
INTRODUCTION
The SRC family of non-receptor protein tyrosine kinases (SFK) consists of 9 members, and can be divided into two groups according to the expression pattern of the individual kinases in the adult. Accordingly, SRC, YES and FYN are ubiquitously expressed, while expression of LCK, FGR, BLK, LYN, YRK and HCK is limited to certain types of cells and tissues. HCK expression is confined to cells of the myeloid and B-lymphocyte lineages and the protein exists as two isoforms. In humans, they comprise the p61HCK and p59HCK isoforms consisting of 525 and 504 amino acids, respectively, while in mice HCK exists as p59HCK (503 amino acids) and p56HCK (482 amino acids). In either species the two isoforms are generated by alternative initiation of protein translation from two in-frame initiation codons found within a single mRNA [1, 2] (Figure 1A). The two isoforms differ from each other by acylation of their N-terminal domain, and as a consequence, exhibit distinct subcellular localizations [1]. The larger isoform is mainly associated with lysosomes due to myristoylation of a glycine (Gly) residue at position +2 relative to the amino terminal methionine (Met). Meanwhile, p59HCK is mostly confined to the plasma membrane as a result of myristoylation (at Gly +2) and of palmitoylation of a cysteine or serine (at Cys/Ser +6) which form part of a Met-Gly-Cys-X-X-Cys/Ser-X consensus sequence [1, 3]. Importantly, SFK members exhibit complementary and often functionally redundant roles in several signal transduction pathways, such as the cooperation of FYN and LCK in T-cell receptor mediated activation [4, 5], and the regulation of macrophage phagocytosis by HCK and FGR [6]. This functional overlap between the SFK members has been shown to play an important role in maintaining relatively normal immune responses in situations of impairment or mutation of one of these kinases [6].
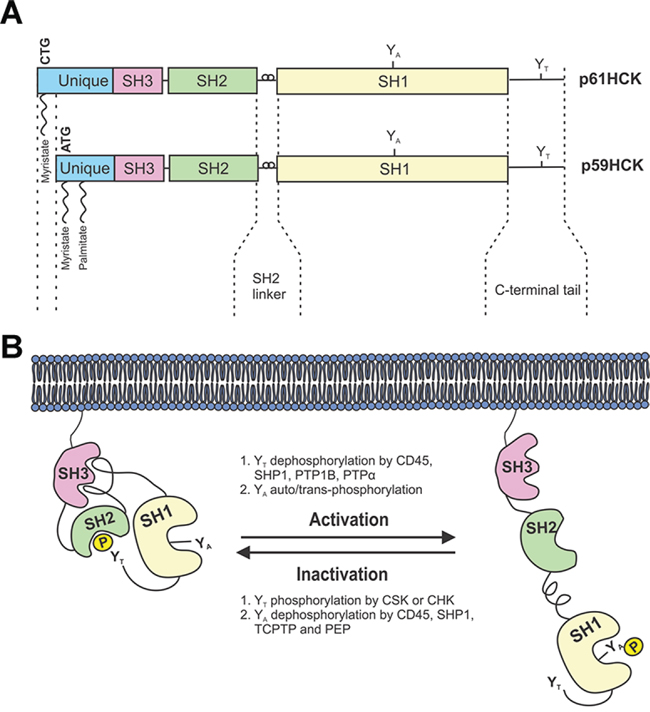
Figure 1: Schematic representation of HCK structure and regulation. A. The HCK protein exists as two isoforms generated from the alternate use of two in-frame initiation codons of translation on a single mRNA. Both protein isoforms are composed of an acylated unique N-terminal domain followed by SRC Homology (SH) domains SH3, SH2 and SH1. The SH1 domain contains the catalytic domain with conserved tyrosine residues (YA), while the very C-terminal tail contains a regulatory tyrosine (YT). B. HCK is maintained in an inactive conformation by the binding of the SH2 linker to the SH3 domain, and by the binding of the phosphorylated tyrosine YT to the SH2 domain. Activation of HCK occurs following dephosphorylation of YT phosphatases, as well as auto/trans-phosphorylation of the YA residue. Adapted from Guiet et. al, 2008.
HCK structure and regulation
HCK shares with all the other SFKs a common architecture of five distinct domains [7, 8] (Figure 1A). They comprise a SFK-family member-specific N-terminal domain required for the aforementioned lipid modifications, followed by three highly conserved SRC Homology domains (SH3, SH2 and SH1) and a C-terminal tail containing a regulatory tyrosine residue (YT) at position 521 in human p61HCK, or 499 in mouse p59HCK, respectively [9]. The regulatory SH3 and SH2 domains bind to a proline-rich Pro-X-X-Pro motif and a phospho-tyrosine pTyr-Glu-Glu-Ile motif, respectively [10]. The catalytic SH1 domain contains two lobes that form the active site of the kinase, which is stabilized upon phosphorylation of a conserved tyrosine residues (YA) at position 410 in human p61HCK (388 in mouse p59HCK) near the ATP binding site [9]. The small N-terminal lobe of the SH1 domain is composed of five β-sheets and a single α-helix responsible for ATP binding, while the second larger lobe is predominantly α-helical and binds to protein substrates [11].
Under steady-state conditions, HCK is maintained in an inactive conformation by two complementing intra-molecular associations (Figure 1B). The first involves binding of the SH2 domain to the phosphorylated YT at the C-terminal tail, while the second involves binding of the SH3 domain to the proline-rich type II helical motif in the linker between the SH2 and SH1 domains [10]. Two enzymatic modifications mediate this inhibitory conformation, namely dephosphorylation of pYA by CD45, a trans-membrane receptor-like tyrosine phosphatase that is expressed exclusively in hematopoietic cells [12], and phosphorylation of YT by either the C-terminal SRC kinase (CSK) or the C-terminal SRC kinase-homologous kinase (CHK) [13]. In contrast to ubiquitously expressed CSK, CHK expression is limited to neurons and hematopoietic cells and may also inhibit SFKs in a non-catalytic manner by forming stable complexes with SFKs [14].
HCK activation is triggered by a variety of stimuli, including binding of bacterial lipopolysaccharide (LPS) to the CD14/Toll-like receptor (TLR)4 protein complex, cytokines such as Interleukin (IL)-2, IL6 and related ligands, and the Granulocyte Macrophage Colony Stimulating Factor (GM-CSF) receptor [15–17] (Table 1). In response to stimulation, the YT residue is dephosphorylated by CD45 or SHP1, and HCK undergoes a conformational change involving the disruption of the SH3-linker interaction which enables “unfolding” of the protein and phosphorylation of the YA residue [10]. However, HCK activation may also occur through non-catalytic mechanisms that disrupt the intra-molecular inhibitory interactions. In particular, the HIV-1 NEF protein can bind directly to the SH3 domain of HCK thereby disrupting the SH3-linker interaction and promoting a significant increase in catalytic activity [17, 18].
Table 1: HCK activators and substrates
Membrane associated activators |
Effects |
References |
Beta 2 integrin |
Cell adhesion |
[101] |
CCR3 |
Chemokine signaling and immune cell migration to inflammatory sites in allergic disease |
[143] |
CD66 |
Adhesion and activation of granulocytes |
|
M-CSFR and G-CSFR |
Cell proliferation, differentiation, and survival |
|
FcγRI and FcγRIIa |
Phagocytosis and antibody cell-mediated cytotoxicity |
|
IL2R |
Cytokine production and secretion |
[149] |
GP130 |
Proliferation, cytokine production and secretion |
|
uPA-R |
Cell migration, adhesion and wound-healing |
|
NEF |
Enhancement of viral infectivity |
[17] |
TLR-4 |
Immune surveillance |
[16] |
Substrates |
||
Bcr/Abl and Tel/Abl |
Myeloid cell transformation and proliferation |
|
CBL |
Cell adhesion and transformation |
|
C3G |
Apoptosis |
[67] |
ELMO1 |
Phagocytosis and cell motility |
|
GAB1 and GAB2 |
Cell proliferation and survival |
[68] |
PAG |
Proliferation and transformation |
[157] |
Paxillin |
Cell migration/podosome formation |
[158] |
p73 |
Cell cycle regulation and apoptosis |
[159] |
RA70 |
Cell differentiation |
[160] |
STAT5 |
Cell proliferation, survival and transformation |
|
VAV1 |
Immune cell activation, generation of reactive oxygen species and cytokine production |
|
WASP |
Cell migration |
Upon activation, HCK engages a range of downstream signaling pathways that mediate various aspects of the immune response (Table 1). Critical roles for HCK in these processes have been demonstrated in mice carrying knock-out alleles (HCKKO) or encoding, as a knock-in mutation, a constitutively active protein with a tyrosine-to-phenylalanine (Y499F) substitution mutation (HCKCA). While HCKKO mice display a relatively mild phenotype, macrophages from these animals exhibit reduced phagocytic ability. Consistent with functional redundancy among SFKs, compound HCKKO;FGRKO knock-out mice show impaired myeloid cell degranulation and migration culminating in increased susceptibility to infection [6, 19]. In contrast, HCKCA animals are highly responsive to LPS, and display an exaggerated innate immune response with the corresponding macrophages and neutrophils exhibiting enhanced effector functions in vitro, including nitric oxide and TNFα production, chemotaxis, and degranulation [20]. As a consequence, HCKCA mice spontaneously acquire a lung pathology characterized by extensive eosinophilic and mononuclear cell infiltration within the lung parenchyma, alveolar airspaces, and around blood vessels, associated with epithelial mucus metaplasia, reminiscent of chronic obstructive pulmonary disorder (COPD) in humans [20].
SFK deregulation in cancer
Deregulation of SFKs has been implicated in disease, including cancer, inflammatory and autoimmune disease [21–23]. The initial recognition of the oncogenic potential of SFKs builds on observations by the pathologist Peyton Rous linking the formation of sarcoma in chickens to (retroviral) infection of these birds [24]. Subsequently, it was shown that the genome of the Rous-sarcoma virus (RSV) encoded for a constitutive active tyrosine kinase referred to as Rous Sarcoma kinase (SRC), which transformed fibroblasts and conferred uncontrolled proliferation [25]. However, it was the seminal work by Harold Varmus and Michael Bishop in 1979, which described the discovery of a mammalian orthologue of the viral (v)-SRC protein, referred to as cellular (c)SRC. Importantly, it was shown that RSV had integrated a genomic sequence encoding for a truncated version of c-SRC that lacked the regulatory carboxy-terminal tail including the regulatory YT at amino acid position 527. Hence, unlike its cellular counterpart c-SRC, by now referred to as a “proto-oncogene”, v-SRC remained constitutively active [26]. These insights resulted in a paradigm shift for cancer not only arising from the acquisition of foreign DNA (i.e. viruses), but more commonly by acquiring mutations in endogenous genes that transformed highly regulated proto-oncogenes to bona fide oncogenes encoding proteins permanently locked in their active conformation. Elevated c-SRC activity and mutations that functionally replicate the v-SRC truncation have now been detected in a wide variety of malignancies. While the oncogenic potential of c-SRC is widely accepted, we discuss here emerging novel mechanisms by which deregulated HCK activity contributes to tumor initiation and progression, including gene amplification and the interaction with regulatory phosphatases and upstream receptor tyrosine kinases.
SFK overexpression
Increased SFK activity and/or gene expression is frequently detected in tumor biopsies [27]. HCK gene amplification is observed in poorly differentiated human gastric cancer [28] and several colorectal cancer cell lines [29]. Increased SRC expression is observed in intestinal adenomas of the ApcMin mouse model of familial adenomatous polyposis, suggesting that the WNT/β-catenin pathway may contribute to increased SRC signaling [30]. Epigenetic changes have also been proposed to induce overexpression of SFKs [21]. Furthermore, microRNA-23b and miRNA-145, which are downregulated in many prostate and colorectal cancers, repress SRC and YES activity [31, 32]. These observations collectively suggest that SFK overexpression in tumor cells can occur as a result of exaggerated activation of upstream signaling pathways as well as aberrant epigenetic modifications to exaggerate SFK activation.
SFK activation by suppression of CSK and Cbp/PAG
Phosphorylation of YT is crucial for SFK inactivation and the loss of this residue confers constitutive activation and transforming capabilities of oncogenic v-SRC and v-YES proteins [33]. Disruption of the interaction between YT and the SH2 domain can result from impaired CSK activity or from pY-residues of growth factor receptors, which compete for binding to the SH2 domain in SFKs, thereby yielding an open and catalytically active conformation [34]. Although CSK was initially thought to act as a tumor suppressor, its contribution to cancer remains less clear. In hepatocellular carcinoma, reduced CSK levels inversely correlate with SFK activity [35]. However, increased CSK expression along with SFK activity has been observed in primary carcinoma [36] and human colorectal cancer cell lines [37], indicating that CSK-dependent regulation of SFK signaling may be specific to some cancers. CSK may also contribute to SFK deregulation by altered trafficking, since CSK-mediated inhibition of SFK relies on membrane-associated CSK binding proteins [38]. Interestingly, this recruitment step is impaired in colorectal cancer, resulting in retention of CSK in the cytoplasm [39]. In accordance with these observations, expression of Cbp/PAG, which traffics CSK to the membrane, is frequently reduced in human colorectal tumors, while introduction of Cbp/PAG into metastatic colorectal cancer cells restores membrane translocation of CSK and reduces invasion of these cells into extracellular matrix [39]. Collectively, these results suggest an important role for Cbp/PAG in ensuring proper CSK localization and associated physiological regulation of SFK activity.
SFK activation by tyrosine phosphatases
SFKs can also act as substrates for protein tyrosine phosphatases other than CD45 and SHP1, which otherwise primarily regulate the activity of SFKs. Increased activity of PTPα, PTPB1, TCPTP and other tyrosine phosphatases may also contribute to elevated SFK activity and sustained signaling [40]. Short hairpin RNA-mediated suppression of PTPα, for instance, reduced SRC activity by up to four-fold in human breast and colon cancer cell lines [41], while overexpression of PTP1B increased SRC activity and promoted anchorage-dependent growth of colon cancer cells [42]. In addition, inhibition of tyrosine phosphatases may also promote SFK activation by increasing the level of the phosphorylated YA residue in their SH1 domain. TCPTP protein levels are reduced in a subset of estrogen receptor, progesterone receptor and HER2 triple-negative human breast cancers, and TCPTP deficient breast cancer cell lines exhibit elevated SFK signaling [43]. Conversely, reintroduction of TCPTP into these cells reduced SFK activity, and impaired cell proliferation and anchorage-independent growth [43]. Likewise, treatment of pancreatic cancer cells with tyrosine phosphatase inhibitors increased pYA of SRC in adherent cells [44], while PTPL1 inactivation in colorectal cancer cells promoted anchorage-independent cell growth [45]. Together, these results suggest that inhibition of tyrosine phosphatases plays a key role in mediating excessive SFK activity in cancer cells.
SFK association with receptor tyrosine kinases
The association of SFKs with receptor tyrosine kinases (RTK) has been implicated in situations of persistent RTK signaling in cancer [46]. SFK activation occurs in response to ligand binding of platelet-derived growth factor receptor (PGDFR), epidermal growth factor receptor (EGFR), fibroblast growth factor receptor (FGFR) and others. In these situations, SFKs are involved in the transduction of signals required for receptor turnover, actin cytoskeletal rearrangement and cell survival [46]. There is evidence that the SH2 domain of SFKs can bind directly to pY599 in PDGFR [47] (Figure 2A). This interaction disrupts intramolecular interaction between the SH2 domain and the pYT residue in SFKs, and exposes their YA residue to become available for phosphorylation [34]. SFK activation can also occur by an indirect mechanism, since EGFR does not contain consensus pY residues to enable binding of the SH2 domain of SFKs (Figure 2A). In epithelial cancers expression of EGFR, ERBB2 and to a lesser extent other EGFR-family members, is increased by mutation and/or gene amplification and this is commonly associated with excessive SRC signaling [48, 49]. Indeed, excessive SFK activity can activate these RTKs in the absence of their ligands [39]. Meanwhile, excessive activation of c-MET, EPHA2 and other RTKs is required for maintaining maximal SRC activity in colorectal cancer cells [50], suggesting that SFK deregulation may create a feedback loop whereby SFK activity increases with RTK activity to fuel tumor promotion.
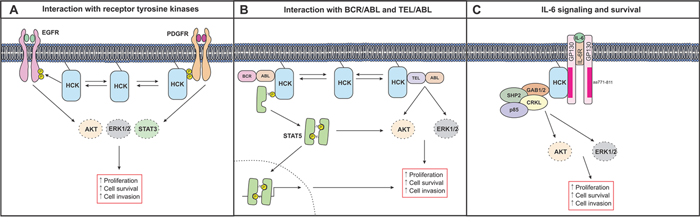
Figure 2: Simplified schematic depiction of HCK signaling in cancer cells. A. The interaction of HCK with receptor tyrosine kinases such as EGFR and PDGFR results in the activation of ERK, AKT and STAT3 signaling pathways to promote cell proliferation in cancer cells. B. Direct association of HCK with BCR/ABL results in the persistent activation of STAT5 and its retention in the cytoplasm. Within the cytoplasm, STAT5 promotes cell growth and survival through the activation of AKT. HCK can also interact with TEL/ABL to promote proliferation, survival and invasion through the activation of AKT and ERK pathways. C. HCK can bind directly to an acidic domain (comprising of amino acids 711–811) of GP130 and mediate ERK and AKT signaling to induce cell survival and proliferation in response to IL6 and related cytokines.
HCK signaling in leukemia
HCK expression is elevated in various types of leukemia (Table 2), and contributes to leukemogenesis through its association with the oncogenic fusion proteins BCR/ABL and TEL/ABL to enhance cell survival. However, decreased HCK activation has also been reported in a small number of acute myeloid leukemia and acute promyelocytic leukemia patients.
Table 2: HCK expression in various forms of leukemia
Type |
HCK expression |
HCK associated cellular effects |
References |
Chronic myeloid leukemia |
Elevated |
Cellular transformation |
|
Acute myeloid leukemia inv(16)(p13q22) and Philadelphia chromosome negative cases |
Reduced |
Downregulated apoptosis |
|
Acute promyelocytic leukemia |
Reduced |
Downregulated apoptosis, increased survival |
[65] |
Multiple myeloma |
Elevated |
Excessive IL6 signaling and cell proliferation and survival |
|
Acute lymphoblastic leukemia |
Elevated |
Cell proliferation and survival |
Interaction with oncogenic fusion proteins
Chronic myeloid leukemia (CML) accounts for 20% of leukemias and is characterized by excessive production of granulocytes that accumulate in the bone-marrow and interfere with normal hematopoiesis. Over time, leukemic cells build up and spill into the bloodstream, eventually accumulating in organs and impairing the normal functions of immune cells. HCK has been implicated in CML through its direct association with, and activation by BCL/ABL [51] (Figure 2B). The BCR/ABL oncogenic fusion protein is derived from translocation of the c-ABL gene on chromosome 9 to the BCR locus on chromosome 22 and is present in a large majority of CML cases, as well as in some acute lymphocytic leukemias [52, 53]. In response to its association with BCR/ABL, HCK mediates persistent activation of STAT5A and the excessive accumulation of this latent transcription factor in the cytoplasm [51], where STAT5A engages PI3K and the adaptor protein GAB2 to activate AKT and to facilitate cell growth and survival [51]. Interestingly, despite the functional redundancy among SFK members, HCK appears to be the most important kinase in BCR/ABL-induced signaling [51, 54]. Overexpression studies demonstrate a physical interaction between HCK and STAT5A that culminates in STAT5A phosphorylation. While similar experiments showed that LYN was also capable of inducing STAT5A phosphorylation, there was no evidence of physical interaction between LYN and STAT5A [54]. Furthermore, co-expression of kinase-defective HCK mutants with BCR/ABL strongly impaired transformation of cytokine-independent growth of myeloid leukemia cells [55], while treatment with the broad-spectrum SFK inhibitor PP2 abrogated BCR/ABL-dependent activation of STAT5A [54]. These studies identify a dominant role for HCK in mediating BCR/ABL-induced activation of STAT5.
Expression of the TEL/ABL oncogenic fusion protein is observed in T- and pre-B cell acute lymphoblastic leukemias and some CML subsets [56–58], and promotes constitutive activation of STAT5, ERK, AKT and other pathways to facilitate the growth and survival of cancer cells [59–61]. HCK activation is increased in TEL/ABL-expressing Ba/F3 cells. Conversely treatment with Imatinib, which also suppresses TEL/ABL activity, inhibits HCK activation as well as STAT5, ERK and AKT signaling [62] (Figure 2B). Likewise, treatment of these cells with the SFK inhibitors PP1, PP2 or SU6656 abrogates TEL/ABL activity and induces growth arrest [62]. Indeed, HCK appears to be a critical regulator of TEL/ABL-dependent growth, since expression of kinase-dead HCK mutants inhibits the growth of TEL/ABL transformed cells and the phosphorylation of ERK1/2 and AKT [62].
Although HCK expression is frequently increased in acute and chronic myeloid leukemia, HCK activity is reduced in acute myeloid leukemia patients with the specific chromosomal rearrangement inv(16)(p13q22) [63]. Likewise, in acute lymphocytic leukemia patients, which lack the BCR/ABL fusion protein, HCK expression is reduced due to aberrant HCK gene methylation [64], while in acute promyelocytic leukemia, HCK transcription is repressed by the fusion protein promyelocytic leukemia-retinoic acid receptor α (PML-RARα). Since the PML-RARα protein promotes survival of myeloid precursor cells, it has been proposed that HCK inhibition may facilitate tumorigenesis by interfering with apoptosis [65]. In line with this, knock-down of HCK by siRNA substantially reduced expression of the anti-apoptotic protein MCL-1, which coincided with increased expression of pro-apoptotic BAX and enhanced death of cancer cells [66]. Meanwhile, the interaction of HCK with the guanine nucleotide exchange factor C3G was shown to indirectly induce apoptosis via a Caspase-1/8/9-dependent mechanism, which depended on the catalytic activity of HCK [67]. However, since the latter observation arose from overexpression in epithelial cells, its physiological relevance remains unclear.
IL6 signaling and survival
Multiple myelomas are characterized by excessive production of plasma cells in the bone-marrow and require IL6 signaling to promote cell growth and survival through activation of the ERK and PI3K signaling cascade [68]. Ligation of IL6 to its receptor results in juxta-positioning of the two IL6R β-chains (also referred to as GP130) and activation of the associated JAK kinases, which in turn confers phosphorylation of conserved cytoplasmic tyrosine residues in GP130. They comprise the membrane proximal pY that provides a docking site for the tyrosine phosphatase SHP2 and the negative regulator SOCS3, and four membrane distal pY, which serve as binding sites for STAT3 and STAT1 [69]. In multiple myeloma cells, various investigators have identified FYN, HCK and LYN in co-immunoprecipitates with GP130, and stimulation of these cells with IL6 results in increased activation of these SFKs [70]. However, the interaction between HCK and GP130 may be functionally more important for mediating proliferative signals than binding of FYN or LYN to GP130 [70]. Surprisingly, HCK was shown to mediate ERK signaling independently of the aforementioned association between GP130 and STAT3 or SHP2, but rather through direct binding of HCK to an acidic domain in GP130 (Figure 2C). Furthermore, deletion of this region (amino acids 771–811) impaired IL6 dependent HCK activation and reduced cell proliferation [71]. In line with this, expression of a kinase-dead HCK mutant in mouse myeloma 7TD1 cells elicited a dominant-negative effect on cell proliferation, thereby providing additional evidence that HCK is required to transmit GP130-dependent growth signals [72]. Similarly, we reported a functional contribution of HCK to the GP130-mediated signals that retain pluripotency of murine embryonic stem cells [15], where HCK undergoes rapid and transient activation in response to cells stimulated with the GP130 ligand leukemia inhibitory factor, LIF [73]. As in multiple myeloma cells, we also found that HCK directly associates with GP130 in embryonic stem cells [15].
The mechanism by which the GP130/HCK-containing complex promotes IL6-induced activation of ERK and PI3K in multiple myeloma cells is dependent on the adaptor proteins GAB1 and GAB2, which are constitutively associated with HCK. These scaffolding proteins are involved in the recruitment of multimeric protein complexes containing SHP2, GRB2, CRKL and the p85 subunit of PI3K, which promote activation of ERK and other downstream signaling molecules [68]. Importantly, kinase-dead HCK mutants, or treatment with the SFK inhibitor PP2, reduced IL6-induced tyrosine phosphorylation of intracellular signaling proteins and significantly decreased myeloma cell survival and proliferation [68]. Together, these results implicate HCK, and possibly its association with the GAB adaptor proteins, as key players in GP130-mediated multiple myeloma pathogenesis.
HCK activation in tumor-associated immune cells
Excessive HCK expression is observed in various solid cancers, including those of the breast [16, 74]. The microenvironment of solid cancers comprises a heterogeneous population of stromal cells that include fibroblasts, endothelial cells, adipocytes, mesenchymal cells and immune cells. All of these cells “communicate” with cancer cells and each other to promote tumor development and progression [75] through the secretion of growth factors [76] and cytokines [77] that promote survival, angiogenesis [78], invasion [79] and metastasis [80, 81]. Stromal cells also play a key role in suppressing anti-tumor immune cell responses and conferring resistance to chemotherapy [82, 83].
Myeloid cells, including macrophages, myeloid-derived suppressor cells and neutrophils make up a major component of the stromal compartment and contribute to the tumor promoting mechanisms described above [84]. In particular, tumor-associated macrophages (TAMs) comprise distinct subsets of alternatively activated macrophage that orchestrate many of the aforementioned hallmarks and enabling characteristics of cancer. Likewise, tumor-associated neutrophils have been shown to facilitate and promote the suppression of immune cell mediated anti-tumor responses [85]. HCK plays a key role in neutrophil phagocytosis [86, 87], and has been shown to co-localize with β-arrestin on the G-protein-coupled IL8 receptor, where HCK traffics to azurophilic granule-rich regions to initiate the release of their content [88]. Similarly, HCK also regulates macrophage activity by promoting their proliferation [89], migration [90] and secretion of IL6, TNFα and other cytokines [16, 91]. Thus, in addition to its direct oncogenic role in (leukemic) cancer cells, excessive HCK activation may be stimulated in neighboring immune cells by incipient tumor cells through cytokine secretion thereby reinforcing a tumor-enabling and tumor-promoting microenvironment (Figure 3).
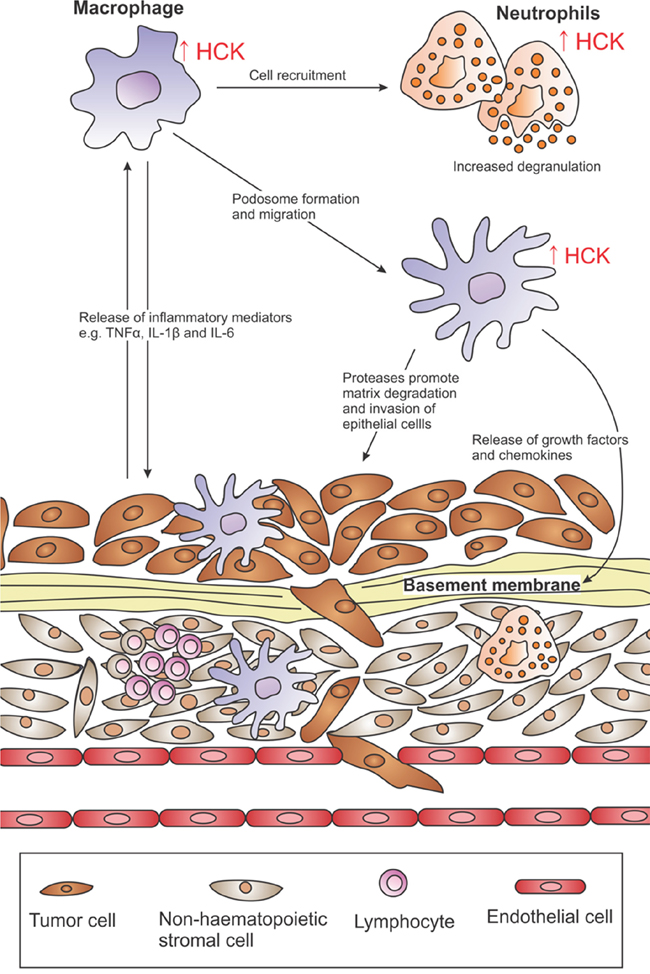
Figure 3: HCK activation in the tumor microenvironment. Excessive HCK activation (↑ HCK) is a common observation in many solid cancers and enhances the recruitment of immune cells into tumors and promotes their proliferative and migratory ability. HCK is also involved in inflammatory signaling and its expression augments the release of TNFα, IL1β and IL6 from macrophages. These cytokines can feedback onto tumor cells to perpetuate inflammation and sustain neoplastic transformation. HCK activation plays a key role in podosome formation in macrophages and promotes their phagocytotic, chemotactic and matrix degradation abilities. This may enhance immune and epithelial cell invasion by facilitating large-scale degradation of the extracellular matrix.
Inflammation
It has been recognized for more than a century that inflammation is an enabling characteristic of many cancers, and increased expression of pro-inflammatory cytokines, chemokines, prostaglandins and associated enzymes are frequently detected in human and murine tumors [92]. In colorectal cancer, for instance, TNFα [93], IL6 [94] and IL1β [95] from tumor infiltrating neutrophils has been shown to assist in neoplastic transformation and expansion of the mutated epithelium. Furthermore, chronic inflammation associated with hepatitis C virus infection results in excessive TGFβ signaling, which promotes cirrhosis and hepatocellular carcinoma [96], while long-term cigarette smoke exposure increases the risk of lung cancer and exacerbates lung inflammation [97]. Neoplastic cells secrete inflammatory molecules such as TNFα [98] and IL1β [99], likely as part of a wound-healing response conserved from their epithelial origins thereby forming a paracrine loop which perpetuates inflammation and sustain the activity of inflammatory immune cells.
HCK is involved in inflammatory signaling, and in human macrophages its increased expression augments TLR4-induced transcription of TNFα and IL6 via the AP-1 transcription factor complex [16, 100]. Consistent with this, genetic ablation of HCK in mice reduced cytokine expression in response to LPS stimulation, and moderately increased the susceptibility of HCKKO mice to infection as a consequence of impaired immunity [6]. Conversely, HCKCA mice display hypersensitivity to LPS-dependent endotoxemia associated with excessive secretion of TNFα and other inflammatory mediators [20]. HCK activation is also critical in integrin-dependent adhesion and immune cell recruitment during systemic inflammation [101, 102]. Thus, HCK provides a critical node that links the extracellular environment to an adequate innate immune cell response characterized by cytokine release and local immune cell recruitment.
Although a functional link between HCK, inflammation and solid cancers still remains somewhat tenuous, we have observed that a small proportion of aging HCKCA mice develop lung adenocarcinomas as a subsequence to their spontaneous lung inflammation with characteristics of COPD [20]. In humans, COPD is a major risk factor for lung cancer and is characterized by increased numbers and activity of polymorphonuclear leukocyte in sputum and bronchial lavage fluid of these patients [103]. Consistent with this, HCK protein levels are significantly elevated in neutrophils from COPD patients [104]. Furthermore, genomic amplification of the HCK locus has been observed in non-small cell lung cancer [105, 106], suggesting a potential role for HCK in promoting COPD-associated inflammation and predisposition to lung cancer.
Migration and invasion
Metastatic spread is the major cause for cancer mortality and occurs when cancer cells dissociate from the primary tumor and seed into distal organs. Highly invasive cancer cells form specialized matrix-degrading membrane protrusions known as invadopodia, which enable cancer cells to migrate through the tumor stroma and to intravasate into blood and lymphatic vessels, from where the cancer cells can spread to distant sites. TAMs form protrusions known as podosomes that are similar to invadopodia in molecular composition and proteolytic activity [107]. Thus, the ECM-remodeling ability of podosomes by TAMs could increase tumor invasiveness by assisting the formation of tunnels in the matrix, thereby enabling the escape of tumor cells independent of their own matrix degrading abilities.
HCK plays a crucial role in podosome formation by for instance phosphorylating the Wiskott-Aldrich syndrome protein (WASP), a hematopoietic cell-specific and actin nucleation promoting factor that regulates actin polymerization and promotes phagocytosis, chemotaxis, podosome formation and matrix degradation [108, 109]. In bone marrow-derived macrophages from HCKKO mice, and in human HCK knock-down macrophage cell lines, tyrosine phosphorylation of WASP in response to CX3CL1 or Fcγ stimulation is significantly impaired, and correlates with reduced phagocytosis, chemotaxis and matrix degradation by these cells [109]. Individual overexpression of the HCK isoforms in murine fibroblasts revealed that p59HCKCA triggered the formation of plasma membrane protrusions, while p61HCKCA resulted in podosomal formation [110]. Importantly, co-expression of both isoforms conferred a transformed phenotype in these cells, manifested by the loss of contact inhibition, shortened cell doubling time and serum independent growth.
Stimulation of macrophages with LPS and/or IFNγ, or with macrophage-colony stimulating factor not only activates HCK, but also promotes formation of podosomes and their arrangement into rosette clusters [111, 112]. Indeed, HCKKO macrophages exhibit reduced rosette forming ability and impaired matrix degradation, consistent with reduced macrophage recruitment and decreased efficiency in promoting invasion of tumor cells [90, 113]. These findings suggest that HCK is involved in podosomal formation and ECM-degradation by macrophages, which may enhance immune and epithelial cell invasion and therefore comprise a key step during tumor progression. Thus, while HCK has a direct role in promoting some blood-derived cancers, its aberrant activation in innate immune cells of the tumor microenvironment facilitates tumorigenesis and enables progression in experimental models and in human cancers.
HCK is associated with reduced chemo-sensitivity and acquired resistance
Reduced sensitivity and resistance to chemotherapy is a major cause of tumor recurrence, and may arise from genetic mutations, amplifications or epigenetic changes that increase the expression or activity of pro-survival factors, or regulate the uptake, metabolism and export of cytotoxic drugs [114]. In the case of breast cancer, tumor cells have been shown to recruit and stimulate macrophages to secrete the TLR4 ligands S100A8/9 thereby reducing chemotherapy-induced apoptosis [115]. Although HCK participates in the transduction of TLR4-dependent signals, this study did not investigate HCK activity in recruited macrophages. However, HCK activity either alone or within the context of other SFKs often reduces the efficacy of recombinant immunotoxins [66], where antibody fragments are tethered to an exotoxin that induces apoptosis upon antigene binding to the surface of tumor cells [116]. Accordingly, co-treatment of cells with the recombinant immunotoxin SS1P and the SFK inhibitor Bosutinib synergistically inhibited the growth of A431/H9 human epithelial carcinoma cells, and also enhanced regression of the corresponding tumor xenografts in mice [66].
HCK may be associated with reduced drug efficacy in the clinic
The mechanism of action of most small molecule kinase inhibitors depends on their binding into the hydrophobic pocket adjacent to the ATP binding site [117]. However, the accessibility of small molecule inhibitors is often contingent on the presence of a small amino acid (e.g. threonine) adjacent to the hydrophobic pocket. Tyrosine kinase mutants that possess larger amino acids (e.g. methionine) at this position can confer clinical resistance, as exemplified in acquired resistance to Imatinib, Gefitinib or Erlotinib [118–121]. Thus, expression of a HCK threonine-to-methionine mutant in BCR/ABL transformed myeloid cells confers resistance against the apoptotic and anti-proliferative effects of the broad-spectrum SFK inhibitor A-419259 [118]. Since HCK also interacts with the BCR/ABL fusion protein, this may have clinical relevance, whereby Imatinib can no longer bind to and inhibit the BCR/ABL fusion protein in CML patients [55, 122].
HCK, alongside other SFKs, can also interfere with drug efficacy by cooperating with receptor kinases that play a major role in oncogenesis. Increased EGFR expression is observed in many human malignancies of epithelial origin, including colorectal cancer, non-small cell lung cancer and brain cancer [123]. EGFR is partially activated upon ligand binding and phosphorylation of its C-terminal tail, but requires interaction with SFKs for full activation. This induces a conformational change in the SFK, resulting in its auto-phosphorylation and transient activation [124]. In turn, SFKs phosphorylate EGFR on an additional tyrosine (Y845), resulting in EGFR-mediated proliferation of breast cancer cells via STAT5B [125]. Increased SFK activity is also observed in the NCI-H226 human lung squamous carcinoma cell line with acquired resistance to the EGFR-inhibitor Cetuximab, and SFK activity has been shown to cooperate with EGFR to enhance survival via HER3 and PI3K/AKT signaling [123]. Consistent with this, treatment of Cetuximab-resistant cells with the SFK kinase and BCR/ABL inhibitor Dasatinib restored Cetuximab sensitivity, while simultaneous inhibition of SFKs and EGFR markedly suppressed HER3 and PI3K/AKT signaling [123]. Together, these findings suggest that SFKs and EGFR cooperate in acquired resistance to Cetuximab, providing a strong rationale for combinatorial strategies against SFKs and EGFR in the treatment of cancer.
Co-targeting SFKs in the clinic
Due to the prevalence of excessive SFK expression in many human malignancies and its correlation with a poorer prognosis, these kinases represent a readily actionable therapeutic target for the treatment of cancer. Several ATP-competitive kinase inhibitors with significant activity against SFKs have been developed (Table 3) [126–128]. However, preclinical tests have revealed that the efficacy of these drugs on primary tumors is variable and dependent on the level of SFK deregulation [29, 129, 130].
Table 3: Anti-tumor activities of SFK inhibitors
Drug |
Examples of targets |
Activities on tumor cells |
Clinical trials |
|||
Proliferation |
Apoptosis |
Invasion, Metastasis |
Drug re-sensitivity |
|||
Bosutinib |
SFKs, ABL, CAMK2G, TEC, STE20 |
Increased [168] |
Restored [171] |
|||
Dasatinib |
SFKs, BCR/ABL, c-Kit, Ephrins, RTKs |
|||||
Saracatinib |
SFKs, BCR/ABL, EGFR |
Increased [132] |
||||
PP2 |
SFKs, RIP2, CK1δ |
Reduced [194] |
Increased [195] |
Reduced [196] |
Restored [197] |
Not tested |
A-419259 |
SFKs |
Increased [118] |
Reduced [198] |
Not tested |
Not tested |
|
RK-20449 |
HCK |
Reduced [142] |
Increased [142] |
Not tested |
Not tested |
Not tested |
SU6656 |
SFKs, PGDF, BRSK2, AMPK, Aurora C, Aurora B, CaMKKβ |
Reduced [199] |
Increased [200] |
Reduced [201] |
Restored [66] |
Not tested |
Given the various roles of SFKs in cells of the tumor stroma, SFK inhibitors have exhibited a great impact in slowing tumor invasion and metastasis. For example, combining the SFK and ABL inhibitor Saracatinib with a MEK inhibitor prevented melanoma cell invasion [131] and the metastatic outgrowth of dormant tumor cells [132]. Likewise, the synergistic activity of Dasatinib with Oxaliplatin promoted shrinkage of metastatic nodules as a consequence of reduced proliferation and angiogenesis [133]. Furthermore, Dasatinib treatment can restore the aforementioned Cetuximab sensitivity of EGFR-positive breast cancers, and also overcome BRAF inhibitor resistance in melanoma cells [134]. It is therefore likely that SFK inhibition may also provide benefit as adjuvant therapy with current standard of care chemotherapies.
Future perspectives
To maximize the therapeutic benefit from incorporating HCK (or SFK) inhibition into existing cancer treatments, several challenges need to be addressed. One major limitation is the lack of appropriate biomarkers to monitor SFK deregulation in patients. This is crucial, since the therapeutic efficacy of SFK inhibitors will be influenced by the extent of SFK activity in tumors [29], as well as other molecular alterations which may impact on the response of cancer cells to SFK inhibitors [135]. To date, preclinical trials using SFK inhibitors have been performed in unselected patients with relatively low overall clinical benefits. However, favorable responses have been observed when patients were stratified, which emphasizes the importance of utilizing predictive biomarkers to identify those individuals who will benefit the most from SFK inhibitor therapy [136, 137].
Secondly, although SFK inhibition is effective in some advanced tumors, particularly in combination with treatments that target other tumor drivers such as HER2, other clinical trials have reported limited benefits [138–140]. For this reason, the use of SFK inhibitors in an early adjuvant setting may be a more effective in delaying the progression of emerging tumors rather than to treat advanced/metastatic disease, since a significant impact of many SFK inhibitors was associated with their capacity to slow down tumor invasion and metastasis [131, 133].
Lastly, new strategies for the design of more conformation specific SFK inhibitors are required, since drugs with high specificity and potency are more effective and exhibit fewer side effects and toxicity. In particular, proteomic tools that interrogate kinase active sites provide a powerful approach for studying protein regulation and for performing screens for highly selective inhibitors for components of complex proteomes [141]. One such new technique involves the use of probes that harbor complimentary functionalities, including distinct moieties to direct interaction to the active site, to facilitate covalent binding once docked at the kinase active site, and to provide chemo-selective tags for reporter conjugation. When applied to HCK, this screening strategies enabled rapid profiling of novel active sites in the kinase domain and the identification of a number of conformation-specific ATP competitive inhibitors [141]. An other approach exploited a library of pyrazolo[3, 4-d]pyrimidine derivatives to identify candidates, which bind to the ATP binding site of HCK [11]. These new ATP-competitive inhibitors have structures distinct from any class of existing HCK inhibitors and showed significant inhibitory and anti-proliferative effects at sub-micromolar concentrations [11]. Other emerging SFK inhibitors with high specificity against HCK include A-419259 [118], which blocks proliferation and induces apoptosis in CML cell lines, as well as RK-20449 [142], which promotes tumor regression in a mouse xenograft model.
CONCLUSIONS
An oncogenic role of HCK has been implicated in human cancers, including leukemia and solid tumors of the breast, colon and stomach. Given the high frequency of excessive HCK activation in these cancers and its correlation with a poor prognosis on one hand, and the relatively mild phenotype displayed by HCKKO mice on the other hand, HCK may be a promising target for cancer treatment. In particular, therapeutic-targeting strategies aimed at inhibiting HCK within the tumor stroma may be part of a paradigm-shifting approach to treating human cancers in the future. Given the role of stromal cells as key enablers and enhancers of many of the hallmarks of cancer, we predict that the inhibition of HCK in tumor-associated innate immune cells will reduce their ability to promote angiogenesis, inflammation, migration and invasion. This will be aided by the emergence of new strategies to identify novel HCK inhibitors with greater specificity and potency, which will not only provide additional insights into the mechanistic by which HCK promotes the progression of tumors, but facilitate translational outcomes for cancer patients in the clinic.
ACKNOWLEDGMENTS
The work in the laboratory of M.E is supported by Ludwig Cancer Research, the Victorian State Government Operational Infrastructure Support, the IRIISS scheme of the National Health and Medical Research Council Australia (NHMRC), and NHMRC grants #1069024, #1067244 and #1081373 awarded to M.E. A.R.P is supported by an Australian Postgraduate Award. R.O’D received project grant support from NHMRC (#1025239) and the Victorian Cancer Agency (ECSG13014). M.E is a NHMRC Principal Research Fellow.
We apologize that due to space limitations only selected original research articles could be cited.
REFERENCES
1. Lock P, Ralph S, Stanley E, Boulet I, Ramsay R, Dunn A. Two isoforms of murine hck, generated by utilization of alternative translational initiation codons, exhibit different patterns of subcellular localization. Molecular and Cell Biology. 1991; 9:4363–4370.
2. Ziegler SF, Marth JD, Lewis DB, Perlmutter R. Novel protein-tyrosine kinase gene (hck) preferentially expressed in cells of hematopoietic origin. Molecular and Cell Biology. 1987; 7:2276–2285.
3. Robbins S, Quintrell N, Bishop J. Myristoylation and differential palmitoylation of the HCK protein-tyrosine kinases govern their attachment to membranes and association with caveolae. Molecular and Cell Biology. 1995; 15:3507–3515.
4. Stein P, Lee H, Rich S, Soriano P. pp59fyn and mutant mice display differential signaling in thymocytes and peripheral T cells. Cell. 1992; 70:741–750.
5. Straus D, Weiss A. Genetic evidence for the involvement of lck tyrosine kinase in signal transduction through the T-cell antigen receptor. Cell. 1992; 70:585–593.
6. Lowell CA, Soriano P, Varmus HE. Functional overlap in the src gene family: inactivation of hck and fgr impairs natural immunity. Genes and Development. 1994; 8:387–398.
7. Boggon T, Erk M. Structure and regulation of Src family kinases. Oncogene. 2004; 48:7918–7927.
8. Ingley E. Src family kinases: Regulation of their activities, levels and identification of new pathways. BBA. 2007; 1784:56–65.
9. Sicheri F, Moarefi I, Kuriyan J. Crystal structure of the Src family tyrosine kinase Hck. Nature. 1997; 385:602–609.
10. Guiet R, Poincloux R, Castandet J, Marois L, Labrousse A, Cabec VL, Maridonneau-Parini I. Hematopoietic cell kinase (Hck) isoforms and phagocytic duties - From signaling and actin reorganization to migration and phagocytosis. European Journal of Cell Biology. 2008; 87:527–542.
11. Tintori C, Laurenzana I, Rocca FL, Falci F, Carraro F, Ruiz A, Este JA, Kissova M, Crespan E, Maga G, Biava M, Brullo C, Schenone S, Botta M. Identification of Hck inhibitors as hits for the development of antileukemia and anti-HIV agents. ChemMedChem. 2012; 8:1353–1360.
12. Roach T, Slater S, Koval M, White L, McFarland E, Okumura M, Thomas M, Brown E. CD45 regulates Src family member kinase activity associated with macrophage integrin-mediated adhesion. Curr Biol. 1997; 7:408–417.
13. Chong Y, Mulhern T, Cheng H. C-terminal Src kinase (CSK) and CSK-homologous (CHK) - endogenous negative regulators of Src-family protein kinases. Growth Factors. 2005; 23:233–244.
14. Chong Y, Mulhern T, Zhu H, Fukita D, Bjorge J, Tantiongco J, Sotirellis N, Lio D, Scholz G, Cheng H. A novel non-catalytic mechanism employed by the C-terminal Src-homologous kinase to inhibit Src-family kinase activity. J Biol Chem. 2004; 279:20752–20766.
15. Ernst M, Gearing D, Dunn A. Functional and biochemical association of Hck with the LIF/IL-6 receptor signal transducing subunit gp130 in embryonic stem cells. EMBO. 1994; 13:1574–1584.
16. Smolinska MJ, Page TH, Urbaniak AM, Mutch BE, Horwood NJ. Hck tyrosine kinase regulates TLR4-induced TNF and IL-6 production via AP-1. The Journal of Immunology. 2011; 187:6043–6051.
17. Pene-Dumitrescu T, Shu S, Wales T, Alvarado J, Shi H, Narute P, Moroco J, Yeh J, Engen J, Smithgall T. HIV-1 Nef interaction influences the ATP-binding site of the Src-family kinase, Hck. BMC Chem Biol. 2012; 12:1–12.
18. Moarefi I, LaFevre-Bernt M, Sicheri F, Huse M, Lee C, Kuriyan J, Miller W. Activation of the Src-family tyrosine kinase Hck by SH3 domain displacement. Nature. 1997; 650–3.
19. Lowell C, Benton G. Resistance to endotoxic shock and reduced neutrophil migration in mice deficient for the Src-family kinases Hck and Fgr. PNAS. 1998; 95:7580–7584.
20. Ernst M, Inglese M, Scholz GM, Harder KW, Clay Fj, Bozinovski S, Waring P, Darwiche R, Kay T, Sly P, Collins R, Turner D, Hibbs ML, Anderson GP, Dunn AR. Constitutive activation of the Src family kinase Hck results in spontaneous pulmonary inflammatory and an enhanced innate immune response. Journal of Experimental Medicine. 2002; 196:589–604.
21. Sirvent A, Benistant C, Roche S. Oncogenic signaling by tyrosine kinases of the SRC family in advanced colorectal cancer. Am J Cancer Res. 2012; 2:357–371.
22. Miklós Kovács TN, Zoltán Jakus, Cassian Sitaru, Edina Simon, Krisztina Futosi, Bálint Botz, Zsuzsanna Helyes, Clifford A. Lowell, Attila Mócsai. The Src family kinases Hck, Fgr, and Lyn are critical for the generation of the in vivo inflammatory environment without a direct role in leukocyte recruitment. Journal of Experimental Medicine. 2014; 211:1993–2011.
23. Lamagna C, Hu Y, DeFranco A, Lowell C. B cell-specific loss of Lyn kinase leads to autoimmunity. Journal of Immunology. 2014; 192:919–928.
24. Rous P. A transmissible avian neoplasm (sarcoma of the common fowl). J Exp Med. 1910; 12:696–705.
25. Stehelin D, Fujita D, Padgett T, Varmus H, Bishop J. Detection and enumeration of transformation-defective strains of avian sarcoma virus with molecular hybridization. Virology. 1977; 76:675–684.
26. Oppermann H, Levinson A, Varmus H, Levintow L, Bishop J. Uninfected vertebrate cells contain a protein that is closely related to the production of the avian sarcoma virus transforming gene (src). PNAS. 1979; 76:1804–1808.
27. Kiyose S, Nagura K, Tao H, Iagarashi H, Yamada H, Goto M, Maeda M, Kurabe N, Suzuki M, Tsuboi M, Kahyo T, Shinmura K, Hattori N, Sugimura H. Detection of kinase amplifications in gastric cancer archives using fluorescence in situ hybridization. Pathol Int. 2012; 62:477–484.
28. Kubo T, Kuroda Y, Shimizu H, Kokubu A, Okada N, Hosoda F, Arai Y, Nakamura Y, Taniguchi H, Yanagihara K, Imoto I, Inazawa J, Hirohashi S, Shibata T. Resequencing and copy number analysis of the human tyrosine kinase gene family in poorly differentiated gastric cancer. Carcinogenesis. 2009; 30:1857–1864.
29. Arcaroli J, Touban B, Tan A, Varella-Garcia M, Powell R, Eckhardt S, Elvin P, Gao D, Messersmith W. Gene array and fluorescence in situ hybridization biomarkers of activity of saracatinib (AZD0530), a Src inhibitor, in a preclinical model of colorectal cancer. Clin Cancer Res. 2010; 16:4165–4177.
30. Carothers A, Javid S, Moran A, Hunt D, Redston M, Bertagnolli M. Deficient C-cadherin adhesion in C57BL/6J-Min/+ mice is associated with increased tyrosine kinase activation and RhoA-dependent actomyosin contractility. Exp Cell Res. 2006; 312:387–400.
31. Majid S, Dar A, Saini S, Arora S, Shahryari V, Zaman M, Chang I, Yamamura S, Tanaka Y, Deng G, Dahiya R. MicroRNA-23b represses proto-oncogene Src kinase and functions as methylation-silenced tumor suppressor with diagnostic and prognostic significance in prostate cancer. Cancer Research. 2012; 72:6435–6446.
32. Gregersen L, Jacobsen A, Frankel L, Wen J, Krogh A, Lund A. MicroRNA-145 targets YES and STAT1 in colon cancer cells. PLoS One. 2010; 5:e8836.
33. Sen B, Johnson FM. Regulation of src family kinases in human cancers. Journal of Signal Transduction. 2011; 2011:1–14.
34. Mori S, Ronnstrand L, Yokote K, Engstrom A, Courtneidge S, Claesson-Welsh L, Heldin C. Identification of two juxtamembrane autophosphorylation sites in the PDGF beta-receptor; involvement in the interaction with Src family tyrosine kinases. EMBO. 1993; 12:2257–2264.
35. Masaki T, Okada M, Tokuda M, Shiratori Y, Hatase O, Shirai M, Nishioka M, Omata M. Reduced C-terminal Src kinase (Csk) activities in hepatocellular carcinoma. Hepatology. 1999; 29:379–384.
36. Benistant C, Bourgaux J, Chapuis H, Mottet N, Roche S, Bali J. The COOH-terminal Src kinase Csk is a tumor antigen in human carcinoma. Cancer Research. 2001; 61:1415–1420.
37. Watanabe N, Matsuda S, Kuramochi S, Tzuku J, Yamamoto T, Endo K. Expression of C-terminal src kinase in human colorectal cancer cell lines. Jpn J Clin Oncol. 1995; 25:5–9.
38. Kawabuchi M, Satomi Y, Takao T, Shimonishi Y, Nada S, Nagai K, Tarakhovsky A, Okada M. Transmembrane phosphoprotein Cbp regulates the activities of Src-family tyrosine kinases. Nature. 2000; 404:999–1003.
39. Sirvent A, Benistant C, Pannequin J, Veracini L, Simon V, Bourgaux J, Hollande F, Cruzalegui F, Roche S. Src family tyrosine kinases-driven colon cancer cell invasion is induced by Csk membrane delocalization. Oncogene. 2010; 29:1303–1315.
40. Jr. RR Src kinase regulation by phosphorylation and dephosphorylation. Biochemical and Biophysical Research Communications. 2005; 331:1–14.
41. Zheng X, Resnick R, Shalloway D. Apoptosis of estrogen-receptor negative breast cancer and colon cancer cell lines by PTP alpha and src RNAi. Int J Cancer. 2008; 122:1999–2007.
42. Zhu S, Bjorge J, Fujita D. PTP1B contributes to the oncogenic properties of colon cancer cells through Src activation. Cancer Research. 2007; 67:10129–10137.
43. Shields B, Wiede F, Gurzov E, Wee K, Hauser C, Zhu H, Molloy T, O'Toole S, Daly R, Sutherland R, Mitchell C, McLean C, Tiganis T. TCPTP regulates SFK and STAT3 signaling and is lost in triple-negative breast cancers. Mol Cell Biol. 2013; 33:557–570.
44. Connelly S, Isley B, Baker C, Gallick G, Summy J. Loss of tyrosine phosphatase-dependent inhibition promotes activation of tyrosine kinase c-Src in detached pancreatic cells. Mol Carcinog. 2010; 49:1007–1021.
45. Zhang Y, Tu Y, Zhao J, Chen K, Wu C. Reversion-induced LIM interaction with Src reveals a novel Src inactivation cycle. J Cell Biol. 2009; 184:785–792.
46. Bromann B, Korkaya H, Courtneidge S. The interplay between Src family kinases and receptor tyrosine kinases. Oncogene. 2004; 23:7959–7968.
47. Kypta R, Goldberg Y, Ulug E, Courtneidge S. Association between the PDGF receptor and members of the src family of tyrosine kinases. Cell. 1990; 62:481–492.
48. Biscardi J, Ishizawar R, Silva C, Parsons S. Tyrosine kinase signalling in breast cancer: epidermal growth factor receptor and c-Src interactions in breast cancer. Breast Cancer Research. 2000; 2:203–210.
49. Yarom N, Jonker D. The role of the epidermal growth factor receptor in the mechanism and treatment of colorectal cancer. Discov Med. 2011; 11:95–105.
50. Leroy C, Fialin C, Sirvent A, Simon V, Urbach S, Poncet J, Robert B, Jouin P, Roche S. Quantitative phosphoproteomics reveals a cluster of tyrosine kinases that mediates SRC invasive activity in advanced colon carcinoma cells. Cancer Research. 2009; 69:2279–2286.
51. Chatain N, Zieglar P, Fahrenkamp D, Jost E, Moriggl R, Leur HS-Vd, Muller-Newen G. Src family kinases mediate cytoplasmic retention of activated STAT5 in BCL-ABL positive cells. Oncogene. 2013; 32:3587–3597.
52. Clark S, McLaughlin J, Timmons M, Pendergast A, Ben-Neriah Y, Dow L, Crist W, Rovera G, Smith S, Witte O. Expression of a distinctive BCR-ABL oncogene in Ph1-positive acute lymphocytic leukemia (ALL). Science. 1988; 239:775–777.
53. Shtivelman E, Lifshitz B, Gale R, Roe B, Canaani E. Alternative splicing of RNAs transcribed from the human abl gene and from the bcr-abl fused gene. Cell. 1986; 47:277–284.
54. Klejman A, Schreiner SJ, Nieborowska-Skorska M, Slupianek A, Wilson M, Smithgall TE, Skorski T. The Src family kinase Hck couples BCR-ABL to STAT5 activation in myeloid leukemia cells. The EMBO Journal. 2002; 21:5766–5774.
55. Lionberger J, Wilson M, Smithgall T. Transformation of myeloid leukemia cells to cytokine independence by Bcr-Abl is supressed by kinase defective Hck. Journal of Biological Chemistry. 2000; 275:18581–18585.
56. Limbergen HV, Beverloo H, Drunen Ev, Janssens A, Hahlen J, Poppe B, Roy NV, Marynen P, Paepe AD, Slater R, Speleman F. Molecular cytogenetic and clinical findings in ETV6/ABL1-positive leukemia. Genes Chromosomes Cancer. 2001; 30:274–282.
57. Keung Y, Beaty M, Steward W, Jackle B, Pettnati M. Chronic myelocytic leukemia with eosinophilia, t(9;12)(q34;p13), and ETV6-ABL gene rearrangement: case report and review of the literature. Genet Cytogenet. 2002; 138:139–142.
58. Hannemann J, McManus D, Kabarowski J, Wiedemann L. Haemopoietic transformation by the TEL/ABL oncogene. Br J Haematol. 1998; 102:475–485.
59. Voss J, Posern G, Hannemann J, Wiedemann L, Turhan A, Poirel H, Bernard O, Adermann K, Kardinal C, Feller S. The leukaemic oncoproteins Bcr-Abl and Tel-Abl (ETV6/Abl) have altered substrate preferences and activate similar intracellular pathways. Oncogene. 2000; 19:1684–1690.
60. Spiejermann K, Pau M, Schwab R, Schmieja J, Franzrahe S, Hiddemann W. Constitutive activation of STAT3 and STAT5 is induced by leukemic fusion proteins with protein tyrosine kinase activity and is sufficient for transformation of haematopoietic precursor cells. Exp Hematol. 2002; 30:262–271.
61. Santos S, Lacronique V, Bouchaert I, Monni R, Bernard O, Gisselbrecht S, Gouilleux F. Constitutive active STAT5 variants induce growth and survival of hematopoietic cells through a PI3-kinase/AKT denepdent pathway. Oncogene. 2001; 20:2080–2090.
62. Pecquet C, Nyga R, Pernard-Lacronique V, Smithgall T, Murakami H, Regnier A, Lassoued K, Gouilleux F. The Src tyrosine kinase Hck is required for Tel-Abl- but not for Tel-Jak2-induced cell transformation. Oncogene. 2007; 26:1577–1585.
63. Sun X, Zhang W, Ramdas L, Stivers DN, Jones DM, Kantarjian HM, Estey EH, Vadhan-Raj S, Medeiros J, Bueso-Ramos CE. Comparative analysis of genes regulated in acute myelomonocytic leukemia with and without inv(16)(p13q22) using microarray techniques, real-time PCR, immunohistochemistry, and flow cytometry immunophenotyping. Nature Modern Pathology. 2007; 20:811–820.
64. Hoshino K, Quintas-Cardama A, Yang H, Sanchez-Gonzalez B, Garcia-Manero G. Aberrant DNA methylation of the Src kinase Hck, but not of Lyn, in Philadelphia chronosome negative acute lymphocytic leukemia. Nature Leukemia. 2007; 21:906–911.
65. Zou D, Yang X, Tan Y, Wang P, Zhu X, Yang W, Kia X, Zhang J, Wang K. Regulation of the hematopoietic cell kinase (HCK) by PML/RARa and PU.1 in acute promyelocytic leukemia. Leukemia Research. 2012; 36:219–223.
66. Liu X-F, Xiang L, Fitzgerald DJ, Pastan I. Antitumor effects of immunotoxins are enhanced by lowering HCK or treatment with Src kinase inhibitors. Molecular Cancer Therapeutics. 2014; 13:82–89.
67. Shivakrupa R, Radha V, Sudhakar C, Swarup G. Physical and functional interaction between Hck tyroskine kinase and guanine neucleotide exchange factor C3G results in apoptosis, which is independent of the C3G catalytic domain. Journal of Biological Chemistry. 2003; 278:52188–52194.
68. Podar K, Mostoslavsky G, Sattler M, Tai Y-T, Yatashi T, Catley LP, Hideshima T, Mulligan RC, Chauhan D, Anderson KC. Critical role for hematopoietic cell kinase (Hck)-mediated phosphorylation of Gab1 and Gab2 docking proteins in Interleukin 6-induced proliferation and survival of multiple myeloma cells. Journal of Biological Chemistry. 2004; 279:21658–21665.
69. Taga T, Hibi M, Hirata Y, Yamasaki K, Yasukawa K, Matsuda T, Hirano T, Kishimoto T. Interleukin-6 triggers the association of its receptor with a possible signal transducer, gp130. Cell. 1989; 3:573–581.
70. Hallek M, Neumann C, Schaffer S, Danhauser-Riedl M, Bubnoff Nv, Vos Gd, Druker B, Yasukawa K, Griffin J, Emmerich B. Signal transduction of interleukin-6 involves tyrosine phosphorylation of multiple cytosolic proteins and activation of Src-family kinases Fyn, Hck and Lyn in multiple myeloma cells. Exp Hematol. 1997; 25:1367–1377.
71. Schaeffer M, Schneiderbauer M, Weidler S, Tavares R, Warmuth M, Vos GD, Hallek M. Signaling through a novel domain of gp130 mediates cell proliferation and activation of Hck and Erk kinases. Molecular and Cell Biology. 2001; 21:8068–8081.
72. Hausherr A, Tavares R, Schaffer M, Obermeier A, Miksch C, Mitina O, Ellwart J, Hallek M, Krause G. Inhibition of IL-6 dependent growth of myeloma cells by an acidic peptide repressing the gp130-mediated activation of Src family kinases. Oncogene. 2007; 26:4987–98.
73. Ernst M, Oates A, Dunn A. gp130-mediated signal transduction in embryonic stem cells involves activation of Jak and Ras/mitogen-activated protein kinase pathways. J Biol Chem. 1996; 271:30136–30143.
74. Achim Rody UH, Laos Pusztai, Cornelia Liedtke, Regine Gaetje, Eugen Ruckhaeberle, Christine Solbach, Lars Hanker, Andre Ahr, Dirk Metzler, Knut Engels, Thomas Karn, Manfred Kaufmann. T-cell metagene predicts a favorable prognosis in estrogen receptor-negative and HER2-positive breast cancers. Breast Cancer Research. 2009; 11:1–15.
75. Hanahan D, Weinberg RA. Hallmarks of Cancer: The next generation. Cell. 2011; 144:647–674.
76. Kahlert C, Pecqueux M, Halama N, Dienemann H, Muley T, Pfannschmidt J, Lasitschka F, Klupp F, Schmidt T, Rahbari N, Reissfelder C, Kunz C, Benner A, Falk C, Weitz J, Koch M. Tumour-site-dependent expression profile of angiogenic factors in tumour-associated stroma of primary colorectal cancer and metastases. British Journal of Cancer. 2013; 110:441–449.
77. Bettuma IJ, Vasiliauskaitea K, Nygaarda V, Clancya T, Pettersena SJ, Tenstada E, Mælandsmoa GM, Prasmickaitea L.Metastasis-associated protein S100A4 induces a network of inflammatory cytokines that activate stromal cells to acquire pro-tumorigenic properties. Cancer Letters. 2014; 344:28–39.
78. Barbera-Guillem E, Nyhus JK, Wolford CC, Friece CR, Sampsel JW. Vascular endothelial growth factor secretion by tumor-infiltrating macrophages essentially supports tumor angiogenesis, and IgG immune complexes potentiate the process. Cancer Research. 2002; 62:7042–7049.
79. Kitamura T, Kometani K, Hashida H, Matsunaga A, Miyoshi H, Hosogi H, Aoki M, Oshima M, Hattori M, Takabayashi A, Minato N, Taketo M. SMAD-4 deficient intestinal tumors recruit CCR1+ myeloid cells that promote invasion. Nature Genetics. 2007; 39:467–475.
80. Vasiljeva O, Papazoglou A, Krüger A, Brodoefel H, Korovin M, Deussing J, Augustin N, Nielsen B, Almholt K, Bogyo M, Peters C, Reinheckel T. Tumor cell-derived and macrophage-derived cathepsin B promotes progression and lung metastasis of mammary cancer. Cancer Research. 2006; 66:5242–50.
81. DeNardo D, Barreto J, Andreu P, Vasquez L, Tawfik D, Kolhatkar N, Coussens L. CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell. 2009; 16:91–102.
82. Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE, Belaygorod L, Carpenter D, Collins L, Piwnica-Worms D, Hewitt S, Udupi GM, Gallagher WM, Wegner C, West BL, Wang-Gillam A, et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression and improves chemotherapeutic responses. Cancer Research. 2013; 73:1128–1141.
83. Mitchem J, Brennan D, Knolhoff B, Belt B, Zhu Y, Sanford D, Belaygorod L, Carpenter D, Collins L, Piwnica-Worms D, Hewitt S, Udupi G, Gallagher W, Wegner C, West B, Wang-Gillam A, et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Research. 2013; 73:1128–1141.
84. Lanskron G, Fuente MDl, Thuwajit P, Thuwajit C, Hermoso M. Chronic inflammation and cytokines in the tumour microenvironment. J Immunol Res. 2014; 2014:1–19.
85. Mishalian I, Bayuh R, Eruslanov E, Michaeli J, Levy L, Zolotarov L, Singhal S, Albelda S, Granot Z, Fridlender Z. Neutrophils recruit regulatory T-cells into tumors via secretion of CCL17—a new mechanism of impaired antitumor immunity. Int J Cancer. 2014; 135:1178–1186.
86. Mohn H, Cabec VL, Fischer S, Maridonneau-Parini I. The src-family protein-tyrosine kinase p59hck is located on the secretory granules in human neutrophils and translocates the phagosome during activation. Biochem J. 1995; 309:657–665.
87. Welch H, Mauran C, Maridonneau-Parini I. Nonreceptor protein-tyrosine kinases in neutrophil activation. Methods. 1996; 9:607–618.
88. Barlic J, Andrews J, Kelvin A, Bosinger S, DeVries M, Xu L, Dobransky T, Felfman R, Ferguson S, Kelvin D. Regulation of tyrosine kinase activation and granule release through beta-arrestin by CXCRI. Nature Immunology. 2000; 1:227–233.
89. Suh H, Kim M, Lee S. Inhibition of granulocyte-macrophage colony-stimulating factor signaling and microglial proliferation by anti-CD45RO: role of Hck tyrosine kinase and phosphatidylinositol 3-kinase/Akt. J Immunol. 2005; 174:2712–2719.
90. Cougoule C, Cabec VL, Poincloux R, Saati TA, Mege J-L, Tabouret G, Lowell CA, Laviolette-Malirat N, Maridonneau-Parini I. Three-dimensional migration of macrophages requires Hck for podosome organization and extracellular matrix-proteolysis. Blood. 2010; 115:1444–1452.
91. English BK, Ihle JN, Myracle A, Yi T. Hck tyroskine kinase activity modulates tumor necrosis factor production by murine macrophages. Journal of Experimental Medicine. 1993; 179:1017–1022.
92. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation and cancer. Cell. 2010; 140:883–899.
93. Popivanova B, Kitamura K, Wu Y, Kondo T, Kagaya T, Kaneko S, Oshima M, Fujii C, Mukaida N. Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J Clin Invest. 2008; 118:560–570.
94. Wang Y, Wang K, Han G, Wang R, Xiao H, Hou C, Guo R, Dou Y, Shen B, Li Y, Chen G. Neutrophil infiltration favors colitis-associated tumorigenesis by activating the interleukin-1 (IL-1)/IL-6 axis. Mucosal Immunology. 2014; 7:1106–1115.
95. Ning C, Li Y, Y Wang, Han G, Wang R, Xiao H, Li X, Hou C, Ma Y, Sheng D, Shen B, Feng J, Guo R, Li Y and Chen G: Complement activation promotes colitis-associated carcinogenesis through activating intestinal IL-1β/IL-17A axis. Mucosal Immunology. 2015; Epub ahead of print.
96. Matsuzaki K, Murata M, Yoshida K, Sekimoto G, Uemura Y, Sakaida N, Kaibori M, Kamiyama Y, Nishizawa M, Fujisawa J, Okazaki K, Seki T. Chronic inflammation associated with hepatitis C virus infection perturbs hepatic transforming growth factor beta signaling, promoting cirrhosis and hepatocellular carcinoma. Hepatology. 2007; 46:48–57.
97. Doll R, Hill B. Smoking and carcinoma of the lung. Br Med J. 1950; 2:739–748.
98. Zins K, Abraham D, Sioud M, Aharinejad S. Colon cancer cell-derived tumor necrosis factor-alpha mediates the tumor growth-promoting response in macrophages by up-regulating the colony-stimulating factor-1 pathway. Cancer Research. 2007; 67:1038–1045.
99. Hou Z, Falcone D, Subbaramaiah K, Danneberg A. Macrophages induce COX-2 expression in breast cancer cells: role of IL-1b autoamplification. Carcinogenesis. 2011; 32:695–702.
100. Smolinska MJ, Horwood NJ, Page TH, Smallie T, Foxwell BMJ. Chemical inhibition of Src family kinases affects major LPS-activated pathways in primary human macrophages. Molecular Immunology. 2008; 45:990–1000.
101. Giagulli C, Ottoboni L, Caveggion E, Rossi B, Lowell C, Constantin G, Laudanna C, Berton G. The Src family kinases Hck and Fgr are dispensible for inside-out, chemoattractant-induced signalling regulating B2 integrin affinity and valency in neutrophils, but are required for B2 integrin-mediated outside-in signaling involved in sustained adhesion. The Journal of Immunology. 2006; 177:604–611.
102. Suen P, LLic D, Caveggion E, Berton G, Damsky C, Lowell C. Impaired integrin-mediated signal transduction, altered cytoskeletal structure and reduced motility in Hck/Fgr deficient macrophages. Journal of Cell Science. 1999; 112:4067–4078.
103. Raviv S, Hawkins KA, DeCamp MM, Kalhan R. Lung cancer in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010; 183:1138–1146.
104. Zhang X, Mahmudi-Azer S, Connett J, Anthonisen N, He J-Q, Pare P, Sandford A. Association of Hck genetic polymorphisms with gene expression and COPD. Hum Gent. 1007; 120:681–690.
105. Ding L, Getz G, Wheeler D, Mardis E, McLellan M, Cibulskis K, Sougnez C, Greulich H, Muzny D, Morgan M, Fulton L, Fulton R, Zhang Q, Wendl M, Lawrence M, Larson D, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008; 455:1069–75.
106. Yanagisawa S, Sugiura H, Yokoyama T, Yamagata T, Ichikawa T, Akamats K, Koarai A, Hirano T, Nakanishi M, Matsunaga K, Minakata Y, Ichinose M. The possible role of hematopoietic cell kinase in the pathophysiology of COPD. Chest. 2009; 135:95–101.
107. Yamaguchi H, Pixley P, Condeelis J. Invadopodia and podosomes in tumor invasion. European Journal of Cell Biology. 2006; 85:213–218.
108. Takenawa T, Suetsugu S. The WASP-WAVE protein network: connecting the membrane to the cytoskeleton. Nat Rev Mol Cell Biol. 2007; 1:37–48.
109. Park H, Dovas A, Hanna S, Lastrucci C, Coucoule C, Guiet R, Maridonneau-Parini I, Cox D. Tyrosine phosphorylation of Wiskott-Aldrich Syndrome Protein (WASP) by Hck regulates macrophage function. Journal of Biological Chemistry. 2014; 289:7897–7906.
110. Poincloux R, Saati TA, Maridonneau-Parini I, Cabec VL. The oncogenic activity of the Src family kinase Hck requires the coopoerative action of the plasma membrane and lysosome-associated isoforms. European Journal of Cancer. 2009; 45:321–327.
111. Cougoule C, Carréno S, Castandet J, Labrousse A, Astarie-Dequeker C, Poincloux R, Cabec VL, Maridonneau-Parini I. Activation of the lysosome-associated p61Hck isoform triggers the biogenesis of podosomes. Traffic. 2005; 6:682–694.
112. Poincloux R, Vincent C, Labrousse A, Castandet J, Rigo M, Cougoule C, Bordier C, Cabec VL, Maridonneau-Parini I. Re-arrangements of podosome structures are observed when Hck is activated in myeloid cells. European Journal of Cell Biology. 2006; 85:327–332.
113. Guiet R, Goethem EV, Cougoule C, Balor S, Valette A, Saati TA, Lowell CA, Cabec VL, Maridonneau-Parini I. The process of macrophage migration promotes matrix metalloproteinase-independent invasion by tumour cells. The Journal of Immunology. 2011; 187:3806–3814.
114. Tredan O, Galmarini B, Patel K, Tannock I. Drug resistance and the solid tumour microenvironment. J Natl Cancer Inst. 2007; 19:1441–1454.
115. Acharyya A, Oskarsson T, Vanharanta S, Malladi S, Kim J, Morris P, Manova-Todorova K, Leversha M, Hogg N, Seshan V, Norton L, Brogi E, Massaque K. A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell. 2012; 150:165–178.
116. Pastan I, Hassan R, FitzGerald D, Kreitman R. Immunotoxin treatment of cancer. Annu Rev Med. 2007; 25:221–237.
117. Liu Y, Shah K, Yang F, Witucki L, Shokat K. A molecular gate which controls unnatural ATP analogue recognition by the tyrosine kinase v-Src. Bioorg Med Chem. 1998; 8:1219–1226.
118. Pene-Dumitrescu T, Peterson L, Donato N, Smithgall T. An inhibitor-resistant mutant of Hck protects CML cells against the antiproliferative and apoptotic effects of the broadspectrum Src family kinase inhibitor A-419259. Oncogene. 2008; 27:7055–7069.
119. Gorre M, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao P, Sawyers C. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001; 293:876–880.
120. Kobayashi S, Boggon T, Dayaram T, Jänne P, Kocher O, Meyerson M, Johnson B, Eck M, Tenen D, Halmos B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. New England Journal of Medicine. 2005; 352:786–792.
121. Blencke S, Zech B, Engkvist O, Greff Z, Orfi L, Horváth Z, Kéri G, Ullrich A, Daub H. Characterization of a conserved structural determinant controlling protein kinase sensitivity to selective inhibitors. Chemical Biology. 2004; 5:691–701.
122. Chen Y. Are SRC family kinases responsible for imatinib- and dasatinib-resistant chromic myeloid leukemias? Leukemia Research. 2011; 35:27–29.
123. Wheeler D, Lida M, Kruser T, Nechrebecki M, Dunn E, Warmstrong E, Huang S, Harari P. Epidermal growth factor receptor cooperates with Src family kinases in acquired resistance to cetuximab. Cancer Biol Ther. 2009; 8:696–703.
124. Osherov N, Levitzki A. Epidermal-growth-factor-dependent activation of the src-family kinases. Eur J Biochem. 1994; 225:1047–1053.
125. Kloth M, Laughlin K, Biscardi J, Boerner J, Parsons S. STAT5b, a mediator of synergism between c-Src and the epidermal growth factor receptor. J Biol Chem. 2003; 278:1671–1679.
126. Montero JC, Seoane S, Ocana A, Pandiella A. Inhibition of Src family kinases and receptor tyrosine kinases by dasatinib: possible combinations in solid tumours. Clinical Cancer Research. 2007; 2011:17.
127. Jallal H, Valentino M, Chen C, Boschelli F, Ali S, Rabbani S. A Src/Abl kinase inhibitor, SKI-606, blocks breast cancer invasion, grwoth and metastasis in vitro and in vivo. Cancer Research. 2007; 67:1580–1588.
128. Nam H, Im S, Oh D, Elvin P, Kim H, Yook Y, Min A, Song S, Han S, Kim T, Bang Y. Antitumor activity of saracatinib (AZD0530), a c-Src/Abl kinase inhibitor, alone or in combination with chemotherapeutic agents in gastric cance. Mol Cancer Ther. 2013; 12:16–26.
129. Serrels A, Macpherson I, Evans T, Lee F, Clark E, Snsom O, Ashton G, Frame M, Brunton V. Identification of potential bio-markers for measuring inhibition of Src kinase activity in colon cancer cells following treatment with dasatinib. Mol Cancer Ther. 2006; 5:3014–3022.
130. Nam S, Williams A, Vultur A, List A, Bhalla K, Smith D, Lee F, Jove R. Dasatinib (BMS-354825) inhibits Stat5 signaling associated with apoptosis in chronic myelogenous leukemia cells. Mol Cancer Ther. 2007; 6:1400–1405.
131. Ferguson J, Arozarena I, Ehrhardt M, Wellbrock C. Combination of MEK and SRC inhibition suppresses melanoma cell growth and invasion. Oncogene. 2013; 32:86–96.
132. Touny LE, Vieira A, Mendoza A, Khanna C, Hoenerhoff M, Green J. Combined SFK/MEK inhibition prevents metastatic outgrowth of dormant tumour cells. Journal of Clinical Investigation. 2014; 124:156–168.
133. Kopetz S, Lesslie D, Dallas N, Park S, Johnson M, Parikh N, Kim M, Abbruzzese A, Ellis L, Chandra J, Gallick G. Synergistic activity of the SRC family kinase inhibitor dasatinib and oxaliplatin in colon carcinoma cells is mediated by oxidative stress. Cancer Research. 2009; 69:3842–3849.
134. Maria R. Girotti, Malin Pedersen, Berta Sanchez-Laorden, Amaya Viros, Samra Turajlic, Dan Niculescu-Duvaz, Alfonso Zambon, John Sinclair, Andrew Hayes, Martin Gore, Paul Lorigan, Caroline Springer, James Larkin, Claus Jorgensen, Richard Marais. Inhibiting EGF receptor or Src family kinase signaling overcomes BRAF inhibitor resistance in melanoma. Cancer Discovery. 2012; 3:158–167.
135. Okamoto W, Okamoto I, Yoshida T, Okamoto K, Takezawa K, Hatashita E, Yamada Y, Kuwata K, Aro T, Yanagihara K, Fukuoka M, Nishio K, Nakagawa K. Identification of c-Src as a potential therapeutic target for gastric cancer and of MET activation as a cause of resistance to c-Src inhibition. Mol Cancer Ther. 2010; 9:1188–1197.
136. Haura E, Tanvetyanon T, Chiappori A, Williams C, Simon G, Antonia S, Gray J, Litschauer S, Tetteh L, Neuger A, Song L, Rawal B, Schell M, Bepler G. Phase I/II study of the Src inhibitor dasatinib in combination with erlotinib in advanced non-small-cell lung cancer. J Clin Oncol. 2010; 28:1387–1394.
137. Johnson F, Bekele B, Feng L, Wistuba I, Tang X, Tran H, Erasmus J, Hwang L, Takebe N, Blumenschein G, Lippman S, Stewert D. Phase II study of dasatinib in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2010; 28:4609–4615.
138. Fury M, Baxi S, Shen R, Kelly K, Lipson B, Carlson D, Stambuck H, Haque S, Pfister D. Phase II study of saracatinib (AZD0530) for patients with recurrent or metastatic head and neck squamos cell carcinoma (HNSCC). Anticancer Res. 2011; 31:249–254.
139. Sharma M, Wrobleski K, Polite B, Knost J, Wallace J, Modi S, Sleckman B, Taber D, Vokes E, Stadler W, Kindler H. Dasatinib in previously treated metastatic colorectal cancer: a phase II trial of the University of Chicago Phase II Consortium. Investigational New Drugs. 2012; 30:1211–1215.
140. Gucalp A, Sparano J, Caravelli J, Santamauro J, Patil S, Abbruzzi A, Pellegrino C, Bromberg J, Dang C, Theodoulou M, Massague J, Norton L, Hudis C, Traina T. Phase II trial of saracatinib (AZD0530), an oral src-inhibitor for the treatment of patients with hormone receptor negative metastatic breast cancer. Clin Breast Cancer. 2011; 11:306–311.
141. Krishnamurty R, Brigham J, Leonard S, Rankitkar P, Larson E, Dale E, Merritt E, Maly D. Active site profiling reveals coupling between domains in SRC-family kinases. Nature Chemical Biology. 2012; 9:43–51.
142. Saito Y, Yuki H, Kuratani M, Hashizume Y, Takagi S, Honma T, Tanaka A, Shirouzu M, Mikuni J, Handa N, Ogahara I, Sone A, Najima Y, Tomabechi Y, Wakiyama M, Uchida N, et al. A Pyrrolo-Pyrimidine Derivative Targets Human Primary AML Stem Cells in Vivo. Science. 2013; 5:1–15.
143. El-Shazly A, Yamaguchi N, Masuyama K, Suda T, Ishikawa T. Novel association of the src family kinases, hck and c-fgr, with CCR3 receptor stimulation: A possible mechanism for eotaxin-induced human eosinophil chemotaxis. Biochemical and Biophysical Research Communications. 1999; 264:163–170.
144. Yoon J, Terada A, Kita H. CD66b regulates adhesion and activation of human eosinophils. J Immunol. 2007; 179:8454–62.
145. Nair K, Zingde S. Adhesion of neutrophils to fibronectin: role of the CD66 antigens. Cell Immunol. 2001; 208:96–106.
146. Marks D, Csar X, Wilson N, Novak U, Ward A, Kanagasundarum V, Hoffman B, Hamilton J. Expression of a Y559F mutant CSF-1 receptor in M1 myeloid cells: A role for Src kinases in CSF-1 receptor mediated differentiation. Mol Cell Biol Res Commun. 1999; 1:144–152.
147. Durden D, Kim H, Liu Y. The Fc gamma RI receptor signals through the activation of hck and MAP kinase. J Immunol. 1995; 154:4039–47.
148. Suzuki T, Kono H, Hirose N, Okada M, Yamamoto T, Yamamoto K, Honda Z. Differential involvement of Src family kinases in Fc gamma receptor-mediated phagocytosis. J Immunol. 2000; 165:473–482.
149. Bosco M, Curiel R, Zea A, Malabarba M, Ortaldo J, Espinoza-Delgado I. IL-2 signalling in human monocytes involves the phosphoryation and activation of p59 hck 1. The Journal of Immunology. 2000; 164:4575–4585.
150. Bohuslav J, Horejsi V, Hansmann C, Stockl J, Weidle U, Majdic O, Bartke I, Knapp W, Stockinger H. Urokinase plasminogen activator receptor, beta 2-integrins, and Src-kinases within a single receptor complex of human monocytes. J Exp Med. 1995; 181:1381–1390.
151. Trigwell S, Wood L, Jones P. Soluble urokinase receptor promotes cell adhesion and requires tyrosine-92 for activation of p56/59(hck) Biochemical and Biophysical Research Communications. 2000; 278:440–446.
152. Danhauser-Riedl S, Warmuth M, Druker B, Emmerich B, Hallek M. Activation of Src kinases p53/p56lyn and p59hck by p210bcr/abl in myeloid cells. Cancer Research. 1996; 56:3589–3596.
153. Scholz G, Cartledge K, Dunn A. Hck enhances the adherence of lipopolysaccharide-stimulated macrophages via Cbl and phosphatidylinositol 3-kinase. J Biol Chem. 2000; 275:14615–14623.
154. Howlett C, Bisson B, Resek M, Tigley A, Robbins S. The proto-oncogene p120 (Cbl) is a downstream substrate of the Hck protein-tyrosine kinase. Biochemical and Biophysical Research Communications. 1999; 257:129–38.
155. Scott M, Zappacosta F, Kim E, Annan R, Miller W. Identification of novel SH3 domain ligands for the Src family kinase Hck Wiskott-Aldrich syndrome protein (WASP), WASP-interacting protein (WIP) and ELMO1. J Biol Chem. 2002; 277:28238–28246.
156. Yokoyama N, Bakker Cd, Zappacosta F, Huddleston M, Annan R, Ravichandran K, Miller W. Identification of tyrosine residues on ELMO1 that are phosphorylated by the Src-family kinase Hck. Biochemistry. 2005; 44:8841–8849.
157. Baumgartner M, Angelisova P, Setterblad N, Mooney N, Werling D, Horejsi V, Langsley G. Constitutive exclusion of Csk from Hck-positive membrane microdomains permits Src kinase dependent proliferation of Theileria-transformed B-lymphocytes Blood. 2003; 101:1874–1881.
158. Achuthan A, Elsegood C, Msendycz P, Hamilton J, Scholz G. CpG DNA enhances macrophage cell spreading by promoting the Src-family kinase mediated phosphorylation of paxillin. Cell Signal. 2006; 18:2252–2261.
159. Paliwal P, Radha V, Swarup G. Regulation of p73 by Hck through kinase-dependent and independent mechanisms. BMC Mol Biol. 2007; 8:45.
160. Kouroku Y, Soyama A, Fujita E, Urase K, Tsukahara T, Momoi T. RA70 is a src kinase-associated protein expressed ubiquitously. Biochemical and Biophysical Research Communications. 1998; 252:738–742.
161. Poincloux R, Coucoule C, Daubon T, Maridonneau-Parini I, Cabec VL. Tyrosine-phosphorylated STAT5 accumulates on podosomes in Hck-transformed fibroblasts and chronic myeloid leukemia cells. Journal of Cellular Physiology. 2007; 213:212–220.
162. Fumagalli L, Campa C, Germena G, Lowell C, Hirsch E, Berton G. Class I phosphoinositide-3-kinases and SRC kinases play a nonredundant role in regulation of adhesion-independent and -dependent neutrophil reactive oxygen species generation. J Immunol. 2013; 190:3648–60.
163. English B, Orlicek S, Mei Z, Meals E. Bacterial LPS and IFN-gamma trigger the tyrosine phosphorylation of vav in macrophages: evidence for involvement of the hck tyrosine kinase. J Leukoc Biol. 1997; 62:859–864.
164. Pene-Dumitrescu T, Smithgall T. Expression of a Src family kinase in chronic myelogenous leukemia cells induces resistance to imatinib in a kinse-dependent manner. Journal of Biological Chemistry. 2010; 285:21446–21457.
165. Hu Y, Liu Y, Pelletier S, Buchdunger E, Warmuth M, Fabbro D, Hallek M, Etten RAV, Li S. Requirement of Src kinases Lyn, Hck and Fgr for BCR-ABL-induced B-lymphoblastic leukemia but not chronic myeloid leukemia. Nature Genetics. 2004; 36:453–461.
166. Golas J, Arndt K, Etienne C, Lucas J, Nardin D, Gibbons J, Frost P, Ye F, Boschelli D, Boschelli F. SKI-606, a 4-anilino-3-quinolinecarbonitrile dual inhibitor of Src and Abl kinases, is a potent antiproliferative agent against chronic myelogenous leukemia cells in culture and causes regression of K562 xenografts in nude mice. Cancer Res. 2003; 63:375–381.
167. Hebbard L, Cecena G, Golas J, Sawada J, Ellies L, Charbono A, Williams R, Jimenez R, Wankell M, Arndt K, DeJoy S, Rollins R, Diesl V, Follettie M, L LC. Control of mammary tumor differentiation by SKI-606 (bosutinib). Oncogene. 2011; 30:301–312.
168. Nguyen T, Hawkins E, Kolluri A, Kmieciak M, Park H, Lin H, Grant S. Synergism between bosutinib (SKI-606) and the Chk1 inhibitor (PF-00477736) in highly imatinib-resistant BCR/ABL(+) leukemia cells. Leuk Res. 2015; 39:65–71.
169. Rabbani S, Valentino M, Arakelian A, Ali S, Boschelli F. SKI-606 (Bosutinib) blocks prostate cancer invasion, growth, and metastasis in vitro and in vivo through regulation of genes involved in cancer growth and skeletal metastasis. Mol Cancer Ther. 2010; 9:1147–1157.
170. Vultur A, Buettner R, Kowolik C, Liang W, Smith D, Boschelli F, Jove R. SKI-606 (bosutinib), a novel Src kinase inhibitor, suppresses migration and invasion of human breast cancer cells. Mol Cancer Ther. 2008; 7:1185–1194.
171. Recaelli S, Boschelli F, Perini P, Pirola A, Viltadi M, Gambacorti-Passerini C. Synergistic activity of the Src/Abl inhibitor bosutinib in combination with imatinib. Leukemia. 2010; 24:1223–1227.
172. Gambacorti-Passerini C, Brümmendorf T, Kim D, Turkina A, Masszi T, Assouline S, Durrant S, Kantarjian H, Khoury H, Zaritskey A, Shen Z, Jin J, Vellenga E, Pasquini R, V VM. Bosutinib efficacy and safety in chronic phase chronic myeloid leukemia after imatinib resistance or intolerance: Minimum 24-month follow-up. Am J Hematol. 2014; 89:732–742.
173. Moy B, Neven P, Lebrun F, Bellet M, Xu B, Sarosiek T, Chow L, Goss P, Zacharchuk C, Leip E, Turnbull K, Bardy-Bouxin N, Duvillie L, Lang I. Bosutinib in combination with the aromatase inhibitor exemestane: a phase II trial in postmenopausal women with previously treated locally advanced or metastatic hormone receptor-positive/HER2-negative breast cancer. Oncologist. 2014; 19:346–347.
174. Finn R, Dering J, Ginther C, Wilson C, Glaspy P, Tchekmedyian N, Slamon D. Dasatinib, and orally active small molecule inhibitor of both the src and abl kinases, selectively inhibits growth of basal-type/triple negative breast cancer cell lines growing in vitro. Breast Cancer Res Treat. 2007; 105:319–326.
175. Okamoto W, Okamoto I, Yoshida T, Okamoto K, Takezawa K, Hashita E, Yamada Y, Kuwata K, Arao T, Yanagihara K, Fukuoka M, Nishio K, Nakagawa K. Identification of c-Src as a potential therapeutic target for gastric cancer and of MET activation as a cause of resistance to c-Src inhibition. Mol Cancer Ther. 2010; 9:1188–1197.
176. Nagaraj N, Smith J, Revetta F, Washington M, Merchant N. Targeted inhibition of SRC kinase signaling attenuages pancreatic tumourigenesis. Mol Cancer Ther. 2010; 9:2322–2332.
177. Guerrouahen B, Futami M, Vaklavas C, Kanerva J, Whichard Z, Nwawka K, Blanchard E, Lee F, Robinson L, Arceci R, Kornblau S, Wieder E, Cayre Y, Corey S. Dasatinib inhibits the growth of molecularly heterogeneous myeloid leukemias. Clin Cancer Res. 2010; 16:1149–1158.
178. Johnson F, Saigal B, Talpaz M, Donato N. Dasatinib (BMS-354825) tyrosine kinase inhibitor suppresses invasion and induces cell cycle arrest and apoptosis of head and neck squamous cell carcinoma and non-small cell lung caancer cells. Clin Cancer Res. 2005; 11:6924–6932.
179. Shor A, Keschman E, Lee F, Muro-Cacho C, Letson G, Trent J, Pledger W, Jove R. Dasatinib inhibits migration and invasion in diverse human sarcoma cell lines and induces apoptosis in bone sarcoma cells dependent on SRC kinase for survival. Cancer Res. 2007; 67:2800–8.
180. Song Z, Lu P, Furman R, Leonard J, Martin P, Tyrell L, Lee F, Knowles D, Coleman M, Wang Y. Activities of SYK and PLCgamma2 predict apoptotic response of CLL cells to SRC tyrosine kinase inhibitor dasatinib. Clin Cancer Res. 2010; 16:587–599.
181. Park S, Zhang J, Phillips K, Araujo J, Najjar A, Volgin A, Gelovani J, Kim S, Wang Z, Gallick G. Targeting SRC family kinases inhibits growth and lymph node metastases of prostate cancer in an orthotopic nude mouse model. Cancer Res. 2008; 68:3323–3333.
182. Trevino J, Summy J, Lesslie D, Parikh N, Hong D, Lee F, Donato N, Abbruzzese J, Baker C, Gallick G. Inhibition of SRC expression and activity inhibits tumor progression and metastasis of human pancreatic adenocarcinoma cells in an orthotopic nude mouse model. Am J Path. 2006; 168:962–972.
183. Araujo J, Mathew P, Armstrong A, EL EB, Posadas E, Lonberg M, Gallick G, Trudel G, P Paliwal P, Agrawal S, Logothetis C. Dasatinib combined with docetaxel for castration-resistant prostate cancer: results from a phase 1-2 study. Cancer. 2012; 118:63–71.
184. Somlo G, Atzori F, Strauss L, Geese W, Specht J, Gradishar W, Rybicki A, Sy O, Vahdat L, Cortes J. Dasatinib plus capecitabine for advanced breast cancer: safety and efficacy in phase I study CA180004. Clin Cancer Res. 2013; 19:1884–1893.
185. Fornier M, Morris P, Abbruzzi A, D'Andrea G, Gilewski T, Bromberg J, Dang C, Dickler M, Modi S, Seidman A, Sklarin K, Chang J, Norton L, Hudis C. A phase I study of dasatinib and weekly paclitaxel for metastatic breast cancer. Ann Oncol. 2011; 12:2575–2581.
186. Cavalloni G, Peraldo-Neia C, Sarotto I, Gammaitoni L, Migliardi G, Soster M, Marchiò S, Aglietta M, Leone F. Antitumor activity of Src inhibitor saracatinib (AZD-0530) in preclinical models of biliary tract carcinomas. Mol Cancer Ther. 2012; 11:1528–1538.
187. Nam H, Im S, Oh D, Elvin P, Kim H, Yoon Y, Min A, Song S, Han S, Kim T, Bang Y. Antitumor activity of saracatinib (AZD0530), a c-Src/Abl kinase inhibitor, alone or in combination with chemotherapeutic agents in gastric cancer. Mol Cancer Ther. 2013; 12:16–26.
188. Yamaguchi H, Takanashi M, Yoshida N, Ito Y, Kamata R, Fukami K, Yanagihara K, Sakai R. Saracatinib impairs the peritoneal dissemination of diffuse-type gastric carcinoma cells resistant to Met and fibroblast growth factor receptor inhibitors. Cancer Science. 2015; 105:528–536.
189. Dong M, Rice L, Lepler S, Pampo C, Siemann D. Impact of the Src inhibitor saracatinib on the metastatic phenotype of a fibrosarcoma (KHT) tumor model. Anticancer Res. 2010; 30:4405–4413.
190. Bai L, Yang J, Ok J, Mack P, Kung H, Evans C. Simultaneous targeting of Src kinase and receptor tyrosine kinase results in synergistic inhibition of renal cell carcinoma proliferation and migration. Int J Cancer. 2012; 130:2693–2702.
191. Luca AD, D'Alessio A, Gallo M, Maiello M, Bode A, Normanno N. Src and CXCR4 are involved in the invasiveness of breast cancer cells with acquired resistance to lapatinib. Cell Cycle. 2014; 13:148–156.
192. Gangadhar T, Clark J, Karrison T, Gajewski T. Phase II study of the Src kinase inhibitor saracatinib (AZD0530) in metastatic melanoma. Investigational New Drugs. 2013; 3:769–793.
193. Laurie S, Goss G, Shepherd F, Reaume M, Nicholas G, Philip L, Wang L, Schock J, Hirsh V, Oza A, Tsao M, Wright J, Leigh N. A phase II trial of saracatinib, an inhibitor of src kinases, in previously-treated advanced non-small-cell lung cancer: the princess margaret hospital phase II consortium. Clin Lung Cancer. 2014; 15:52–57.
194. Kaulfuß S, Seemann H, Kampe R, Meyer J, Dressel R, König B, Scharf J, Burfeind P. Blockade of the PDGFR family together with SRC leads to diminished proliferation of colorectal cancer cells. Oncotarget. 2013; 4:1037–1049.
195. Yoon S, Cheong H, Kim S, Kim K, Lee S, Lee N, Park H, Won J. Src Family Kinase Inhibitor PP2 Has Different Effects on All-Trans-Retinoic Acid or Arsenic Trioxide-Induced Differentiation of an Acute Promyelocytic Leukemia Cell Line. Cancer Res Treat. 2013; 45:126–133.
196. Nam J, Ino Y, Sakamoto M, Hirohashi S. Src family kinase inhibitor PP2 restores the E-cadherin/catenin cell adhesion system in human cancer cells and reduces cancer metastasis. Clin Cancer Res. 2002; 8:2430–2436.
197. Ahn J, Lee M. Suppression of autophagy sensitizes multidrug resistant cells towards Src tyrosine kinase specific inhibitor PP2. Cancer Letters. 2011; 310:188–197.
198. Zhang Q, Thomas S, Xi S, Smithgall T, Siegfried J, Kamens J, Gooding W, Grandis J. SRC family kinases mediate epidermal growth factor receptor ligand cleavage, proliferation, and invasion of head and neck cancer cells. Cancer Res. 2004; 64:6166–6173.
199. Henderson Y, Toro-Serra R, Chen Y, Ryu J, Frederick M, Zhou G, Gallick G, Lai S, Clayman G. Src inhibitors in suppression of papillary thyroid carcinoma growth. Head Neck. 2014; 36:375–384.
200. Kim S, Kang J, Kim C, Ihm S, Choi M, Yoo H, Lee S. Inhibition of p21 and Akt potentiates SU6656-induced caspase-independent cell death in FRO anaplastic thyroid carcinoma cells. Horm Metab Res. 2013; 45:408–414.
201. Tureckova J, Vojtechova M, Krausova M, Sloncova E, Korinek V. Focal adhesion kinase functions as an akt downstream target in migration of colorectal cancer cells. Transl Oncol. 2009; 2:281–290.