary antibodies. Normalized fold-change values in the ratio of p-AKT/AKT, PML/ACTB, BCL2/ACTB and BCL2L1/ACTB are shown. B–C. The AKT-mediated ITPR3 phosphorylation was investigated in primary cells obtained from healthy and MPM-affected individuals (B) and in commercial cell lines (C) Cell lysates were immunoprecipitated with ITPR3 antibody. The immunoprecipitates were immunoblotted with AKT-S antibody or ITPR3 antibody.
Next, immunoprecipitated fractions were assayed for phosphorylation using an AKT-substrate (AKT-S) antibody. The data reported in Figure 4B–4C show that the endogenous type III ITPR was highly phosphorylated by AKT in mesothelioma cells.
The recovery of Ca2+ homeostasis enhances [Ca2+]m uptake, thereby promoting apoptotic activity
To evaluate if the disruption of Ca2+ homeostasis is a major contributor to the absence of the apoptotic process that is typical of MPM, we attempted to force Ca2+-influx into mesothelioma cells because higher [Ca2+]i should increase mitochondrial Ca2+ uptake, thereby activating the apoptotic machinery. MPP89 cells were pretreated with a high external [Ca2+] (10 mM) or with sodium orthovanadate (Na3VO4, NaV, 1 μM), a general inhibitor of P-type ATPases, in particular ATP2Bs [38].
After 5 days of incubation in [Ca2+] or NaV, the growth capacity of the cells was affected (Figure 5A): a crystal violet cell proliferation assay indicated that the mesothelioma cells were less proliferative in the presence of high external [Ca2+] or NaV. This reduction in cell number suggested that the sensitivity of these cells to apoptosis was restored. Indeed, after 5 days of pretreatment with either compound, the MPP89 cells exhibited reactivation of the apoptotic machinery (Figure 5B).
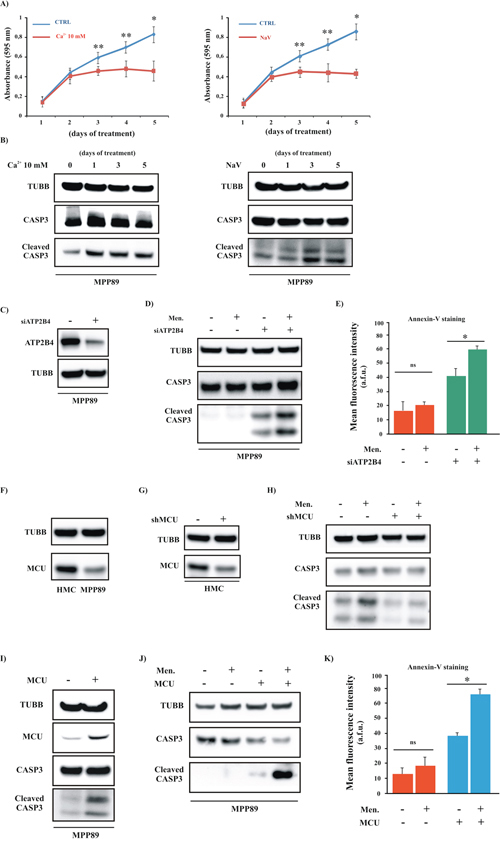
Figure 5: Positive modulation of Ca2+ handling restores the apoptotic machinery in MPM cell lines. A. Growth curves of MPP89 cells after 1, 2, 3, 4 and 5 days of treatment with 10 mM Ca2+ (left) or NaV (right) ([CTRL]: 1 day: 0.14 ± 0.049, 2 day: 0.42 ± 0.069, 3 day: 0.57 ± 0.067, 4 day: 0.66 ± 0.056 and 5 day 0.80 ± 0.103; [Ca2+ 10 mM]: 1 day: 0.13 ± 0.072, 2 day: 0.39 ± 0.091, 3 day: 0.44 ± 0.037, 4 day: 0.46 ± 0.108 and 5 day 0.44 ± 0.112; [NaV]: 1 day: 0.13 ± 0.029 2 day: 0.38 ± 0.044, 3 day: 0.43 ± 0.039, 4 day: 0.42 ± 0.108 and 5 day 0.41 ± 0.031, n = 4). B. Mesothelioma cell lines were cultured in complete medium and treated with 10 mM Ca2+ (left) or NaV (right) for different periods (1, 2, 3, 4 and 5 days) and immunoblotted. C–E. Representative immunoblot (C–D) and Annexin-V assay (E) in siRNA-ATP2B4 (siATP2B4)-expressing cells. For both experiments, cells were transfected with siATP2B4. After 36 h, the cells were harvested and immunoblotted for the indicated proteins or loaded with Annexin-V. Where indicated, the cells were treated with 30 μM Men for 30 min. (Annexin-V fluorescence intensity: 15.78 ± 6.75 a.f.u [CTRL], 19.22 ± 3.26 a.f.u [Men], 41.27 ± 4.62 [siATP2B4], 59.45 ± 1.87 a.f.u. [siATP2B4+Men]). F. Representative immunoblot showing the protein expression level of MCU in normal and cancerous cells. G–H. HMC cells were transfected with shRNA-MCU (shMCU). After 36 h of transfection, the cells were lysed for immunoblot and primary antibodies for TUBB and CASP3 were used. Where indicated, the cells were treated with 30 μM Men for 30 min. I–K. Western blot and Annexin-V assays in MCU-expressing cells. For both experiments, cells were transfected with MCU plasmid. After 36 h, the cells were harvested and immunoblotted for the indicated proteins (I–J) or loaded with Annexin-V (K) Where indicated, the cells were treated with 30 μM Men for 30 min. (Annexin-V fluorescence intensity: 12.78 ± 3.89 a.f.u [CTRL], 16.27 ± 4.11 a.f.u. [Men], 37.92 ± 1.53 a.f.u. [MCU], 64.76 ± 2.34 a.f.u. [MCU+Men]). *p < 0.01, **p < 0.05.
The inhibition of ATP2Bs promoted by NaV and the high external [Ca2+] should increase the [Ca2+]i levels and, in turn, [Ca2+]m uptake. Thus, we investigated the effect of NaV and 10 mM [Ca2+] on mitochondrial Ca2+ homeostasis. As illustrated in Figure 5B, a relevant increase in the cleavage of CASP3 was detected after just 3 days; this effect was maintained through the fifth day. Based on this finding, we measured the ability of the mitochondria to absorb Ca2+ after 3 days of treatment. Our hypotheses were confirmed: pretreated MPP89 cells exhibited significant augmentation of [Ca2+]m uptake (Supplementary Figure S4A). In addition to this, we found that these compounds also considerably modulated the overall intracellular Ca2+ signaling (Supplementary Figure S4B–S4C).
Even if NaV is reported to be a potent inhibitor of ATP2B4, we cannot be sure that the observed effects are due to a selective and unique interference with ATP2B4 activity.
Thus, to rule out the possibility that we were observing spurious effects, we evaluated the impact of silencing ATP2B4 on mitochondrial Ca2+ uptake and the apoptotic process using RNA interference (RNAi). Silencing ATP2B4 in MPM cells expressing a mtAEQ restores the [Ca2+]m uptake (Supplementary Figure S4A). In addition, the RNAi rescues the sensitivity of mesothelioma cells to apoptotic stimuli (Figure 5C–5E).
Because increasing the intracellular Ca2+ levels restored the ability of the mitochondria to take up Ca2+ and to initiate the apoptotic process in MPM cells, we examined whether the direct and positive modulation of [Ca2+]m could rescue the sensitivity of mesothelioma cells to apoptotic stimuli.
Recent publications report the identification of the complex responsible for [Ca2+]m uptake: the mitochondrial Ca2+ uniporter (MCU) complex [39]. From its molecular discovery, several studies have investigated the significance of mitochondrial Ca2+ uptake on specific cellular processes, including carcinogenesis, through the modulation of MCU expression levels [40]. For example, a recent study identified MCU down-regulation as a characteristic feature of some human colon and prostate cancer cells [40]. No studies have yet assessed MCU in the context of mesothelioma. Based on these data, we investigated the MCU protein levels in normal and mesothelioma cells. Interestingly, we found that MPM cells displayed a significant reduction in the basal MCU protein levels (Figure 5F). Next, we explored whether the modulation of MCU activity could modulate apoptotic cell death via regulation of [Ca2+]m uptake in normal and MPM cells. HMCs were transfected using a specific shRNA to silence MCU expression. Compared to the shRNA control, cells transfected with shRNA-MCU (shMCU) showed a significant decrease in the MCU protein levels (Figure 5G) and a significant reduction of [Ca2+]m in HMCs (Supplementary Figure S4D). Furthermore, silencing MCU induced a marked reduction in the apoptotic machinery and in the sensitivity to pro-apoptotic stimuli (Figure 5H).
Similar experiments were carried out in MPM cells. Contrary to HMCs, we investigated the effects induced by MCU overexpression in MPM samples.
First, we evaluated the ability of the MCU plasmid to modulate MCU expression. As expected, a marked increase in the MCU protein levels was found upon MCU overexpression (Figure 5I). Next, we tested the effectiveness of MCU in modulating [Ca2+]m. The data (Supplementary Figure S4A) demonstrated that MCU caused a marked enhancement of the increase in [Ca2+]m.
Then, we investigated the possible activation of the apoptotic cascade. Mesothelioma cells with forced increases in the expression of MCU protein displayed elevated apoptotic activity (Figure 5I). Finally, we explored whether the augmentation of MCU expression sensitized mesothelioma cells to apoptotic stimuli. As reported in Figure 5J–5K, we detected a prominent activation of the apoptotic machinery in MCU-expressing cells that were exposed to a Ca2+-dependent apoptotic stimulus.
Increased [Ca2+]m transport restores apoptosis in primary MPM cells
We attempted to reproduce this last set of experiments using primary cells obtained from biopsies of MPM-affected patients. MPM cells were pretreated with a high external [Ca2+] (10 mM) or with 1 μM NaV for 5 days, and the growth capacity of the cells was assessed. We found that similar to the mesothelioma cell lines, the primary mesothelioma short-term cultures were less proliferative in the presence of high external [Ca2+] or NaV after 5 days of incubation (Figure 6A). In parallel with the demonstrated involvement of the apoptotic machinery, the amount of cleaved CASP3 was increased (Figure 6B). Similar results were obtained after ATP2B4 silencing. In fact, compared to the siRNA control, cells transfected with siRNA-ATP2B4 (siATP2B4) showed a significant decrease in protein expression (Figure 6C), accompanied with a prominent increase in [Ca2+]m (Supplementary Figure S4A). We also evaluated the effects of ATP2B4 silencing in modulating the apoptotic response. To assess whether ATP2B4 silencing modified the apoptotic process, we challenged siATP2B4-transfected MPM cells with the Ca2+-dependent apoptotic stimulus menadione. As result, the silencing of ATP2B4 promoted reactivation of the apoptotic machinery and sensitized mesothelioma cells to apoptotic stimuli (Figure 6D–6E).
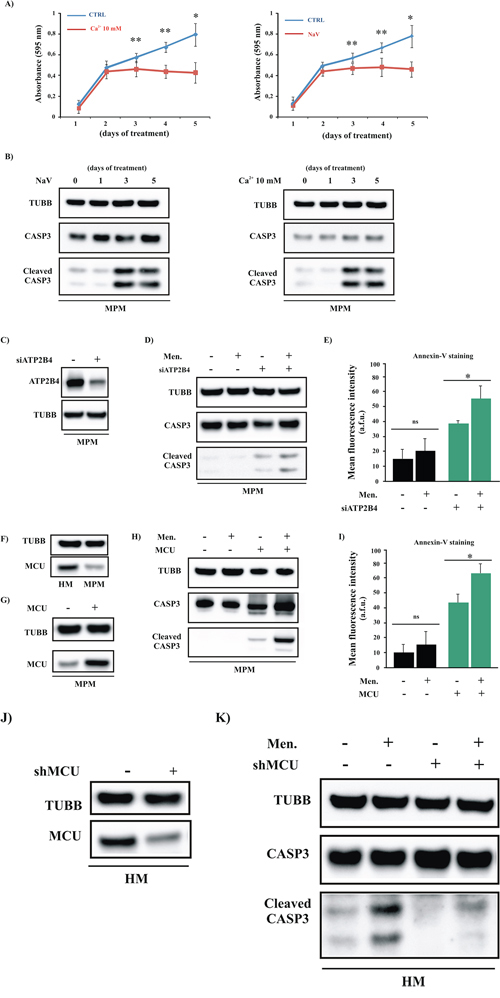
Figure 6: Increased mitochondrial Ca2+ transport restores the apoptotic process in primary MPM cells. A. Primary cells obtained from MPM-affected patients were cultured in complete medium and exposed to a high external [Ca2+] (10 mM) or to NaV for different periods. After 1, 2, 3, 4 or 5 days of treatment, the cells were fixed for crystal violet cell proliferation assays ([CTRL]: 1 day 0.16 ± 0.021, 2 day: 0.53 ± 0.051, 3 day: 0.61 ± 0.029, 4 day: 0.70 ± 0.032 and 5 day 0.80 ± 0.126; [Ca2+ 10 mM, left panel]: 1 day 0.12 ± 0.023, 2 day: 0.45 ± 0.061, 3 day: 0.47 ± 0.058, 4 day: 0.45 ± 0.043 and 5 day 0.44 ± 0.115; [NaV, right panel]: 1 day 0.14 ± 0.038, 2 day: 0.45 ± 0.043, 3 day: 0.48 ± 0.058, 4 day: 0.49 ± 0.078 and 5 day 0.47 ± 0.068, n = 4). B. Primary cells obtained from MPM-affected patients (MPM) were cultured in complete medium and exposed to a high external [Ca2+] (10 mM) or to NaV for different periods. After 1, 2, 3, 4 or 5 days of treatment, the cells were lysed for immunoblotting. C–D. Representative immunoblot showing the cleavage of the apoptotic marker CASP3 in siRNA-ATP2B4 (siATP2B4)-expressing cells. E. Apoptotic measurements in siATP2B4-transfected MPM cells were also carried out by loading cells with Annexin-V. Where indicated cells were treated with Men for 30 min. (Annexin-V fluorescence intensity: 13.93 ± 4.98 a.f.u. [CTRL], 20.12 ± 8.92 a.f.u. [MEN], 39.05 ± 2.21 a.f.u. [siATP2B4], 55.01 ± 8.03 a.f.u. [siATP2B4 +Men] F. Immunoblotting showing the protein expression levels of MCU in HM and MPM cells. G–I. MPM cells were transfected with MCU plasmid and immunoblotted for the indicated proteins (G–H) or loaded with Annexin-V (I) to investigate the amount of PS on the outer leaflet of the PM (Annexin-V fluorescence intensity: 9.85 ± 4.63 a.f.u. [CTRL], 14.71 ± 5.38 a.f.u. [MEN], 42.75 ± 3.43 a.f.u. [MCU], 64.11 ± 4.73 a.f.u. [MCU+Men]). J–K. Downregulation of the MCU protein in HM cells after transfection with shRNA-MCU (shMCU) was confirmed by western blot analysis (K) Immunoblot showing the amount of cleaved CASP3 in total HM cell transfected with shMCU. TUBB was used as a loading control. Where indicated, the cells were transfected with shMCU (+) or control shRNA (–) and loaded with 30 μM Men for 30 min. *p < 0.01, **p < 0.05.
Next, to examine the relationship between Ca2+ signaling and the apoptotic process, we evaluated whether direct modulation of [Ca2+]m signaling was useful and necessary to enhance the sensitivity of cancerous cells to apoptosis-sensitizing cofactors. As first, and similarly to the mesothelioma cell lines, we found that cells obtained from biopsies of MPM-affected patients displayed lower amounts of MCU protein than HMCs (Figure 6F). This evidence confirms our hypothesis that MCU is a key player in mesothelioma disease. Thus, to increase the ability of mitochondria to absorb Ca2+, MPM cells were transfected with the MCU plasmid and treated with the Ca2+-dependent apoptotic stimulus menadione. The results obtained were very promising: the amount of active CASP3 and the fluorescence intensity of Annexin-V after the addition of menadione were significantly increased when the cells were transfected with MCU (Figure 6G–6I). Next, we aimed to confirm the importance of MCU during the Ca2+-dependent apoptotic process by promoting MCU-silencing in primary HMC samples. As expected, silencing of MCU reduces the ability of mitochondria to take up Ca2+ (Supplementary Figure S4D) and the MCU protein levels (Figure 6J). Of particular interest, we confirmed that shRNA-MCU (shMCU)-transfected HMC samples were less sensitive to apoptosis-sensitizing treatments than samples transfected with the control shRNA (Figure 6J–6K).
Overall these results highlight the role of MCU during MPM onset and demonstrate that by modulating Ca2+ handling, it could be possible to develop novel therapeutic approaches against this disease. Interestingly, treatments with NaV and high external [Ca2+] restore Ca2+ signaling and apoptosis without affecting MCU expression (Supplementary Figure S5A–5B), demonstrating the significance of correct ER-mitochondrial Ca2+ cross-talk as a primary contributor to the apoptotic program in mesothelioma.
DISCUSSION
Few research studies have examined the importance of Ca2+ in MPM. Several investigations have indicated roles for different proteins in executing the mitochondrial (intrinsic) apoptotic pathway, and although many of these proteins are involved in Ca2+-handling, the direct relationship between Ca2+ and the regulation of apoptosis has yet to be addressed.
Among these proteins, BCL2 family members and the oncogene AKT were found to be dysregulated in mesothelioma cells [41–43]. Notably, it is widely appreciated that these proteins are also fundamental cofactors of the Ca2+-dependent pathway of apoptosis [9, 44]. Given these findings, our results associate the lack of apoptosis that is characteristic of mesothelioma cells with the core mechanism of the Ca2+-dependent apoptotic pathway. Notably, we found that MPM cells display a significant alteration in the expression levels of the apoptosis-related proteins reported above, including BCL2, BCL2L1 and AKT. Of relevance, in several tumor types the kinase AKT may exert its antiapoptotic function by regulating the type III ITPRs. Our results suggest that this feature is also preserved in mesothelioma disease. We found that the AKT-mediated ITPR3 phosphorylation was higher than in normal mesothelial cells. Furthermore, the levels of the oncosuppressor PML were reduced in MPM cells. PML was recently found to trigger the apoptotic program by modulating Ca2+ release from the ER [33]. This finding may support the hypothesis that MPM cells exhibit dysregulated Ca2+ signaling. The results reported in Figure 1 and Supplementary Figure S2 robustly support this model and demonstrate that the overall Ca2+ homeostasis of MPM cells is strongly affected.
Considering these results, we wondered whether Ca2+ is involved in the lack of apoptosis that is characteristic of mesothelioma cells. To address this question, we analyzed whether pro-apoptotic stimuli promote the apoptotic process via the employment of Ca2+ as an apoptosis-sensitizing cofactor. When normal mesothelial cells were exposed to apoptotic stimuli, we detected the progressive release of Ca2+ from intracellular stores, along with consequent alterations in mitochondrial energy and structure and, subsequently, the activation of caspases. In contrast, MPM cells were insensitive to these stimuli. Thus, these findings indicate that the observed reduction in Ca2+ homeostasis represents a pivotal mediator of the unresponsiveness of MPM cells to apoptotic stimuli. Studies are currently underway to elucidate the exact molecular mechanisms that underlie the downregulation of Ca2+ signaling which occurs in mesothelioma. Considering our results, we hypothesize that the PML-AKT-Ca2+ axis is directly involved in mesothelioma because a close relationship between AKT-PML and Ca2+-dependent apoptosis has already been demonstrated in other tumor types [33]. Furthermore, based on the identification of relevant alterations in Ca2+ flux across the PM and in the expression of specific Ca2+ channels and pumps, we also propose that these molecules actively participate in the pathogenesis of MPM. Consistent with this, silencing PMCA by RNA interference or NaV treatment restores the Ca2+ homeostasis and rescues the sensitivity of mesothelioma to apoptotic stimuli. Of relevance, the use of vanadate compounds as therapeutic agents has been reported [45, 46].
However, we do not exclude the role of other molecular pathways in the pathogenesis of MPM.
Indeed, the key role of BCL2/L1 in mesothelioma has been well demonstrated, and the use of pharmacological inhibitors of BCL2 family members is promising. Unfortunately, the development of these inhibitors requires further refinement [47]. Other studies have suggested the involvement of receptor tyrosine kinases (RTKs) in mesothelioma. Once activated, these kinases lead to the upregulation of two major downstream cell signaling cascades: the RAF1-MEK-RAS-ERK and PI3K-AKT-PTEN-MTOR pathways. Based on these findings, small molecule inhibitors of specific RTKs were applied in clinical studies, but they displayed only limited activity in advanced MPM patients [48, 49]. It is clear that several studies have identified molecules that mediate MPM, but a well-established molecular pathway and an effective molecular approach to combat this disease have yet to be discovered and developed.
Recent evidence has shown that specific cellular processes regulated by mitochondrial Ca2+ uptake, including carcinogenesis, are strictly supervised through the modulation and expression of the recent discovery channel (MCU) responsible for a correct [Ca2+]m handling. For example, MCU silencing attenuates [Ca2+]m [9, 10], and reduced [Ca2+]m levels suppress cell death in a variety of cancerous cell lines, including those derived from the cervix, central nervous system and lymph tissues [44, 50, 51]. Furthermore, reduced MCU levels have been found in human samples of colon and prostate cells [40]. Nevertheless, the potential role for MCU to modulate cell death and other events in mesothelioma has not yet been addressed. Thus, having observed the critical dysregulation in overall Ca2+ handling and Ca2+-dependent apoptosis, we hypothesized that MCU could be affected in mesothelioma cells. The results reported in Figure 5F and 6F support this hypothesis and show that MPM samples display an overall reduction in MCU expression.
Based on these findings, we evaluated the involvement of the MCU in regulating the [Ca2+]m levels and survival as a possible fundamental aspect of the pathology of MPM. The results were surprising. We detected a remarkable reduction in cell growth accompanied by an increase in the apoptotic response and a restored sensitivity to proapoptotic agents.
Interestingly, one of the most recent advances in cancer research is the demonstration that microRNAs (miRs) cause a variety of human disorders, particularly cancer [52, 53], and regulate the expression of genes encoding key regulatory molecules of Ca2+ signaling. Specifically, miR-25 was found to decrease mitochondrial Ca2+ uptake via specific MCU downregulation, conferring resistance to apoptotic stimuli [40]. One intriguing finding is that a comparative analysis of miR expression demonstrated that miR-25 was markedly upregulated in MPM cells [54]. Therefore, we sought to verify whether miR-25 may be a feature during the progression of MPM. Preliminary results (data not shown) suggest that inhibition of miR-25 may modulate apoptotic cell death via the enhancement of [Ca2+]m uptake. In addition, a marked increase in the levels of MCU protein was found following miR-25 inhibition (data not shown).
Nevertheless it may be possible to consider miR-25 as a possible diagnostic marker and a novel therapeutic target, it is well known that a single miR can target multiple downstream transcripts and that many transcripts can be regulated by multiple miRs. Thus, further investigations are ongoing to discern the exact relationship between MPM, MCU and miR-25.
In conclusion, i) intracellular Ca2+ signaling plays an important role in MPM; ii) the downregulation of Ca2+ handling exerts an adverse effect on the execution of the apoptotic process and iii) on the sensitivity to apoptotic stimuli; iv) the recovery of Ca2+ homeostasis enhances Ca2+ uptake by the mitochondria, thereby promoting apoptotic activity; and v) MCU is a key mediator of MPM, and vi) its pharmacological modulation restores the rate of apoptosis and the sensitivity to apoptotic stimuli.
MATERIALS AND METHODS
Cell culture and transfection
Human normal mesothelial cells (HMs) were obtained from biopsies collected from young non-oncologic patients (age 29–35 ys) affected by pneumothorax at the Surgical Clinic of the University/Hospital, Ferrara.
MPM cell lines of commercial origin, such as MPP89, were from our cell line collections [54], and primary MPM cells were established from tumor biopsies obtained from patients affected by MPM [55]. HMCs were grown in RPMI medium 1640 (Euroclone), 10% FBS (Life Technologies), 2 mM L-Glutamine, and MPM cells were grown in D-MEM Ham’s F12 (Euroclone), 10% FBS (Life Technologies). Cells were transfected using Lipofectamine 2000 or Lipofectamine LTX (Life Technologies) with the following plasmids: MCU and mtGFP. For aequorin measurements, cells were transfected with aequorin-targeted probes (mtAEQ, cytAEQ and erAEQ). For immunostaining, mitochondrial morphology analysis, ROS measurements cytochrome c staining and single-cell [Ca2+]i measurements, cells were seeded on 24-mm glass coverslips. For aequorin measurements, the cells were seeded before transfection onto 13-mm glass coverslips and allowed to grow to 50–60% confluence. For the proliferation assay, cells were seeded on 12-well plates. For immunoblotting analysis, cells were seeded on 6-well plates with the same conditions for growth.
HMC and MPM cells were derived from surgical pleural samples of patients. Informed written consent was obtained from the patients to established MPM cell lines. This study was approved by the County Ethical Committee, Ferrara, Italy. Samples were obtained in sterile conditions and put in RPMI medium supplied with 50 U/ml of penicillin (CAMBREX) and 50 mg/ml of streptomycin (CAMBREX). The biopsy samples were cut in small pieces and transferred to 25-cm2 flasks containing RPMI 1640 medium supplemented with 10% FBS (CAMBREX), 2 mM L-glutamine (CAMBREX), 25 U/ml of penicillin (CAMBREX) and 25 mg/ml of streptomycin (CAMBREX) and added with 200U/ml of collagenase type 2 (Wortington). The samples were incubated at 37°C in a 5% CO2-humidified atmosphere over night to separate the cells from the tissue. The next day, the samples were washed with PBS 1X, mixed by pipetting, centrifuged for 10 minutes at 1000 r.p.m. and plated in new 25-cm2 flasks with 5 ml of complete medium. The cultures were maintained at 37°C in a 5% CO2-humidified atmosphere. The medium was changed twice a week, and the cultures were split according to their growth rate
Different morphological and proliferative parameters of HMC and MPM cultures were studied using immunofluorescence techniques. A panel of antibodies panel containing mouse anti-human MSLN HBME-1 (DAKO), goat anti-human CALB2 (SANTA CRUZ), mouse anti-human KRT 8–18 (SANTA CRUZ), and mouse anti-human VIM (SIGMA-ALDRICH) was used to characterized the cell lines. Immunofluorescence was carried out by covering fixed cells with 75 μl of the primary antibody diluted in PBS 1X as follows: mouse anti-human MSLN HBME-1 1:50, goat anti-human CALB2 1:50, mouse anti-human KRT 8-18 1:100, mouse anti-human VIM 1:200. The cells were incubated for 30 min at 37° C and subsequently washed three times in PBS 1X for 10 min. The slides were then dried on blotting paper and covered with 75 μl of secondary antibodies (anti-mouse or anti-goat) diluted in PBS 1X 1:50. The cells were incubated for another 30 min at 37°C and then washed three times in PBS 1X for 10 min. Finally, the samples were assembled on a chamber slide with 2 ml of glycerol-PBS 1X 7:3 supplemented with 0.5 mg/ml DAPI.
Intracellular calcium measurements
The cytosolic Ca2+ response was evaluated using the fluorescent Ca2+ indicator Fura-2/AM (Life Technologies). Cells were grown on 24-mm coverslips and incubated at 37°C for 30 min in 1 mM Ca2+/KRB supplemented with 2.5 mM Fura-2/AM, 0.02% Pluronic F-68 (Sigma), 0.1 mM Sulfinpyrazone (Sigma). The cells were then washed and supplied with 1 mM Ca2+/KRB. To determine the Ca2+ cytosolic response, the cells were placed in an open Leyden chamber on a 37°C thermo-controlled stage and exposed to 340/380 wavelength light using the Olympus Xcellence multiple wavelength high-resolution fluorescence microscopy system equipped with a UAPON 40 × 0340-2 objective, numerical aperture 1.35 (Olympus). The fluorescence data were expressed as emission ratios. Fluorescence was measured every 100 ms, and the [Ca2+]i was calculated by the ratio method using the equation: [Ca2+]c 1/4 Kd (R–Rmin)/(R–Rmax) Sf2/Sf1.
To measure the thapsigargin-releasable Ca2+ levels, cells grown on 24-mm coverslips were incubated for 30 min in 1 mM Ca2+-free medium/KRB supplemented with 2.5 mM Fura-2/AM, 0.02% Pluronic F-68 (Sigma), 0.1 mM Sulfinpyrazone (Sigma) and EGTA 100 μM. The fluorescence was measured every 100 ms. After 2 min., thapsigargin 100 nM was added to the cells, and the amount of Ca2+ released was estimated by normalization of the 340/380 ratio.
Aequorin measurements
All aequorin measurements were performed as previously described [56]. The cells were cultured on 12-mm glass coverslips and co-transfected at 70% confluence with 0.5 μg of aequorin (endoplasmic reticulum-targeted mutated aequorin, erAEQ, cytosolic aequorin, cytAEQ or mitochondria targeted aequorin, mtAEQ) and 1.25 μg of the indicated plasmid. Cells transfected with erAEQ were reconstituted with coelenterazine n (Tebu-Bio, Le-Perray-en-Yvelines, France), after ER Ca2+ depletion by incubation for 1 h at 4°C in KRB supplemented with 100 mM EGTA and 40 mM tBHQ (2,5-Di-tert-butylhydroquinone) (Sigma). The cells were then washed with KRB supplemented with 2% BSA and 1 mM EGTA. Cells that were transfected with cytAEQ and mtAEQ were reconstituted with coelenterazine (Synchem, Felsberg/Altenburg, Germany) for 2 h in KRB supplemented with 1 mM CaCl2. All aequorin measurements were carried out in 1 mM Ca2+/KRB (cytAEQ and mtAEQ) or 100 mM EGTA/KRB (erAEQ). ER refilling was trigger by perfusing with 1 mM Ca2+/KRB until a steady state was reached. An agonist was added to the same medium, as specified in the figures. The experiments were terminated by lysing the cells with 100 mM digitonin in a hypotonic Ca2+-rich solution (10 mM CaCl2 in H2O). The output of the discriminator was captured by a Thorn EMI photon-counting board and stored in an IBM-compatible computer for further analyses. The aequorin luminescence data were calibrated offline into [Ca2+] values using a computer algorithm based on the Ca2+ response curve of wild-type and mutant aequorins.
Antibodies, reagents and immunoblotting
For immunoblot and immunostaining analysis, the following antibodies were used: anti-TUBB (SIGMA-ALDRICH, T8328), anti-GAPDH (Cell Signaling, 14C10), anti-MCU (SIGMA-ALDRICH, HPA016480), anti-CASP3 (Cell Signaling, 9665), anti-PARP1 (Cell Signaling, 9532), anti-TRPC-1 (Santa Cruz, sc20110), anti-TRPC-4 (Santa Cruz, sc15063), anti-ATP2Bs (Santa Cruz, sc201917), anti-HSPD1 (Santa Cruz, sc376240), anti-ATP2B4 (Abcam, ab2783), anti-PML (Santa Cruz, sc966), anti-AKT (Cell Signaling, 9272), anti-pTHR308 AKT (Cell Signaling, 13038), anti-BCL2 (Santa Cruz, sc7382), anti-BCL2L1 (Santa Cruz, sc8392), anti-Cytochrome C (Abcam, ab90529), anti-ITPR3 (BD, 610312), anti-pSER/THR AKT Substrate (Cell Signaling, 9611), anti-ATP2A2 (Cell Signaling, 4388), anti-ACTN (SIGMA-ALDRICH, A1978), anti-CANX (Santa Cruz, sc-11397). Other chemicals used are the following: Sodium orthovanadate (NaV, SIGMA-ALDRICH), menadione (SIGMA-ALDRICH), H2O2 (SIGMA-ALDRICH), C2-ceramide (C2, SIGMA-ALDRICH), thapsigargin (SIGMA-ALDRICH, T9033).
For immunoblotting, cells were scraped into ice-cold phosphate-buffered saline and lysed in a modified 10 mM Tris buffer pH 7.4 containing 150 mM NaCl, 1% Triton X-100, 10% glycerol, 10 mM EDTA and protease inhibitor cocktail. After 30 min of incubation on ice, the lysates were cleared via centrifugation at 12,000 g at 4°C for 10 min. Protein concentrations were determined using the Bio-Rad procedure. Protein extracts, 15 or 25 μg, were separated on 4–12% bis-tris acrylamide and 4–20% tris-glycine gels (Life technologies, NP0323 and EC6026) and electro-transferred to PVDF or nitrocellulose membrane according to standard procedures. Non-specific binding sites were saturated by incubating the membranes with TBS-Tween 20 (0.05%) supplemented with 5% nonfat powdered milk for 1 h. Next, the membranes were incubated overnight with primary antibodies, and the binding was assessed using appropriate HRP-labeled secondary antibodies [Life Technologies, A16104 (goat anti-rabbit) and A24512 (goat anti-mouse)] plus a chemiluminescent substrate (Thermo Scientific, 34080). The chemiluminescence signals were detected using ImageQuant LAS 4010 (GE Healthcare). The immunoblots shown are representative of at least four independent experiments.
Immunoprecipitation
Immunoprecipitations were carried out using protein G-coated sepharose beads (GE Healthcare, Chalfont St. Giles, UK) following the manufacturer’s instructions. For whole-cell extracts, cells were incubated on ice for 20 minutes in the following lysis buffer: 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% NP-40 and supplemented with protease and phosphatase inhibitors (2 mM Na3VO4, 2 mM NaF, 1 mM PMSF and protease inhibitor cocktail). The lysates were clarified by centrifugation for 10 minutes at 13200 rpm before use. The total protein content of the cell lysates was measured with the Lowry assay. The extracted proteins (1000 μg) were incubated overnight with an ITPR3 antibody at 4°C. The next day, beads were added to the lysates. Precipitation of the immune complexes was carried out for 3 h at 4°C. Afterwards, the beads were washed with PBS supplemented with phosphatase inhibitors and PMSF. The samples were proceed by SDS-PAGE and analyzed using a standard western blotting technique.
Immunofluorescence
Images were taken with a Nikon Swept Field Confocal (Nikon). For each condition, at least 25 independent visual fields were counted. Mesothelioma cells were washed with PBS, fixed in 4% formaldehyde for 10 min and washed with PBS. Then, the cells were permeabilized for 10 min with 0.1% Triton X-100 in PBS and blocked in PBS containing 2% BSA and 0.05% Triton X-100 for 1 h. The cells were then incubated with primary antibody (anti-cytochrome c and anti-HSPD1) for 3 h at room temperature and washed three times with PBS. The appropriate isotype-matched, AlexaFluor-conjugated secondary antibodies [Life Technologies, A11008 (488 goat anti-rabbit) and A-21050 (633 goat anti-mouse)] were used. Images were obtained with a Nikon Swept Field confocal equipped with a CFI Plan Apo VC60XH objective (n.a. 1.4) (Nikon Instruments, Melville, NY) and an Andor DU885 EM-CCD camera (Andor Technology Ltd, Belfast, Northern Ireland). Statistical evaluation was performed using the colocalization counter JACOP, available in the Fiji software.
RNA interference (RNAi)
The shRNA targeting MCU (TRCN0000133861) was purchased from Sigma-Aldrich. The siRNA targeting ATP2B4 (GS493) was purchased from Qiagen. shRNA and siRNA were transfected using the HiPerfect® transfection reagent according to the manufacturer’s instructions.
Mitochondrial Ψm
Mitochondrial Ψm was measured by loading cells with 20 nM tetramethyl rhodamine methyl ester (TMRM; Life Technologies) for 30 min at 37°C. The TMRM excitation was performed at 560 nm and emission was collected through a 590 to 650 nm band-pass filter. Images were taken every 5 s with a fixed 20-ms exposure time using a Nikon Swept Field confocal equipped with CFI Plan Apo VC60XH objective (n.a. 1.4) (Nikon Instruments, Melville, NY) and an Andor DU885 EM-CCD camera (Andor Technology Ltd, Belfast, Northern Ireland). FCCP (carbonyl cyanide p-trifluoromethoxyphenylhydrazone, 10 μM), an uncoupler of oxidative phosphorylation, was added after 12 acquisitions to completely eliminate the electrical gradient established by the respiratory chain. Where indicated, cells were pre-treated with menadione. The values are expressed in a.f.u. (arbitrary fluorescence units).
To establish the mitochondrial Ψm levels, the ratio between the residual TMRM fluorescence (obtained after FCCP addition) and the initial TMRM fluorescence was calculated. The values obtained represent the amount of the potential-indicating probe incorporated in the different experimental conditions.
Mitochondrial morphology analysis
Mesothelioma cells were seeded onto 24-mm glass coverslips, allowed to grow to 50–60% confluence and then transfected with 1.5 μg of plasmid DNA mtGFP. After 36 h of expression, the cells were treated as described and then imaged with a Nikon Swept Field confocal microscope equipped with a CFI Plan Apo VC60XH objective (n.a. 1.4) (Nikon Instruments, Melville, NY) and an Andor DU885 EM-CCD camera (Andor Technology Ltd, Belfast, Northern Ireland). When indicated, the cells were treated with the reported compound. The coverslip were placed in an incubation chamber with controlled temperature, CO2 and humidity, and then 51-plane z-stacks were acquired with a voxel dimension of 133×133×200 nm (X Y × Z). The mitochondrial network was described in number and volume using the 3D object counter available in software Fiji (http://fiji.sc/wiki/index.php/Fiji, last accessed June 20, 2011).
Detection of mitochondrial ROS
The cells were seeded onto 24-mm glass coverslips and allowed to grow to 60–70% confluence. The cells were then incubated with MitoSOX-Red indicator (Life Technologies Ltd) for 30 min and washed. When indicated, cells were treated with the pro-apoptotic compound menadione. After washing, the amount of mitochondrial superoxide was determined using a Tali™ image-based cytometer (Life Technologies). The data are presented as a histogram showing the % of the mean intensity of MitoSOX fluorescence.
Growth curve assay
Cell growth curves were generated by crystal violet staining. Cells were seeded with a low cell density (5.000 per well) in 12-well plates in triplicate and allowed to grow for 5 days. At 1, 2, 3, 4 and 5 days, the cells were washed with phosphate-buffered saline, fixed with 4% paraformaldehyde and stained with 0.1% crystal violet. Crystal violet was dissolved with 1 mol/l acetic acid, and the A595 was measured.
Statistical analysis
The results are expressed as the mean ± SEM, and the n refers to the number of independent experiments. The probability of significant differences between experimental groups was determined by Student’s t test.
ACKNOWLEDGMENTS
Italian Association for Cancer Research (AIRC), University of Ferrara, the Italian Ministry of Health and the Italian Ministry of Education, University and Research, the Italian association for Multiple Sclerosis (FISM). The experiments were conceived and designed by S.P., C.G., M.T. and P.P. The experiments were performed by S.P., C.G., S.Ma., S.Mi., P.M., I.B, F.M. The data were analyzed by S.P., C.G., G.C, M.T. P.P. The paper was written by S.P., M.T., P.P. This work was supported by: the Italian Association for Cancer Research (AIRC: project IG-16046 to MT, IG-14442 to PP and MFAG-13521 to CG) and local funds from the University of Ferrara to Paolo Pinton and Carlotta Giorgi; Telethon (GGP11139B), the Italian Ministry of Health, the Italian Ministry of Education, University and Research (COFIN n. 20129JLHSY_002, FIRB n. RBAP11FXBC_002, and Futuro in Ricerca n. RBFR10EGVP_001) to Paolo Pinton. Simone Patergnani was supported by a research fellowship FISM—Fondazione Italiana Sclerosi Multipla—cod. 2014/B/3. This study was supported in parts from grants of the University of Ferrara FAR projects (M.T., F.M.), Fondazione Buzzi UNICEM, Casale Monferrato (M.T.), MIUR PRIN 2008 (F.M.), ASLEM, San Marino (M.T.).
REFERENCES
1. Robinson BW, Musk AW, Lake RA. Malignant mesothelioma. Lancet. 2005; 366:397–408.
2. Yang H, Testa JR, Carbone M. Mesothelioma epidemiology, carcinogenesis, and pathogenesis. Curr Treat Options Oncol. 2008; 9:147–157.
3. Carbone M, Yang H. Molecular pathways: targeting mechanisms of asbestos and erionite carcinogenesis in mesothelioma. Clin Cancer Res. 2012; 18:598–604.
4. Fennell DA, Rudd RM. Defective core-apoptosis signalling in diffuse malignant pleural mesothelioma: opportunities for effective drug development. Lancet Oncol. 2004; 5:354–362.
5. de Assis LV, Locatelli J, Isoldi MC. The role of key genes and pathways involved in the tumorigenesis of Malignant Mesothelioma. Biochim Biophys Acta. 2014; 1845:232–247.
6. Bonora M, Bononi A, De Marchi E, Giorgi C, Lebiedzinska M, Marchi S, Patergnani S, Rimessi A, Suski JM, Wojtala A, Wieckowski MR, Kroemer G, Galluzzi L, Pinton P. Role of the c subunit of the FO ATP synthase in mitochondrial permeability transition. Cell Cycle. 2013; 12:674–683.
7. Akl H, Bultynck G. Altered Ca(2+) signaling in cancer cells: proto-oncogenes and tumor suppressors targeting IP3 receptors. Biochim Biophys Acta. 2013; 1835:180–193.
8. Monteith GR, Davis FM, Roberts-Thomson SJ. Calcium channels and pumps in cancer: changes and consequences. J Biol Chem. 2012; 287:31666–31673.
9. Marchi S, Rimessi A, Giorgi C, Baldini C, Ferroni L, Rizzuto R, Pinton P. Akt kinase reducing endoplasmic reticulum Ca2+ release protects cells from Ca2+-dependent apoptotic stimuli. Biochemical and biophysical research communications. 2008; 375:501–505.
10. Ivanova H, Vervliet T, Missiaen L, Parys JB, De Smedt H, Bultynck G. Inositol 1,4,5-trisphosphate receptor-isoform diversity in cell death and survival. Biochim Biophys Acta. 2014; 1843:2164–2183.
11. Yoon MJ, Lee AR, Jeong SA, Kim YS, Kim JY, Kwon YJ, Choi KS. Release of Ca2+ from the endoplasmic reticulum and its subsequent influx into mitochondria trigger celastrol-induced paraptosis in cancer cells. Oncotarget. 2014; 5:6816–6831.
12. Schmidt S, Liu G, Yang W, Honisch S, Pantelakos S, Stournaras C, Honig A, Lang F. Enhanced Orai1 and STIM1 expression as well as store operated Ca2+ entry in therapy resistant ovary carcinoma cells. Oncotarget. 2014; 5:4799–4810.
13. Patergnani S, Suski JM, Agnoletto C, Bononi A, Bonora M, De Marchi E, Giorgi C, Marchi S, Missiroli S, Poletti F, Rimessi A, Duszynski J, Wieckowski MR, Pinton P. Calcium signaling around Mitochondria Associated Membranes (MAMs). Cell Commun Signal. 2011; 9:19.
14. Shin DM, Muallem S. What the mitochondria see. Mol Cell. 2010; 39:6–7.
15. Rizzuto R, Marchi S, Bonora M, Aguiari P, Bononi A, De Stefani D, Giorgi C, Leo S, Rimessi A, Siviero R, Zecchini E, Pinton P. Ca(2+) transfer from the ER to mitochondria: when, how and why. Biochim Biophys Acta. 2009; 1787:1342–1351.
16. Giorgi C, Baldassari F, Bononi A, Bonora M, De Marchi E, Marchi S, Missiroli S, Patergnani S, Rimessi A, Suski JM, Wieckowski MR, Pinton P. Mitochondrial Ca(2+) and apoptosis. Cell Calcium. 2012; 52:36–43.
17. Akl H, Vervloessem T, Kiviluoto S, Bittremieux M, Parys JB, De Smedt H, Bultynck G. A dual role for the anti-apoptotic Bcl-2 protein in cancer: mitochondria versus endoplasmic reticulum. Biochim Biophys Acta. 2014; 1843:2240–2252.
18. Giorgi C, Bonora M, Missiroli S, Poletti F, Ramirez FG, Morciano G, Morganti C, Pandolfi PP, Mammano F, Pinton P. Intravital imaging reveals p53-dependent cancer cell death induced by phototherapy via calcium signaling. Oncotarget. 2015; 6:1435–1445.
19. Azimi I, Roberts-Thomson SJ, Monteith GR. Calcium influx pathways in breast cancer: opportunities for pharmacological intervention. Br J Pharmacol. 2014; 171:945–960.
20. Dubois C, Vanden Abeele F, Lehen'kyi V, Gkika D, Guarmit B, Lepage G, Slomianny C, Borowiec AS, Bidaux G, Benahmed M, Shuba Y, Prevarskaya N. Remodeling of channel-forming ORAI proteins determines an oncogenic switch in prostate cancer. Cancer Cell. 2014; 26:19–32.
21. Raphael M, Lehen'kyi V, Vandenberghe M, Beck B, Khalimonchyk S, Vanden Abeele F, Farsetti L, Germain E, Bokhobza A, Mihalache A, Gosset P, Romanin C, Clezardin P, Skryma R, Prevarskaya N. TRPV6 calcium channel translocates to the plasma membrane via Orai1-mediated mechanism and controls cancer cell survival. Proc Natl Acad Sci U S A. 2014; 111:E3870–3879.
22. Weidinger C, Shaw PJ, Feske S. STIM1 and STIM2-mediated Ca(2+) influx regulates antitumour immunity by CD8(+) T cells. EMBO Mol Med. 2013; 5:1311–1321.
23. Al-Taweel N, Varghese E, Florea AM, Busselberg D. Cisplatin (CDDP) triggers cell death of MCF-7 cells following disruption of intracellular calcium ([Ca(2+)]i) homeostasis. J Toxicol Sci. 2014; 39:765–774.
24. Kamp DW, Liu G, Cheresh P, Kim SJ, Mueller A, Lam AP, Trejo H, Williams D, Tulasiram S, Baker M, Ridge K, Chandel NS, Beri R. Asbestos-induced alveolar epithelial cell apoptosis. The role of endoplasmic reticulum stress response. Am J Respir Cell Mol Biol. 2013; 49:892–901.
25. Papp T, Schipper H, Pemsel H, Unverricht M, Muller KM, Wiethege T, Schiffmann D, Rahman Q. Mutational analysis of the PTEN/MMAC1 tumour suppressor gene in primary human malignant mesotheliomas. Oncol Rep. 2001; 8:1375–1379.
26. Ni Z, Liu Y, Keshava N, Zhou G, Whong W, Ong T. Analysis of K-ras and p53 mutations in mesotheliomas from humans and rats exposed to asbestos. Mutat Res. 2000; 468:87–92.
27. Mossman BT, Shukla A, Heintz NH, Verschraegen CF, Thomas A, Hassan R. New insights into understanding the mechanisms, pathogenesis, and management of malignant mesotheliomas. Am J Pathol. 2013; 182:1065–1077.
28. Sekido Y. Molecular pathogenesis of malignant mesothelioma. Carcinogenesis. 2013; 34:1413–1419.
29. Rimessi A, Marchi S, Patergnani S, Pinton P. H-Ras-driven tumoral maintenance is sustained through caveolin-1-dependent alterations in calcium signaling. Oncogene. 2014; 33:2329–2340.
30. Marchi S, Patergnani S, Pinton P. The endoplasmic reticulum-mitochondria connection: one touch, multiple functions. Biochim Biophys Acta. 2014; 1837:461–469.
31. Aung CS, Ye W, Plowman G, Peters AA, Monteith GR, Roberts-Thomson SJ. Plasma membrane calcium ATPase 4 and the remodeling of calcium homeostasis in human colon cancer cells. Carcinogenesis. 2009; 30:1962–1969.
32. Bononi A, Bonora M, Marchi S, Missiroli S, Poletti F, Giorgi C, Pandolfi PP, Pinton P. Identification of PTEN at the ER and MAMs and its regulation of Ca(2+) signaling and apoptosis in a protein phosphatase-dependent manner. Cell Death Differ. 2013; 20:1631–1643.
33. Giorgi C, Ito K, Lin HK, Santangelo C, Wieckowski MR, Lebiedzinska M, Bononi A, Bonora M, Duszynski J, Bernardi R, Rizzuto R, Tacchetti C, Pinton P, Pandolfi PP. PML regulates apoptosis at endoplasmic reticulum by modulating calcium release. Science. 2010; 330:1247–1251.
34. Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007; 87:99–163.
35. Giorgi C, Romagnoli A, Pinton P, Rizzuto R. Ca2+ signaling, mitochondria and cell death. Curr Mol Med. 2008; 8:119–130.
36. Khan MT, Wagner L, 2nd, Yule DI, Bhanumathy C, Joseph SK. Akt kinase phosphorylation of inositol 1,4,5-trisphosphate receptors. J Biol Chem. 2006; 281:3731–3737.
37. Marchi S, Marinello M, Bononi A, Bonora M, Giorgi C, Rimessi A, Pinton P. Selective modulation of subtype III IP(3)R by Akt regulates ER Ca(2)(+) release and apoptosis. Cell Death Dis. 2012; 3:e304.
38. Amarjargal N, Mazurek B, Haupt H, Andreeva N, Fuchs J, Gross J. Effects of SERCA and PMCA inhibitors on the survival of rat cochlear hair cells during ischemia in vitro. Physiol Res. 2008; 57:631–638.
39. Marchi S, Pinton P. The mitochondrial calcium uniporter complex: molecular components, structure and physiopathological implications. J Physiol. 2014; 592:829–839.
40. Marchi S, Lupini L, Patergnani S, Rimessi A, Missiroli S, Bonora M, Bononi A, Corra F, Giorgi C, De Marchi E, Poletti F, Gafa R, Lanza G, Negrini M, Rizzuto R, Pinton P. Downregulation of the mitochondrial calcium uniporter by cancer-related miR-25. Curr Biol. 2013; 23:58–63.
41. Cioce M, Canino C, Goparaju C, Yang H, Carbone M, Pass HI. Autocrine CSF-1R signaling drives mesothelioma chemoresistance via AKT activation. Cell Death Dis. 2014; 5:e1167.
42. Varani K, Maniero S, Vincenzi F, Targa M, Stefanelli A, Maniscalco P, Martini F, Tognon M, Borea PA. A(3) receptors are overexpressed in pleura from patients with mesothelioma and reduce cell growth via Akt/nuclear factor-kappaB pathway. Am J Respir Crit Care Med. 2011; 183:522–530.
43. Braun F, de Carne Trecesson S, Bertin-Ciftci J, Juin P. Protect and serve: Bcl-2 proteins as guardians and rulers of cancer cell survival. Cell Cycle. 2013; 12:2937–2947.
44. Pinton P, Ferrari D, Rapizzi E, Di Virgilio F, Pozzan T, Rizzuto R. The Ca2+ concentration of the endoplasmic reticulum is a key determinant of ceramide-induced apoptosis: significance for the molecular mechanism of Bcl-2 action. EMBO J. 2001; 20:2690–2701.
45. Wu Y, Ma Y, Xu Z, Wang D, Zhao B, Pan H, Wang J, Xu D, Zhao X, Pan S, Liu L, Dai W, Jiang H. Sodium orthovanadate inhibits growth of human hepatocellular carcinoma cells in vitro and in an orthotopic model in vivo. Cancer Lett. 2014; 351:108–116.
46. Evangelou AM. Vanadium in cancer treatment. Crit Rev Oncol Hematol. 2002; 42:249–265.
47. Khan KH, Blanco-Codesido M, Molife LR. Cancer therapeutics: Targeting the apoptotic pathway. Crit Rev Oncol Hematol. 2014; 90:200–219.
48. Brevet M, Shimizu S, Bott MJ, Shukla N, Zhou Q, Olshen AB, Rusch V, Ladanyi M. Coactivation of receptor tyrosine kinases in malignant mesothelioma as a rationale for combination targeted therapy. J Thorac Oncol. 2011; 6:864–874.
49. Ou WB, Hubert C, Corson JM, Bueno R, Flynn DL, Sugarbaker DJ, Fletcher JA. Targeted inhibition of multiple receptor tyrosine kinases in mesothelioma. Neoplasia. 2011; 13:12–22.
50. Kruman II, Mattson MP. Pivotal role of mitochondrial calcium uptake in neural cell apoptosis and necrosis. J Neurochem. 1999; 72:529–540.
51. Zaid H, Abu-Hamad S, Israelson A, Nathan I, Shoshan-Barmatz V. The voltage-dependent anion channel-1 modulates apoptotic cell death. Cell Death Differ. 2005; 12:751–760.
52. Tsai LM, Yu D. MicroRNAs in common diseases and potential therapeutic applications. Clin Exp Pharmacol Physiol. 2010; 37:102–107.
53. Kirschner MB, Cheng YY, Badrian B, Kao SC, Creaney J, Edelman JJ, Armstrong NJ, Vallely MP, Musk AW, Robinson BW, McCaughan BC, Klebe S, Mutsaers SE, van Zandwijk N, Reid G. Increased circulating miR-625-3p: a potential biomarker for patients with malignant pleural mesothelioma. J Thorac Oncol. 2012; 7:1184–1191.
54. Balatti V, Maniero S, Ferracin M, Veronese A, Negrini M, Ferrocci G, Martini F, Tognon MG. MicroRNAs dysregulation in human malignant pleural mesothelioma. J Thorac Oncol. 2011; 6:844–851.
55. Cristaudo A, Foddis R, Vivaldi A, Buselli R, Gattini V, Guglielmi G, Cosentino F, Ottenga F, Ciancia E, Libener R, Filiberti R, Neri M, Betta P, Tognon M, Mutti L, Puntoni R. SV40 enhances the risk of malignant mesothelioma among people exposed to asbestos: a molecular epidemiologic case-control study. Cancer Res. 2005; 65:3049–3052.
56. Bonora M, Giorgi C, Bononi A, Marchi S, Patergnani S, Rimessi A, Rizzuto R, Pinton P. Subcellular calcium measurements in mammalian cells using jellyfish photoprotein aequorin-based probes. Nat Protoc. 2013; 8:2105–2118.