
INTRODUCTION
Remarkable progress has been made in the last decade in the identification of oncogenes and tumor suppressors that are causative to the development of cancer. Many of these discoveries resulted from the cloning of fusion genes created by translocations that are characteristic of human leukemia and lymphomas [1-7]. The BCR-ABL fusion gene, associated with the development of chronic myeloid leukemia (CML) [1-6], is the prototype of such a discovery and has ultimately led to the development of one of the first targeted molecular therapies in cancer [4]. The revolution in the treatment of CML patients that has resulted from the specific and potent targeting of the BCR-ABL kinase with STI571/Gleevec/Imatinib Mesylate [8-10] serves as an important reminder of the advances that can come from an understanding of the molecular basis of disease. Retroviral insertional mutagenesis screens have been another powerful and complementary strategy in cancer gene discovery; a number of oncogenes and tumor suppressors that play crucial roles in development of human cancer and leukemia have been identified in this manner [11-15]. Recently, using retroviral-mediated insertional mutagenesis to identify genes that collaborate with oncogenes and tumor suppressors of the Myc, p53, RB, Ras and ABL pathways have resulted in a number of new targets for development of novel cancer therapeutics [13]. Nevertheless, specific molecular causes of many cancers remain unknown, along with the mutations responsible for a large proportion of human cancers. There is growing evidence that leukemogenesis, like the genesis of other malignancies, is a multi-step process requiring the accumulation of several mutations for the development of overt disease. Therefore, continued identification of new genes and characterization of the molecular basis of their transforming activity is critical to the future development of targeted cancer therapies that will be less toxic and more effective.
Ahi-1 (Abelson helper integration site 1) is a novel oncogene commonly activated by provirus insertional mutagenesis in v-abl and myc-induced murine leukemias and lymphomas [16]. It encodes a unique protein with a SH3 domain, multiple SH3 binding sites and a WD40-repeat domain, suggesting that the normal protein has novel signaling activities. Ahi-1/AHI-1 transcript levels are normally down-regulated during both early murine and human hematopoietic cell differentiation and are highly deregulated in certain human leukemic cells, including leukemic stem cells from patients with chronic myeloid leukemia (CML) and leukemic Sezary cells in cutaneous T-cell lymphoma (CTCL) [17, 18]. Interestingly, overexpression of Ahi-1 alone in primitive hematopoietic cells confers a proliferative advantage in vitro and induces a lethal leukemia in vivo; these effects can be enhanced by BCR-ABL, a fusion oncoprotein that plays a major role in the genesis of CML [19]. Importantly, a novel AHI-1-BCR-ABL-JAK2 interaction complex has recently been identified in CML cells, mediating these effects and playing a key role in mediation of tyrosine kinase inhibitor (TKI) response/resistance of primary CML stem/progenitor cells. These findings suggest that AHI-1 could be a potential new therapeutic target in CML stem cells, a population highly resistant to current TKI therapy and thus causing disease relapse. Moreover, mutations in AHI-1 have also been associated with Joubert syndrome, an autosomal recessive brain disorder [20-22]. Abnormal development and axonal decussation occur in individuals with point mutations in AHI-1, particularly within the WD40-repeat and SH3 domains [22]. Ahi-1 can also interact with Huntingtin-associated protein 1 (Hap1) to form a stable complex critical for neonatal development and involved in intracellular trafficking [23]. In addition, AHI-1 isoforms and its mutations also underlie other diseases, including Joubert syndrome-associated nephronophthisis and autism, and metabolic syndromes, including type 2 diabetes [18, 19, 22, 24-27]. Therefore, it is likely that AHI-1 mutations are critical in the development of diseases such as Joubert syndrome and specific types of human leukemia. Here we provide an extensive review of the molecular and cellular functions of AHI-1 and its interacting proteins in the regulation of normal and disease development since its identification as a cooperative oncogene in v-abl-induced murine leukemia in 2002 [16]. Also discussed are potential applications of targeting specific AHI-1 interaction complexes as a new therapeutic strategy for treatment of CML and brain disorders.
IDENTIFICATION OF Ahi-1/AHI-1 GENE BY PROVIRUS INSERTIONAL MUTAGENESIS IN MURINE LEUKEMIA AND LYMPHOMAS
Identification of the Ahi-1 gene in v-abl-induced mouse pre-B cell lymphoma
The Ahi-1 (Abelson helper integration site-1) locus was initially identified as a common helper provirus integration site in 16% of Abelson murine leukemia virus (A-MuLV)-induced pre-B lymphomas (Figure 1) [28]. A-MuLV is a replication-defective murine retrovirus containing the v-abl oncogene which is responsible for its transforming potential [29-33] in murine models of leukemia and lymphomas. A-MuLV requires a non-defective helper MuLV virus to be able to replicate both in vitro and in vivo [34]. It was shown that the expression of v-abl is not sufficient to induce full malignant transformation in several mouse strains and that additional genetic events may be required [35, 36]. Using long-range restriction mapping, the Ahi-1 locus was mapped to a position ~35 kb downstream of the c-myb proto-oncogene on mouse chromosome 10 (Figure 1) [37]. Another MuLV provirus integration site (Mis-2) was also mapped to the same region, 160 kb downstream of c-myb and ~120 kb downstream of Ahi-1 [38]. It was observed that enhanced expression of c-myb in A-MuLV-induced pre-B-lymphomas harboring a provirus inserted within the Ahi-1 locus was not seen [37], suggesting that the locus contained another gene (potentially Ahi-1 gene) whose dysregulation might be involved in regulation of the malignant transformation of hematopoietic cells. In addition, the Ahi-1 locus was also the target of provirus insertional mutagenesis in 14% of the c-myc-induced murine T cell leukemia (Figure 1) [39], 5% of the Moloney MuLV-induced rat thymomas [38], 11% of the Hoxa9/Meis1-induced murine acute myeloid leukemia [40] and in acute myeloid leukemias arising in Nf1 heterozygous mice [41]. These findings suggest that the Ahi-1 locus is the target of provirus insertional mutagenesis and that its deregulation may contribute to multiple types of murine leukemia and lymphomas.
Subsequently, a new gene (Ahi-1) at the site targeted by the provirus insertional mutations within the Ahi-1 locus was then identified using an exon trapping method [16]. Most of the proviral insertions were localized to the 3’ end of the Ahi-1 gene in an inverse transcriptional orientation, primarily near and downstream of the last exon, although some were found within various introns (Figure 1). Cloning of the Ahi-1 cDNA showed that it encodes a 1047 amino acid protein with a number of interesting domains characteristic of a novel signaling protein.
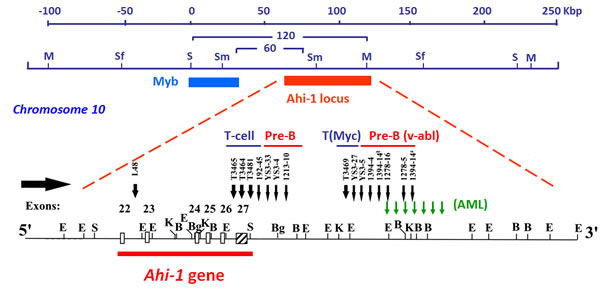
Figure 1: Identification of the Ahi-1 gene by provirus insertional mutagenesis in various murine leukemias and lymphomas. Schematic diagram of the positions of the Ahi-1 and Myb regions on mouse chromosome 10 within a 120 kbp genomic region. Structural organization of the proviruses integrated at the 3’ end of the Ahi-1 gene is indicated in v-abl-induced pre-B lymphomas, Myc-induced T-cell lymphomas and Nf-1-induced murine AML. The localization of the proviruses relative to the exons is shown.
The structure of Ahi-1/AHI-1 protein
The AHI-1/Ahi-1 gene covers more than 200 kb in a region of human chromosome 6 and a minimum of 100 kb on mouse chromosome 10 (Figure 1). Mouse and human Ahi-1/AHI-1 contain at least 27 and 33 exons, respectively [16]. The 1.3 kb region upstream of the ATG start codon of human AHI-1 shows baseline promoter activity and contains two putative TATA boxes and several transcription factor binding sites, such as Oct-1 and c-fos [42]. The Ahi-1/AHI-1 gene is highly conserved in mammals and encodes a unique protein with a Src homology 3 (SH3) domain, multiple SH3 binding sites and multiple WD40-repeats, all known mediators of protein-protein interaction (Figure 2) [16, 43-46]. SH3 domains are often found in proteins containing SH2 domains and are known to bind to proline-rich motifs and to mediate specific protein-protein interactions [43, 44, 47]. The WD40-repeat domain was first identified in the β-subunits of G-proteins [45]. A large number of WD40-repeat-containing proteins have been identified and are known to be involved in aspects of cellular metabolism, including assembling and remodeling of chromosomal proteins [45, 46, 48-51] and regulating the mRNA processing body, including mRNA degradation. They have also been implicated in several inherited diseases, such as Cockayne syndrome, Triple-A syndrome and lissencephaly [51]. Interestingly, the Ahi-1/AHI-1 protein is the only protein thus far identified to contain both WD40 repeats and a SH3 domain. The SH3-binding sites were initially found to bind specifically to the SH3 domain of ABL [52]; many SH3 domain interacting proteins have been characterized as regulators of protein-protein interactions involved in signal transduction, cell cycle control and malignant transformation [43, 44, 46, 47]. Ahi-1/AHI-1 also harbors two potential tyrosine phosphorylation sites, one within the SH3 domain. Phosphotyrosine motifs are known to bind specifically to SH2 domains of proteins [46]. In addition, several PEST sequences, known to mediate protein degradation [53, 54], are located in Ahi-1/AHI-1 protein. Moreover, the human AHI-1 contains a coiled-coil domain in its N-terminal region, also involved in protein-protein interactions [55], which is entirely absent in the mouse and rat Ahi-1 proteins (Figure 2) [16]. Thus Ahi-1 has multiple features of a unique adaptor protein regulating specific signaling pathways.
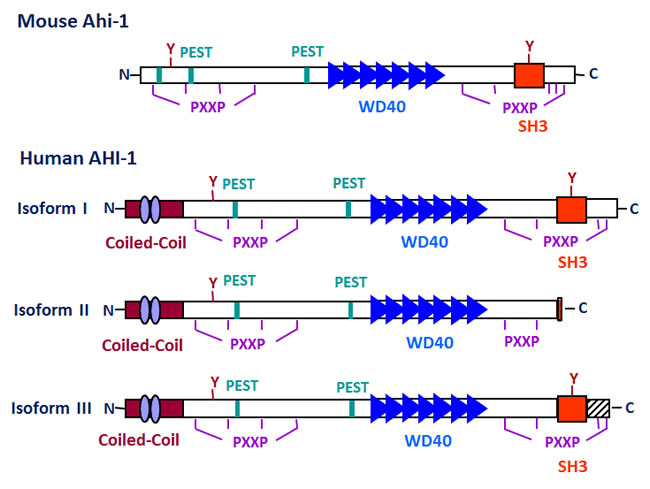
Figure 2: A schematic diagram of the mouse Ahi-1 and human AHI-1 (isoforms I-III) proteins. A summary of the structural motifs present in the Ahi-1/AHI-1 proteins is shown, including: one SH3 domain (red box), seven WD40 repeats (blue triangles), proline-rich motifs (PXXP), PEST sequences (green boxes) and tyrosine kinase phosphorylation sites (Y). Human AHI-1 contains an additional coiled-coil domain which is absent from the mouse Ahi-1 (purple ovals). AHI-1 isoform II, the shortest isoform, lacks the SH3 domain and isoform III, which is still shorter than isoform I, contains additional coding sequences in its C-terminus which are absent from both isoform I and II.
Ahi-1/AHI-1 expression in mouse brain development and hematopoiesis
Murine Ahi-1 encodes two major RNA species (5.0 and 4.2 kb) and several shorter splice variants [16]. Both mouse and human Ahi-1/AHI-1 are highly expressed in brain and testis and have lower expression in liver, lung, thymus, kidney and pancreas [16, 56]. We and others have recently demonstrated that Ahi-1 transcripts are expressed at all stages of mouse embryo development, with increasing expression just prior to birth, suggesting that Ahi-1 expression is developmentally regulated [16, 21, 56]. Mouse Ahi-1 mRNA in the cerebellum has its highest expression at E18 and P5, whereas expression in the cerebral cortex appears maximally at E16 and E18 [21]. However, the expression of mouse Ahi-1 protein in the cerebellum is very low and is only restricted to Purkinje cells and cerebellar nuclei [56, 57]. Moreover, in adult human brain tissue, the highest expression of AHI-1 at both RNA and protein levels has been detected in cerebellum and cerebral cortex [20, 56]. The different cerebellum expression of Ahi-1/AHI-1 in mouse and human may be due to the additional coiled-coil domain present in the human AHI-1 [56]. In addition, Ahi-1 is also found to be abundant in the hypothalamus and amygdala, two important brain region whose dysfunction can lead to emotional and depression phenotypes [57, 58]. Interestingly, it has recently been reported that impaired Wnt-beta-catenin signaling that disrupts renal homeostasis leads to cystic kidney ciliopathy, as demonstrated in Ahi-1 conditional mutant mice [59]. Ahi-1 deficiency can also cause defective Wnt-dependent cerebellar midline fusion that is critical for the development of Joubert syndrome [60]. In addition, Ahi-1 is reported to directly interact with Hap1, a huntingtin-associated protein involved in intracellular trafficking, to regulate cerebellar and brainstem development [57]. It is known that some of these proteins play important roles in regulation of stem cell functions. These results suggest that Ahi-1/AHI-1 interacts with multiple signaling proteins and plays an important role in regulating normal brain functions by interacting with molecular partners in tissue- and disease-specific manners.
It has been shown that expression of Ahi-1/AHI-1 is regulated at multiple stages of hematopoiesis in a highly conserved fashion in mice and humans [17]. Ahi-1/AHI-1 is expressed at its highest level in the most primitive hematopoietic stem cells (HSC) and is rapidly down-regulated as cells differentiate. Interestingly, RNA expression of the mouse Ahi-1 gene is 5-fold higher in the mouse hematopoietic stem cell-enriched population (Sca-1+lin-) purified from normal adult bone marrow (BM) compared to the more differentiated hematopoietic cells (lin+). In addition, within the different lineages of differentiated lin+ cells, Ahi-1 transcript levels are 6- to 7-fold lower in the granulocyte/macrophage lineage compared with the T-lymphoid, erythroid and B-lymphoid lineages [17]. A similar pattern of down-regulated human AHI-1 transcript levels during normal hematopoietic cell differentiation has also been observed in normal adult human BM cells, with an overall 6-fold decrease from the most primitive lin-CD34+CD38- subset to the most mature lin+ CD34- cells. In addition, similar to mouse, human lin+ BM cells from the granulocyte lineage showed significantly reduced expression of AHI-1 compared to T, B and erythroid cell lineages [17]. The conserved pattern of changes in Ahi-1/AHI-1 expression between mice and humans during multi-step hematopoietic cell differentiation suggests that Ahi-1/AHI-1 may play important roles in the regulation of the normal hematopoietic stem cell-renewal program and downstream cell differentiation events.
Ahi-1/AHI-1 isoforms and mutations
Ahi-1/AHI-1 is subject to alternative splicing and both murine and human Ahi-1/AHI-1 genes can encode at least three isoforms (Figure 2). Notably, isoform II, the shortest AHI-1 isoform, lacks the SH3 domain and isoform III contains additional coding sequences not present in isoform I or II [16]. Comparative genetic analysis of the evolution of the human AHI-1 gene indicates that it has undergone positive selection during development of the human species [20]. Thus, changes in AHI-1 are likely to have been important in the evolution of human-specific characteristics; these may include features of the mechanisms regulating early stages of normal hematopoietic cell differentiation.
Involvement of Ahi-1 in mouse models of leukemogenesis is suggested by the high frequency of Ahi-1 mutations observed in certain virus-induced murine lymphomas [16]. Gene-expression analyses of pre-B and T-cell leukemic cells with insertional Ahi-1 mutations has shown both increased expression of Ahi-1 and Ahi-1/viral fused transcripts in the malignant cells, including deletions of the SH3 domain in some cases [16]. Recently, mutations in the human AHI-1 gene have been found to be associated with Joubert syndrome, an autosomal recessive brain disorder [20-22]. Abnormal cerebellar development and axonal decussation were found in individuals with point mutations in AHI-1. These mutations generate stop codons, or amino acid substitutions, or splicing errors within the AHI-1 protein. Truncating mutations are the most frequent type (80%) that abolish completely, or partially, two critical domains of AHI-1: WD40-repeat and SH3 [22]. Altered AHI-1 isoforms and its mutations also underline other diseases, including Joubert syndrome-associated nephronophthisis and autism, and metabolic syndromes [18, 19, 22, 24-27]. Therefore, it is very likely that truncated forms of AHI-1 are critical in development of diseases such as Joubert syndrome and specific types of human leukemia.
BIOLOGICAL FUNCTIONS OF AHI-1 IN HUMAN T-CELL LYMPHOMAS
Deregulated expression of AHI-1 in human cutaneous T-cell lymphoma cells
The first evidence that AHI-1 may be involved in the regulation of human leukemia and lymphoma development is based on an interesting observation that AHI-1 transcripts are significantly higher in a broad spectrum of established human leukemic and lymphoid cell lines, compared to normal human BM. The highest expression level is in two cutaneous T-cell lymphoma (CTCL) cell lines, Hut78, derived from a blood sample of a patient with Sezary Syndrome (SS), and Hut102, derived from the blood of a patient with mycosis fungoides (MF), where increases in AHI-1 transcripts of 40-fold have been detected compared to normal BM [17]. CTCL is a heterogeneous group of T-cell lymphomas that are characterized by malignant T-cells that infiltrate the skin. SS and MF are two major subtypes of CTCL which together account for more than 70% of all CTCL cases [61-64]. SS is the leukemic form of CTCL and is characterized by erythroderma, generalized lymphadenopathy and the presence of malignant mature memory T-helper cells (CD4+CD7-CD45RO+), called Sezary cells, in the skin, lymph nodes and peripheral blood [64-66]. AHI-1 is expressed at significantly higher levels at both RNA and protein levels in primary Sezary cells (CD4+CD7-) from patients with SS compared to normal CD4+ T-cells from normal controls [18, 24]. Particularly, AHI-1 isoform II, lacking the SH3 domain, shows the highest expression in SS samples compared to controls [18, 24]. Little is known about the molecular pathways involved in the development of CTCL; however, the marked deregulation of AHI-1 in CTCL cell lines and primary Sezary cells suggests a potential oncogenic role for AHI-1 in this group of diseases.
Evidence of an oncogenic role of AHI-1 in CTCL cells
To obtain direct evidence that deregulated expression of AHI-1 contributes to the transformed properties of human CTCL cells, knockdown of AHI-1 expression in Hut78 cells was performed using retroviral-mediated RNA interference (RNAi). A screen of nine constructs that produce short hairpin AHI-1 transcripts yielded one that specifically inhibited AHI-1 expression in transduced Hut78 cells by 80%, as evaluated by quantitative real-time RT-PCR (Q-RT-PCR), Northern and Western blot analyses [18, 24]. Hut78 cells are characterized by several interesting transforming properties, including autocrine production of Interleukin (IL)-2, IL-4 and tumor necrosis factor-alpha (TNF-alpha), growth factor independence and the ability to produce tumors in mice [67-70]. Interestingly, retroviral-mediated suppression of AHI-1 reduced autocrine production of IL-2, IL-4 and TNF-alpha in Hut78 cells by up to 85% and caused a significant reduction in their growth factor independence in semi-solid cultures (up to 10-fold) and in single-cell cultures (4-fold) by comparison to cells transduced with a control vector. It was interesting to note that these phenotypes can be restored in vitro in the presence of all three growth factors or IL-4 and TNF-alpha alone, but not IL-2 alone, indicating that AHI-1 expression is important in mediating autocrine production of cytokines that may have a pathogenic role in the progression of disease (Figure 3A). In addition, aberrant expression of IL-2 and TNF-alpha also occurs in primary CD4+CD7- Sezary cells, further supporting the idea that a multi-factorial autocrine mechanism mediated by AHI-1 could be involved in disease development. Importantly, the ability of Hut78 cells to produce tumors in NOD/SCID-beta2microglobulin-/- mice within 4 weeks was also lost when AHI-1 expression was suppressed (Figure 3B) [18]. Thus, lymphomagenic activity of Hut78 cells is somehow dependent on the expression of AHI-1. Taken together, these findings provide strong evidence of the oncogenic activity of AHI-1 in human T-cell lymphomagenesis; its deregulation may contribute to the development of human CTCL, including Sezary syndrome. Further studies will be needed to fully understand the molecular mechanisms, biological functions and clinical role of deregulated AHI-1 expression in CTCL.
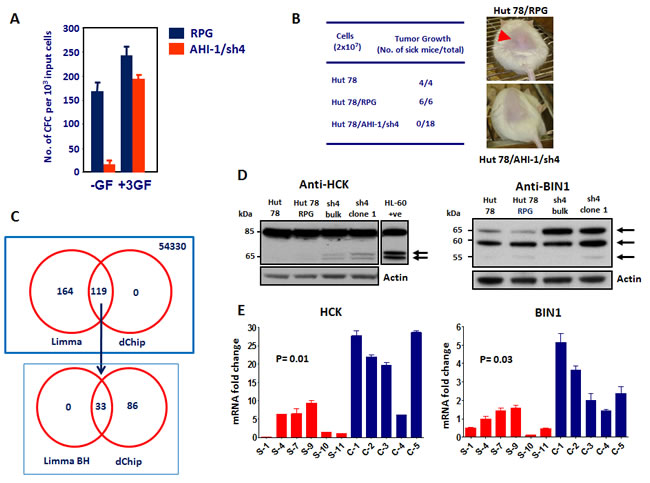
Figure 3: Biological effects of AHI-1 suppression in Hut78 cells and identification of its potential cooperating genes. (A) The number of CFCs able to make colonies in the absence of any growth factor (-GF) is significantly lower in Hut78 cells with AHI-1 suppression (AHI-1/sh4) compared to the empty vector control (RPG). This effect is rescued by adding the three growth factors (IL-2, IL-4 and TNF-α) to the media. (B) Subcutaneous injection of NOD/SCID-β2m immunodeficient mice with Hut78 cells and empty vector controls (Hut78/RPG) results in tumor formation within 4 weeks in all injected mice. AHI-1-suppressed Hut78 cells cannot form any local tumors in these mice even after 20 weeks. (C) Venn diagram of differentially expressed genes from the microarray analysis selected by both Limma and dChip analyses. Affymetrix GeneChip microarray analysis identified several differentially expressed genes in AHI-1-suppressed cells compared to Hut78 and empty vector controls. Initial Limma analysis selected 283 differentially expressed probe sets, which was further refined to 33 with the Benjamini and Hochberg (BH) P value adjustment. (D) Protein expression of HCK (left panel) and BIN1 (right panel) in AHI-1-suppressed cells (sh4-bulk and sh4-clone1) compared to Hut78 and Hut78/RPG controls. The two isoforms of HCK and the 65 kDa isoform of BIN1 show upregulation in the absence of AHI-1. (E) The mRNA expression levels of HCK (left panel) and BIN1 (right panel) are significantly downregulated in six SS patients (red bars) compared to five CD4+ T cell samples from normal controls (blue bars).
Identification of BIN1 and HCK as potential mediators of AHI-1 in CTCL
Microarray analysis using the Affymetrix Human Genome U133 plus 2.0 Arrays which contains over 47,000 transcripts (54,330 probes) recently identified several differentially expressed genes that may play critical roles in AHI-1-mediated leukemic transformation of human CTCL cells (Figure 3C) [24]. Two strong candidates identified in this study are a tyrosine kinase, HCK, and a tumor suppressor, BIN1, which show upregulation at both RNA and protein levels in AHI-1-suppressed CTCL cells (Figure 3D) [24]. HCK (hematopoietic cell kinase) is a member of the Src family tyrosine kinases and its expression is restricted to hematopoietic cells with predominant expression in myeloid lineage cells and B lymphocytes [71]. It has been reported that HCK has oncogenic potential in Philadelphia chromosome-positive (Ph+) leukemia and lymphoma cells [71, 72], however, other studies also demonstrated tumor suppressor functions for HCK in Ph- leukemias [73-75]. In CTCL cells, changes in HCK protein expression and its phosphorylation were observed in AHI-1-suppressed or overexpressed cells, and suppression of activities of Src family kinases, including HCK, by TKI (dasatinib) treatment resulted in reduced or increased growth factor-independent growth of AHI-1-overexpressed or -suppressed cells in a dose-dependent fashion [24]. These results thus suggest that HCK could be a critical player and potential target in AHI-1-mediated CTCL cell transformation.
BIN1 (bridging integrator 1) is a nucleocytoplasmic adaptor protein that was first identified through its interaction with MYC oncoprotein, where it inhibits its transforming activity [76]. MYC is involved in the development of many cancers, where its overexpression is associated with poor prognosis. BIN1 attenuation is frequently described in several cancers, including lung, breast and prostate cancer [77-80]. Alternative splicing can yield more than 10 isoforms of BIN1 with diverse patterns of distribution between tissues, subcellular localization and protein interactions [76, 78, 81, 82]. Notably, only nuclear-localizing isoforms of BIN1 have tumor suppressor activities that can restrict proliferation, survival, and immune escape of oncogenically transformed cells (Figure 4) [24, 83-85]. In particular, aberrant splicing of a brain-specific exon (exon 12A, Figure 4) in malignant cells can abolish the tumor-suppressor activity of BIN1 by interfering with MYC binding [77], which is regulated by phosphorylation of MYC at Ser62 [86]. It has recently been documented that Bin1 loss can promote immune escape in cancer by deregulating the immunomodulatory enzyme indoleamine 2, 3-dioxygenase (IDO); IDO inhibitors have also been found to potentiate cancer chemotherapy [87]. Moreover, it has been shown that Bin1 interacts with the c-ABL tyrosine kinase in an SH3-dependent manner [88]. Similarly, AHI-1 has recently been found to physically interact with BCR-ABL to mediate malignant transformation of CML stem/progenitor cells [19]. Interestingly, characterization of BIN1 in normal and leukemic hematopoietic cells has not been previously described; nevertheless, induced up-regulation of BIN1 is observed in AHI-1 suppressed cells and down-regulation of BIN1 is found in SS patient samples (Figure 3D &E). Knockdown of AHI-1 expression in CTCL cells can normalize their transforming activity and this effect seems to be associated with induction of up-regulation of BIN1 and down-regulation of its interacting oncoprotein, MYC [24]. Thus AHI-1 may directly or indirectly inhibit expression of BIN1 to enhance its oncogenic activity, possibly through interaction with MYC. These findings strongly suggest that AHI-1 may cooperate with BIN1 in the loss of its tumor suppressor activity through its interacting oncoprotein MYC to mediate cellular proliferation and apoptosis control of human CTCL cells and drive human CTCL pathogenesis.
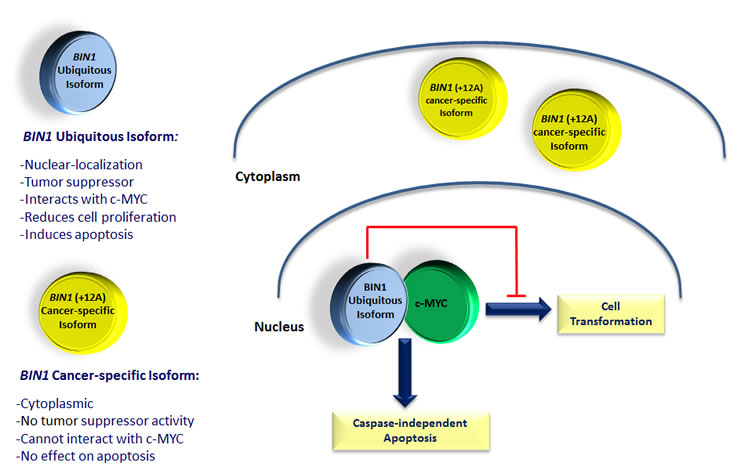
Figure 4: Schematic diagram of ubiquitous and cancer-specific isoforms of BIN1, their cellular localization and biological functions. Ubiquitous isoform of BIN1 is localized in the cell nucleus. It can physically interact with the c-MYC oncogene, inhibiting its transforming activity and increasing the caspase-independent apoptosis in cancerous cells. The cancer-specific isoform of BIN1, with the inclusion of exon 12A, is localized in the cytoplasm, cannot interact with c-MYC and does not have any tumor suppressor activity in cancerous cells.
BIOLOGICAL FUNCTIONS OF AHI-1 IN HUMAN CHRONIC MYELOID LEUKEMIA
Clinical challenges in the treatment of CML
CML has long served as a paradigm for generating new insights into the cellular origin, pathogenesis and improved treatment approaches for many types of human cancer. It is a clonal, multi-step, multi-lineage myeloproliferative disease that typically evolves through three distinct stages: chronic phase (CP), accelerated phases (AP) and blast crisis (BC) [1, 2]. The feature that uniquely defines CML is a clone-specific BCR-ABL fusion gene that encodes an oncoprotein with constitutively elevated tyrosine kinase (TK) activity, driving the pathogenesis of the disease [4-6]. Activation of BCR-ABL deregulates cellular proliferation, apoptosis, and genomic stability of primitive CML cells through effects on multiple intracellular signaling pathways such as the JAK2-STAT, RAS and phosphatidylinositol 3-kinase (PI3K) pathways [5, 6]. Recognition of the consistent molecular and genetic alteration of BCR-ABL in CML patients’ leukemic cells led to the development of TKIs with selectivity for the BCR-ABL kinase [4, 89, 90]. Imatinib (IM) was the first TKI developed and has proven effective for treatment of early phase CML [8-10]. However, early relapses, acquired drug resistance and persistence of leukemic stem cells remain significant issues in many CML patients [10, 91, 92]. The major clinical limitations encountered with IM therapy are: (1) IM must be given continually for many years and its discontinuation always results in rapid reappearance of large numbers of leukemic cells [93-96]; (2) Even in treated patients, 15-25% of patients in early CP and up 40% with AP disease will fail treatment, indicating a need for alternatives [10, 92, 97, 98]; and (3) Relapses are frequently associated with mutations in the BCR-ABL kinase domain [91, 92, 99]. Dasatinib (DA) and Nilotinib (NL) are the second generation of TKIs [89, 90]. Initial clinical experience with both DA and NL indicates that they may also elicit inadequate responses and fail to prevent early disease progression in some patients [100, 101]. In particular, the T315I mutation, found in patients with IM-resistant disease, has been shown to also mediate resistance to DA and NL [102, 103]. Clinical evidence thus suggests that single agent molecularly-targeted therapy may not cure most patients as molecular remissions are rare. These observations emphasize the need for development of new agents and new strategies to prevent continuous development of resistant subclones in primitive CML cells.
Distinct features of CML stem cells resulting in drug resistance
Accumulating evidence indicates that primitive quiescent CML cells are relatively unresponsive to TKIs [104, 105]. We and others have recently discovered that CML stem cells are insensitive to IM, with multiple unique features that would be expected to promote intrinsic and acquired resistance to BCR-ABL-targeted therapeutics [2, 106-109]. These include elevated BCR-ABL expression and TK activity (Figure 5A), deregulated expression of several transporter genes (OCT1, ABCB1 and ABCG2) and a high degree of genetic instability. Thus, leukemic stem cells are a critical source of disease recurrence and a significant reservoir for the emergence of drug-resistant subclones and it is therefore critical to identify other therapies targeting CML stem cells to overcome resistance.
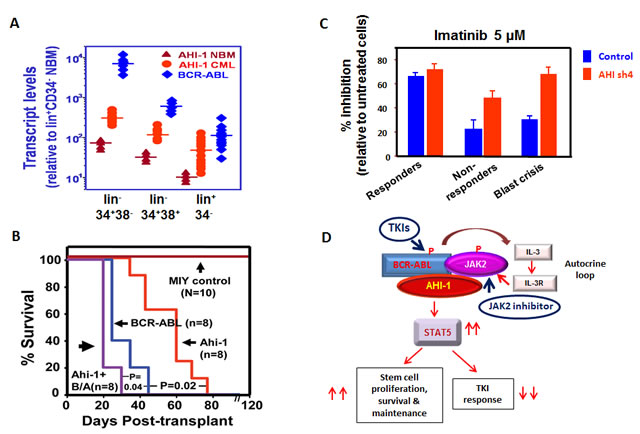
Figure 5: Model of the targeting of the AHI-1-BCR-ABL-JAK2 complex in CML stem cells by combination treatment with TKI and JAK2 inhibitors. (A) Detection of increased transcript levels of AHI-1 and BCR-ABL in a CML stem cell-enriched population (lin-CD34+CD38-), progenitor cells (lin-CD34+CD38+) and their differentiated cells (lin+CD34-) as compared to purified normal bone marrow cells using Q-RT-PCR. (B) Overexpression of Ahi-1 induces a lethal leukemia in vivo and enhances the effects of BCR-ABL. Survival curves of NOD/SCID-β2M-/- mice injected with BaF3 cells transduced with control vector, Ahi-1, BCR-ABL and Ahi-1 plus BCR-ABL (B/A). (C) Inhibition of colony formation in semi-solid media by Imatinib in lin-CD34+ CML progenitor cells from IM-responders, non-responders and blast crisis patient samples transduced with either a control vector or AHI-1/sh4 vector with suppression of AHI-1. (D) Schematic diagram of the AHI-1-BCR-ABL-JAK2 interaction complex that regulates constitutive activation of BCR-ABL and JAK2/STAT5 and results in increased proliferation, survival and a reduced TKI response in CML stem and progenitor cells. Targeting both BCR-ABL and JAK2 activities to destabilize this protein interaction complex may present a potential therapeutic option for CML.
Deregulated AHI-1 expression in CML stem cells and its enhanced transforming activity in conjunction with BCR-ABL in vitro and in vivo
Interestingly, overexpression of either Ahi-1 or BCR-ABL alone in a murine IL-3 dependent pro-B cell line, BaF3, increases proliferation and viability of these cells in the absence and presence of IL-3. However, overexpression of both of these genes simultaneously enhances these effects [19]. Moreover, intravenous injection of NOD/SCID-beta2m immunodeficient mice with either Ahi-1- or BCR-ABL-transduced BaF3 cells causes lethal leukemia within 70 and 40 days, respectively (Figure 5B). However, leukemogenic activities of co-transduced Ahi-1 and BCR-ABL are further enhanced, producing a shorter latency of 26 days [19]. In addition, overexpression of either Ahi-1 or BCR-ABL alone in primitive murine hematopoietic stem cells (HSC) can increase their proliferation in liquid media, as well as elevating the number of colonies from both the colony forming cell (CFC) assay, an assay used to measure the activity of progenitor cells, and the long term culture-initiating cell (LTC-IC) assay, an assay for measuring stem cell activities in vitro [110-112]. Importantly, in murine HSCs co-transduced with Ahi-1 and BCR-ABL, all of these effects are enhanced [19]. Thus, overexpression of Ahi-1/AHI-1 alone can transform primitive hematopoietic cells in vitro and in vivo; these effects are enhanced by BCR-ABL. In addition to overexpression studies, stable suppression of AHI-1 by small interfering RNA in primary CML stem/progenitor cells reduces growth autonomy. Furthermore, co-expression of Ahi-1 in BCR-ABL inducible cells reverses growth deficiencies exhibited by down-regulation of BCR-ABL and results in sustained tyrosine phosphorylation of BCR-ABL and enhanced activation of the JAK2/STAT5 pathway [19]. Importantly, co-immunoprecipitation assays have identified a new AHI-1-BCR-ABL-JAK2 interaction complex at endogenous levels in CML cells [19]. JAK2 is known to directly interact with the C-terminus of BCR-ABL and to be a critical target of BCR-ABL in CML [113-115]. Therefore, through an AHI-1-BCR-ABL-JAK2 interaction complex, AHI-1 regulates transforming activities of BCR-ABL transformed cells associated with sustained tyrosine phosphorylation of BCR-ABL and enhanced activation of JAK2/STAT5 (Figure 5D).
Regulation of IM sensitivity of CML stem/progenitor cells by AHI-1
As described above, IM is an inhibitor of the BCR-ABL tyrosine kinase and the first-line therapy for the CML patients [4, 8, 9]. However, early relapses and IM-resistance are two major problems in some IM-treated CML patients [10]. Studies have indicated that CML stem cells, especially those that are in the quiescent G0 stage of the cell cycle, are less responsive to IM, and are therefore a critical target population for IM resistance [104-106]. Thus, identification of other therapies targeting CML stem cells, as well as development of complementary therapies that target molecular events downstream of BCR-ABL are two major challenges in the treatment of CML.
There are several close biological connections between AHI-1 and BCR-ABL. Both AHI-1 and BCR-ABL transcript levels are significantly increased in the CML stem cell-enriched population (lin-CD34+CD38-) as compared to normal BM (Figure 5A) [17, 106, 107] and AHI-1 and BCR-ABL transcript levels are significantly higher in CML stem/progenitor cells from IM-nonresponders and blast crisis patients compared to the same cells isolated from IM-responders [19]. Interestingly, the AHI-1-BCR-ABL-JAK2 complex was reported to play a key role in regulating IM response/resistance in BCR-ABL-transduced cells and primary CML stem/progenitors. Overexpression of AHI-1 in human K562 cells resulted in greater resistance to IM, while suppression of AHI-1 resulted in increased sensitivity to IM [19]. Altering AHI-1 expression in K562 cells mediates changes in phosphorylation and protein expression of BCR-ABL, JAK2 and STAT5, with enhanced activity and expression of these proteins when AHI-1 is overexpressed, reduced activity and expression when AHI-1 is suppressed, and restored effects in AHI-1 suppressed cells with overexpression of AHI-1. Strikingly, suppression of AHI-1 expression in primary CML stem/progenitor cells also resulted in increased TKI sensitivity (Figure 5C). Particularly, CML stem/progenitor cells from IM-nonresponders and blast crisis patients became more sensitive to TKIs when AHI-1 expression was suppressed. Together, these findings provide strong evidence that AHI-1-BCR-ABL-JAK2 complex modulates response/resistance to TKIs in CML stem/progenitor cells (Figure 5D).
Interestingly, recent studies have demonstrated that JAK2 inhibitors (TG101209, WP1193) and a dual kinase inhibitor of JAK2 and ABL kinases (ON044580) induce apoptosis in IM-sensitive and IM-resistant CML cell lines [116, 117] and that treatment with TKIs in combination with TG101209 results in greater inhibition of CML stem and progenitor cells, compared to treatment with either TKIs or TG101209 alone or a combination of TKIs (Chen, DeGeer and Jiang, unpublished data). Thus, targeting both BCR-ABL and JAK2 in CML stem/progenitor cells will provide a rational strategy for improving the treatment outcome of CML patients (Figure 5D).
AHI-1 IS DISRUPTED IN JOUBERT SYNDROME AND OTHER RELATED BRAIN DISORDERS
In addition to the role of Ahi-1/AHI-1 in the development of leukemia and lymphoma in mice and humans, studies have also demonstrated its function in brain disorders such as Joubert syndrome (JS) and related disorders (JSRD), schizophrenia and autism [20, 21, 118-121]. JSRD are autosomal recessive disorders, characterized by a developmental mid-hindbrain malformation. ‘‘Molar tooth sign’’ is the characteristic feature on a cerebral MRI, which defines the cerebellar vermis and brainstem anomalies and is the diagnostic marker of Joubert syndrome [122-125]. JSRD is considered a multisystem disease, with extra-neurological features including retinal degeneration and cystic kidney disease (e.g. nephronophthisis) [126, 127].
Interestingly, genetic pedigree analysis has demonstrated a high linkage and association between the AHI-1 locus and JS, a rare autosomal recessive disorder characterized by abnormal brain development and mental retardation [20, 21, 118]. A high frequency of AHI-1 mutations can be identified in patients with JS, with most mutations being frameshift or nonsense mutations which result in truncated N-terminal AHI-1 or loss of the WD40-repeat and/or SH3 domains [20, 21]. Recently, several linkage and association studies have further identified AHI-1 as a susceptibility gene for schizophrenia, a major neuropsychiatric disorders associated with depression [119-121, 128, 129]. It is worth noting that several features of autism spectrum disorder (ASD), such as deficits in social behaviour, language dysfunction and repetitive behaviours, have also been described in up to 40% of patients with JS [130, 131]. A three-stage family-based association study has demonstrated evidence of an associated haplotype in AHI-1 with ASD in a region of the genes that has also been associated with schizophrenia [25]. These findings suggest an important role for AHI1 in common brain disorders affecting human cognition and behaviour.
Despite a strong genetic association of the AHI-1 gene with susceptibility to several neuronal diseases, the functions of AHI-1 in regulating normal brain development and disease pathogenesis remain largely unknown. It has recently been reported that Ahi-1 forms a stable complex with huntingtin-associated protein 1 (Hap1) in mouse brains [57], a protein which is important for neonatal development and involved in intracellular trafficking [23, 132-135]. Ahi-1 and Hap1 stabilize each other; Hap1 knockout mice display a significant reduction in Ahi-1 expression levels, defective cerebellar development and abnormal axonal decussation. Similarly, suppression of Ahi-1 in cerebellar neurons from postnatal mouse brains decreases Hap1 levels. In addition, truncated Ahi-1, which corresponds to the mutations in JS, prevents neurite outgrowth in neuronal culture and is unable to stabilize Hap1 [57]. Furthermore, reduction in either Ahi-1 or Hap1 in cerebellar neurons from postnatal mouse brains reduces the protein level of tyrosine kinase receptor B (TrkB) [57], which is critical for neuronal differentiation and brain development [136, 137]. Similarly, Ahi-1 deficiency in mouse brain alters TrkB signaling by promoting the degradation of endocytic TrkB, reducing TrkB signaling in neuronal cells and resulting in depressive phenotypes, which can be alleviated with antidepressant drugs or by overexpression of TrkB in the amygdala [58]. These results provide evidence for the involvement of Ahi-1 deficiency in depression, which occurs in JSRD and has been found to be associated with the AHI-1 gene locus.
JSRD has additional neurological features, such as nephronophthisis and retinal degeneration [126, 127]. Ahi-1-null mice with complete loss of the Ahi-1 protein have normal embryonic development; however, the mice show postnatal runting and the majority do not survive to adulthood [59, 138]. The brain morphology of these mice is highly preserved, suggesting that other effects outside of the nervous system influence the survival of these mice [138]. One study has demonstrated the development of the cystic kidney disease nephronophthisis as a potential cause of death in Ahi-1-null mice [59]. Furthermore, mouse models with conditional Ahi-1 knockout in the kidneys have demonstrated a significant decrease in basal Wnt activity [59]. The Wnt signalling pathway functions in a broad array of cellular processes, and mutations in this pathway have been identified in variety of diseases, from developmental disorders to cancer [139, 140]. In addition, Wnt activity is upregulated in mouse renal injury, suggesting its potential role in adult renal homeostatic injury repair [141]. Interestingly, Ahi-1 interacts with beta-catenin, an integral component in the Wnt signaling pathway, and facilitates its translocation and accumulation in the nucleus, resulting in positive modulation of downstream transcription [59]. In vivo studies have shown that Ahi-1 is also required for the Wnt response to injury and renal tubule repair, a function that is abrogated in Ahi-1-knockout kidneys, leading to renal cystogenesis [59]. Moreover, other studies have demonstrated that Ahi-1 knockout mice fail to form photoreceptor sensory cilia and photoreceptor outer segments. The retinal degeneration in Ahi-1 knockout mice resembles the retinal phenotype observed in patients with JSRD [138, 142]. In summary, the use of Ahi-1-deficient mouse models has facilitated our understanding of the molecular mechanisms and the pathogenesis of JS and its related JSRD, and enabled identification of potential Ahi-1/AHI-1 interacting proteins critical in the development of these diseases.
CONCLUSION
The discovery of ABL tyrosine kinase inhibitors has marked a major advance in the cancer therapy field. It has provided an excellent example of how a specific molecular abnormality can be targeted therapeutically to transform a life-threatening malignancy into a chronic disease. However, TKI therapy is not curative and does not eliminate leukemia stem cells, which remain a potential of relapse. Resistance to single TKI therapy is increasingly being recognized, and more effective strategies using combination therapies are needed to combat the emergence of disease resistance by effectively eradicating leukemic stem cells. Understanding the unique biological properties of cancer stem cells and identifying oncogenes and tumor suppressors and the molecular networks that regulate these properties continue to provide new insights into the complex processes of malignant transformation and disease progression and to uncover improved therapeutic options to eradicate these critical cells and develop molecular cures. Additional insights of broader applicability and benefit will also be forthcoming from such studies.
ACKNOWLEDGMENTS
The preparation of this manuscript was enabled by support from research grants from the Canadian Cancer Society (Grant# 700289), the leukemia & Lymphoma Society of Canada and the Cancer Research Society (X.J). S. Esmailzadeh is a recipient of CIHR-Skin Research Training Graduate Studentship. X. Jiang is a Michael Smith Foundation for Health Research Scholar. We thank Ashley Ringrose for her critical reading of this manuscript.
REFERENCES
1. Goldman JM, Melo JV. Chronic myeloid leukemia--advances in biology and new approaches to treatment. The New England journal of medicine. 2003; 349:1451-1464.
2. Jiang X, Smith C, Eaves A, Eaves C. The challenges of targeting chronic myeloid leukemia stem cells. Clinical lymphoma & myeloma. 2007; 7 Suppl 2:S71-80.
3. Lugo TG, Pendergast AM, Muller AJ, Witte ON. Tyrosine kinase activity and transformation potency of bcr-abl oncogene products. Science. 1990; 247:1079-1082.
4. Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, Zimmermann J, Lydon NB. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nature medicine. 1996; 2:561-566.
5. Sattler M, Griffin JD. Molecular mechanisms of transformation by the BCR-ABL oncogene. Seminars in hematology. 2003; 40:4-10.
6. Van Etten RA. Mechanisms of transformation by the BCR-ABL oncogene: new perspectives in the post-imatinib era. Leukemia research. 2004; 28 Suppl 1:S21-28.
7. Tan J, Muntean AG, Hess JL. PAFc, a key player in MLL-rearranged leukemogenesis. Oncotarget. 2010; 1:461-465.
8. Kantarjian H, Sawyers C, Hochhaus A, Guilhot F, Schiffer C, Gambacorti-Passerini C, Niederwieser D, Resta D, Capdeville R, Zoellner U, Talpaz M, Druker B, Goldman J, O’Brien SG, Russell N, Fischer T et al. Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia. The New England journal of medicine. 2002; 346:645-652.
9. O’Brien SG, Guilhot F, Larson RA, Gathmann I, Baccarani M, Cervantes F, Cornelissen JJ, Fischer T, Hochhaus A, Hughes T, Lechner K, Nielsen JL, Rousselot P, Reiffers J, Saglio G, Shepherd J et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. The New England journal of medicine. 2003; 348:994-1004.
10. O’Hare T, Corbin AS, Druker BJ. Targeted CML therapy: controlling drug resistance, seeking cure. Current opinion in genetics & development. 2006; 16:92-99.
11. Suzuki T, Shen H, Akagi K, Morse HC, Malley JD, Naiman DQ, Jenkins NA, Copeland NG. New genes involved in cancer identified by retroviral tagging. Nature genetics. 2002; 32:166-174.
12. Uren AG, Kool J, Berns A, van Lohuizen M. Retroviral insertional mutagenesis: past, present and future. Oncogene. 2005; 24:7656-7672.
13. Kool J, Berns A. High-throughput insertional mutagenesis screens in mice to identify oncogenic networks. Nature reviews Cancer. 2009; 9:389-399.
14. Sashida G, Bazzoli E, Menendez S, Liu Y, Nimer SD. The oncogenic role of the ETS transcription factors MEF and ERG. Cell Cycle. 2010; 9:3457-3459.
15. Jeang KT. Human T cell leukemia virus type 1 (HTLV-1) and oncogene or oncomiR addiction? Oncotarget. 2010; 1:453-456.
16. Jiang X, Hanna Z, Kaouass M, Girard L, Jolicoeur P. Ahi-1, a novel gene encoding a modular protein with WD40-repeat and SH3 domains, is targeted by the Ahi-1 and Mis-2 provirus integrations. Journal of virology. 2002; 76:9046-9059.
17. Jiang X, Zhao Y, Chan WY, Vercauteren S, Pang E, Kennedy S, Nicolini F, Eaves A, Eaves C. Deregulated expression in Ph+ human leukemias of AHI-1, a gene activated by insertional mutagenesis in mouse models of leukemia. Blood. 2004; 103:3897-3904.
18. Ringrose A, Zhou Y, Pang E, Zhou L, Lin AE, Sheng G, Li XJ, Weng A, Su MW, Pittelkow MR, Jiang X. Evidence for an oncogenic role of AHI-1 in Sezary syndrome, a leukemic variant of human cutaneous T-cell lymphomas. Leukemia. 2006; 20:1593-1601.
19. Zhou LL, Zhao Y, Ringrose A, DeGeer D, Kennah E, Lin AE, Sheng G, Li XJ, Turhan A, Jiang X. AHI-1 interacts with BCR-ABL and modulates BCR-ABL transforming activity and imatinib response of CML stem/progenitor cells. The Journal of experimental medicine. 2008; 205:2657-2671.
20. Ferland RJ, Eyaid W, Collura RV, Tully LD, Hill RS, Al-Nouri D, Al-Rumayyan A, Topcu M, Gascon G, Bodell A, Shugart YY, Ruvolo M, Walsh CA. Abnormal cerebellar development and axonal decussation due to mutations in AHI1 in Joubert syndrome. Nature genetics. 2004; 36:1008-1013.
21. Dixon-Salazar T, Silhavy JL, Marsh SE, Louie CM, Scott LC, Gururaj A, Al-Gazali L, Al-Tawari AA, Kayserili H, Sztriha L, Gleeson JG. Mutations in the AHI1 gene, encoding jouberin, cause Joubert syndrome with cortical polymicrogyria. American journal of human genetics. 2004; 75:979-987.
22. Valente EM, Brancati F, Silhavy JL, Castori M, Marsh SE, Barrano G, Bertini E, Boltshauser E, Zaki MS, Abdel-Aleem A, Abdel-Salam GM, Bellacchio E, Battini R, Cruse RP, Dobyns WB, Krishnamoorthy KS et al. AHI1 gene mutations cause specific forms of Joubert syndrome-related disorders. Annals of neurology. 2006; 59:527-534.
23. Li XJ, Li SH, Sharp AH, Nucifora FC, Jr., Schilling G, Lanahan A, Worley P, Snyder SH, Ross CA. A huntingtin-associated protein enriched in brain with implications for pathology. Nature. 1995; 378:398-402.
24. Kennah E, Ringrose A, Zhou LL, Esmailzadeh S, Qian H, Su MW, Zhou Y, Jiang X. Identification of tyrosine kinase, HCK, and tumor suppressor, BIN1, as potential mediators of AHI-1 oncogene in primary and transformed CTCL cells. Blood. 2009; 113:4646-4655.
25. Alvarez Retuerto AI, Cantor RM, Gleeson JG, Ustaszewska A, Schackwitz WS, Pennacchio LA, Geschwind DH. Association of common variants in the Joubert syndrome gene (AHI1) with autism. Human molecular genetics. 2008; 17:3887-3896.
26. Salonen JT, Uimari P, Aalto JM, Pirskanen M, Kaikkonen J, Todorova B, Hypponen J, Korhonen VP, Asikainen J, Devine C, Tuomainen TP, Luedemann J, Nauck M, Kerner W, Stephens RH, New JP et al. Type 2 diabetes whole-genome association study in four populations: the DiaGen consortium. American journal of human genetics. 2007; 81:338-345.
27. Prior MJ, Foletta VC, Jowett JB, Segal DH, Carless MA, Curran JE, Dyer TD, Moses EK, McAinch AJ, Konstantopoulos N, Bozaoglu K, Collier GR, Cameron-Smith D, Blangero J, Walder KR. The characterization of Abelson helper integration site-1 in skeletal muscle and its links to the metabolic syndrome. Metabolism: clinical and experimental. 2010; 59:1057-1064.
28. Poirier Y, Kozak C, Jolicoeur P. Identification of a common helper provirus integration site in Abelson murine leukemia virus-induced lymphoma DNA. Journal of virology. 1988; 62:3985-3992.
29. Abelson HT, Rabstein LS. Lymphosarcoma: virus-induced thymic-independent disease in mice. Cancer research. 1970; 30:2213-2222.
30. Goff SP, Gilboa E, Witte ON, Baltimore D. Structure of the Abelson murine leukemia virus genome and the homologous cellular gene: studies with cloned viral DNA. Cell. 1980; 22:777-785.
31. Goff SP, Witte ON, Gilboa E, Rosenberg N, Baltimore D. Genome structure of Abelson murine leukemia virus variants: proviruses in fibroblasts and lymphoid cells. Journal of virology. 1981; 38:460-468.
32. Prywes R, Foulkes JG, Rosenberg N, Baltimore D. Sequences of the A-MuLV protein needed for fibroblast and lymphoid cell transformation. Cell. 1983; 34:569-579.
33. Risser R. The pathogenesis of Abelson virus lymphomas of the mouse. Biochimica et biophysica acta. 1982; 651:213-244.
34. Scher CD, Siegler R. Direct transformation of 3T3 cells by Abelson murine leukaemia virus. Nature. 1975; 253:729-731.
35. Green PL, Kaehler DA, Risser R. Cell transformation and tumor induction by Abelson murine leukemia virus in the absence of helper virus. Proceedings of the National Academy of Sciences of the United States of America. 1987; 84:5932-5936.
36. Savard P, DesGroseillers L, Rassart E, Poirier Y, Jolicoeur P. Important role of the long terminal repeat of the helper Moloney murine leukemia virus in Abelson virus-induced lymphoma. Journal of virology. 1987; 61:3266-3275.
37. Jiang X, Villeneuve L, Turmel C, Kozak CA, Jolicoeur P. The Myb and Ahi-1 genes are physically very closely linked on mouse chromosome 10. Mammalian genome. 1994; 5:142-148.
38. Villeneuve L, Jiang X, Turmel C, Kozak CA, Jolicoeur P. Long-range mapping of Mis-2, a common provirus integration site identified in murine leukemia virus-induced thymomas and located 160 kilobase pairs downstream of Myb. Journal of virology. 1993; 67:5733-5739.
39. Girard L, Hanna Z, Beaulieu N, Hoemann CD, Simard C, Kozak CA, Jolicoeur P. Frequent provirus insertional mutagenesis of Notch1 in thymomas of MMTVD/myc transgenic mice suggests a collaboration of c-myc and Notch1 for oncogenesis. Genes & development. 1996; 10:1930-1944.
40. Jin G, Yamazaki Y, Takuwa M, Takahara T, Kaneko K, Kuwata T, Miyata S, Nakamura T. Trib1 and Evi1 cooperate with Hoxa and Meis1 in myeloid leukemogenesis. Blood. 2007; 109:3998-4005.
41. Blaydes SM, Kogan SC, Truong BT, Gilbert DJ, Jenkins NA, Copeland NG, Largaespada DA, Brannan CI. Retroviral integration at the Epi1 locus cooperates with Nf1 gene loss in the progression to acute myeloid leukemia. Journal of virology. 2001; 75:9427-9434.
42. Meng XF, Luo Y, Xiao W, Li M, Shi J. Cloning and characterization of the promoter of the human AHI1 gene. Biochemical genetics. 2009; 47:427-438.
43. Pawson T, Gish GD. SH2 and SH3 domains: from structure to function. Cell. 1992; 71:359-362.
44. Kishan KV, Agrawal V. SH3-like fold proteins are structurally conserved and functionally divergent. Current protein & peptide science. 2005; 6:143-150.
45. Neer EJ, Schmidt CJ, Nambudripad R, Smith TF. The ancient regulatory-protein family of WD-repeat proteins. Nature. 1994; 371:297-300.
46. Seet BT, Dikic I, Zhou MM, Pawson T. Reading protein modifications with interaction domains. Nature reviews Molecular cell biology. 2006; 7:473-483.
47. Koch CA, Anderson D, Moran MF, Ellis C, Pawson T. SH2 and SH3 domains: elements that control interactions of cytoplasmic signaling proteins. Science. 1991; 252:668-674.
48. Smith TF, Gaitatzes C, Saxena K, Neer EJ. The WD repeat: a common architecture for diverse functions. Trends in biochemical sciences. 1999; 24:181-185.
49. Hennig L, Taranto P, Walser M, Schonrock N, Gruissem W. Arabidopsis MSI1 is required for epigenetic maintenance of reproductive development. Development. 2003; 130:2555-2565.
50. Henikoff S. Versatile assembler. Nature. 2003; 423:814-815, 817.
51. Li D, Roberts R. WD-repeat proteins: structure characteristics, biological function, and their involvement in human diseases. Cellular and molecular life sciences : CMLS. 2001; 58:2085-2097.
52. Cicchetti P, Mayer BJ, Thiel G, Baltimore D. Identification of a protein that binds to the SH3 region of Abl and is similar to Bcr and GAP-rho. Science. 1992; 257:803-806.
53. Rogers S, Wells R, Rechsteiner M. Amino acid sequences common to rapidly degraded proteins: the PEST hypothesis. Science. 1986; 234:364-368.
54. Rechsteiner M, Rogers SW. PEST sequences and regulation by proteolysis. Trends in biochemical sciences. 1996; 21:267-271.
55. Mason JM, Arndt KM. Coiled coil domains: stability, specificity, and biological implications. Chembiochem : a European journal of chemical biology. 2004; 5:170-176.
56. Doering JE, Kane K, Hsiao YC, Yao C, Shi B, Slowik AD, Dhagat B, Scott DD, Ault JG, Page-McCaw PS, Ferland RJ. Species differences in the expression of Ahi1, a protein implicated in the neurodevelopmental disorder Joubert syndrome, with preferential accumulation to stigmoid bodies. The Journal of comparative neurology. 2008; 511:238-256.
57. Sheng G, Xu X, Lin YF, Wang CE, Rong J, Cheng D, Peng J, Jiang X, Li SH, Li XJ. Huntingtin-associated protein 1 interacts with Ahi1 to regulate cerebellar and brainstem development in mice. The Journal of clinical investigation. 2008; 118:2785-2795.
58. Xu X, Yang H, Lin YF, Li X, Cape A, Ressler KJ, Li S, Li XJ. Neuronal Abelson helper integration site-1 (Ahi1) deficiency in mice alters TrkB signaling with a depressive phenotype. Proceedings of the National Academy of Sciences of the United States of America. 2010; 107:19126-19131.
59. Lancaster MA, Louie CM, Silhavy JL, Sintasath L, Decambre M, Nigam SK, Willert K, Gleeson JG. Impaired Wnt-beta-catenin signaling disrupts adult renal homeostasis and leads to cystic kidney ciliopathy. Nature medicine. 2009; 15:1046-1054.
60. Lancaster MA, Gopal DJ, Kim J, Saleem SN, Silhavy JL, Louie CM, Thacker BE, Williams Y, Zaki MS, Gleeson JG. Defective Wnt-dependent cerebellar midline fusion in a mouse model of Joubert syndrome. Nature medicine. 2011; 17:726-731.
61. Klemke CD, Goerdt S, Schrama D, Becker JC. New insights into the molecular biology and targeted therapy of cutaneous T-cell lymphomas. Journal der Deutschen Dermatologischen Gesellschaft = Journal of the German Society of Dermatology : JDDG. 2006; 4:395-406.
62. Assaf C, Hummel M, Zemlin M, Steinhoff M, Geilen CC, Stein H, Orfanos CE. Transition of Sezary syndrome into mycosis fungoides after complete clinical and molecular remission under extracorporeal photophoresis. Journal of clinical pathology. 2004; 57:1325-1328.
63. Booken N, Gratchev A, Utikal J, Weiss C, Yu X, Qadoumi M, Schmuth M, Sepp N, Nashan D, Rass K, Tuting T, Assaf C, Dippel E, Stadler R, Klemke CD, Goerdt S. Sezary syndrome is a unique cutaneous T-cell lymphoma as identified by an expanded gene signature including diagnostic marker molecules CDO1 and DNM3. Leukemia. 2008; 22:393-399.
64. Wilcox RA. Cutaneous T-cell lymphoma: 2011 update on diagnosis, risk-stratification, and management. American journal of hematology. 2011; 86:928-948.
65. Kari L, Loboda A, Nebozhyn M, Rook AH, Vonderheid EC, Nichols C, Virok D, Chang C, Horng WH, Johnston J, Wysocka M, Showe MK, Showe LC. Classification and prediction of survival in patients with the leukemic phase of cutaneous T cell lymphoma. The Journal of experimental medicine. 2003; 197:1477-1488.
66. Hwang ST, Janik JE, Jaffe ES, Wilson WH. Mycosis fungoides and Sezary syndrome. Lancet. 2008; 371:945-957.
67. Bunn PA, Jr., Foss FM. T-cell lymphoma cell lines (HUT102 and HUT78) established at the National Cancer Institute: history and importance to understanding the biology, clinical features, and therapy of cutaneous T-cell lymphomas (CTCL) and adult T-cell leukemia-lymphomas (ATLL). Journal of cellular biochemistry Supplement. 1996; 24:12-23.
68. Gootenberg JE, Ruscetti FW, Mier JW, Gazdar A, Gallo RC. Human cutaneous T cell lymphoma and leukemia cell lines produce and respond to T cell growth factor. The Journal of experimental medicine. 1981; 154:1403-1418.
69. Gazdar AF, Carney DN, Bunn PA, Russell EK, Jaffe ES, Schechter GP, Guccion JG. Mitogen requirements for the in vitro propagation of cutaneous T-cell lymphomas. Blood. 1980; 55:409-417.
70. O’Connell MA, Cleere R, Long A, O’Neill LA, Kelleher D. Cellular proliferation and activation of NF kappa B are induced by autocrine production of tumor necrosis factor alpha in the human T lymphoma line HuT 78. The Journal of biological chemistry. 1995; 270:7399-7404.
71. Corey SJ, Anderson SM. Src-related protein tyrosine kinases in hematopoiesis. Blood. 1999; 93:1-14.
72. Hu Y, Liu Y, Pelletier S, Buchdunger E, Warmuth M, Fabbro D, Hallek M, Van Etten RA, Li S. Requirement of Src kinases Lyn, Hck and Fgr for BCR-ABL1-induced B-lymphoblastic leukemia but not chronic myeloid leukemia. Nature genetics. 2004; 36:453-461.
73. Shivakrupa R, Radha V, Sudhakar C, Swarup G. Physical and functional interaction between Hck tyrosine kinase and guanine nucleotide exchange factor C3G results in apoptosis, which is independent of C3G catalytic domain. The Journal of biological chemistry. 2003; 278:52188-52194.
74. Hoshino K, Quintas-Cardama A, Yang H, Sanchez-Gonzalez B, Garcia-Manero G. Aberrant DNA methylation of the Src kinase Hck, but not of Lyn, in Philadelphia chromosome negative acute lymphocytic leukemia. Leukemia. 2007; 21:906-911.
75. Radha V, Sudhakar C, Ray P, Swarup G. Induction of cytochrome c release and apoptosis by Hck-SH3 domain-mediated signalling requires caspase-3. Apoptosis. 2002; 7:195-207.
76. Sakamuro D, Elliott KJ, Wechsler-Reya R, Prendergast GC. BIN1 is a novel MYC-interacting protein with features of a tumour suppressor. Nature genetics. 1996; 14:69-77.
77. Ge K, Duhadaway J, Sakamuro D, Wechsler-Reya R, Reynolds C, Prendergast GC. Losses of the tumor suppressor BIN1 in breast carcinoma are frequent and reflect deficits in programmed cell death capacity. International journal of cancer Journal international du cancer. 2000; 85:376-383.
78. DuHadaway JB, Lynch FJ, Brisbay S, Bueso-Ramos C, Troncoso P, McDonnell T, Prendergast GC. Immunohistochemical analysis of Bin1/Amphiphysin II in human tissues: diverse sites of nuclear expression and losses in prostate cancer. Journal of cellular biochemistry. 2003; 88:635-642.
79. Tajiri T, Liu X, Thompson PM, Tanaka S, Suita S, Zhao H, Maris JM, Prendergast GC, Hogarty MD. Expression of a MYCN-interacting isoform of the tumor suppressor BIN1 is reduced in neuroblastomas with unfavorable biological features. Clinical cancer research : an official journal of the American Association for Cancer Research. 2003; 9:3345-3355.
80. Chang MY, Boulden J, Katz JB, Wang L, Meyer TJ, Soler AP, Muller AJ, Prendergast GC. Bin1 ablation increases susceptibility to cancer during aging, particularly lung cancer. Cancer research. 2007; 67:7605-7612.
81. Wechsler-Reya R, Sakamuro D, Zhang J, Duhadaway J, Prendergast GC. Structural analysis of the human BIN1 gene. Evidence for tissue-specific transcriptional regulation and alternate RNA splicing. The Journal of biological chemistry. 1997; 272:31453-31458.
82. Elliott K, Ge K, Du W, Prendergast GC. The c-Myc-interacting adaptor protein Bin1 activates a caspase-independent cell death program. Oncogene. 2000; 19:4669-4684.
83. Wechsler-Reya RJ, Elliott KJ, Prendergast GC. A role for the putative tumor suppressor Bin1 in muscle cell differentiation. Molecular and cellular biology. 1998; 18:566-575.
84. Elliott K, Sakamuro D, Basu A, Du W, Wunner W, Staller P, Gaubatz S, Zhang H, Prochownik E, Eilers M, Prendergast GC. Bin1 functionally interacts with Myc and inhibits cell proliferation via multiple mechanisms. Oncogene. 1999; 18:3564-3573.
85. Prendergast GC, Muller AJ, Ramalingam A, Chang MY. BAR the door: cancer suppression by amphiphysin-like genes. Biochimica et biophysica acta. 2009; 1795:25-36.
86. Pineda-Lucena A, Ho CS, Mao DY, Sheng Y, Laister RC, Muhandiram R, Lu Y, Seet BT, Katz S, Szyperski T, Penn LZ, Arrowsmith CH. A structure-based model of the c-Myc/Bin1 protein interaction shows alternative splicing of Bin1 and c-Myc phosphorylation are key binding determinants. Journal of molecular biology. 2005; 351:182-194.
87. Muller AJ, DuHadaway JB, Donover PS, Sutanto-Ward E, Prendergast GC. Inhibition of indoleamine 2,3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nature medicine. 2005; 11:312-319.
88. Kadlec L, Pendergast AM. The amphiphysin-like protein 1 (ALP1) interacts functionally with the cABL tyrosine kinase and may play a role in cytoskeletal regulation. Proceedings of the National Academy of Sciences of the United States of America. 1997; 94:12390-12395.
89. Shah NP, Tran C, Lee FY, Chen P, Norris D, Sawyers CL. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science. 2004; 305:399-401.
90. Weisberg E, Manley PW, Breitenstein W, Bruggen J, Cowan-Jacob SW, Ray A, Huntly B, Fabbro D, Fendrich G, Hall-Meyers E, Kung AL, Mestan J, Daley GQ, Callahan L, Catley L, Cavazza C et al. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer cell. 2005; 7:129-141.
91. Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, Sawyers CL. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001; 293:876-880.
92. Deininger M, Buchdunger E, Druker BJ. The development of imatinib as a therapeutic agent for chronic myeloid leukemia. Blood. 2005; 105:2640-2653.
93. Rousselot P, Huguet F, Rea D, Legros L, Cayuela JM, Maarek O, Blanchet O, Marit G, Gluckman E, Reiffers J, Gardembas M, Mahon FX. Imatinib mesylate discontinuation in patients with chronic myelogenous leukemia in complete molecular remission for more than 2 years. Blood. 2007; 109:58-60.
94. Ross DM, Branford S, Seymour JF, Schwarer AP, Arthur C, Bartley PA, Slader C, Field C, Dang P, Filshie RJ, Mills AK, Grigg AP, Melo JV, Hughes TP. Patients with chronic myeloid leukemia who maintain a complete molecular response after stopping imatinib treatment have evidence of persistent leukemia by DNA PCR. Leukemia. 2010; 24:1719-1724.
95. Sobrinho-Simoes M, Wilczek V, Score J, Cross NC, Apperley JF, Melo JV. In search of the original leukemic clone in chronic myeloid leukemia patients in complete molecular remission after stem cell transplantation or imatinib. Blood. 2010; 116:1329-1335.
96. Mahon FX, Rea D, Guilhot J, Guilhot F, Huguet F, Nicolini F, Legros L, Charbonnier A, Guerci A, Varet B, Etienne G, Reiffers J, Rousselot P. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: the prospective, multicentre Stop Imatinib (STIM) trial. The lancet oncology. 2010; 11:1029-1035.
97. Forrest DL, Jiang X, Eaves CJ, Smith CL. An approach to the management of chronic myeloid leukemia in British Columbia. Curr Oncol. 2008; 15:90-97.
98. Kantarjian H, O’Brien S, Cortes J, Giles F, Thomas D, Kornblau S, Shan J, Beth Rios M, Keating M, Freireich E, Talpaz M. Sudden onset of the blastic phase of chronic myelogenous leukemia: patterns and implications. Cancer. 2003; 98:81-85.
99. Shah NP, Nicoll JM, Nagar B, Gorre ME, Paquette RL, Kuriyan J, Sawyers CL. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer cell. 2002; 2:117-125.
100. Talpaz M, Shah NP, Kantarjian H, Donato N, Nicoll J, Paquette R, Cortes J, O’Brien S, Nicaise C, Bleickardt E, Blackwood-Chirchir MA, Iyer V, Chen TT, Huang F, Decillis AP, Sawyers CL. Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. The New England journal of medicine. 2006; 354:2531-2541.
101. Kantarjian H, Giles F, Wunderle L, Bhalla K, O’Brien S, Wassmann B, Tanaka C, Manley P, Rae P, Mietlowski W, Bochinski K, Hochhaus A, Griffin JD, Hoelzer D, Albitar M, Dugan M et al. Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positive ALL. The New England journal of medicine. 2006; 354:2542-2551.
102. Apperley JF. Part I: mechanisms of resistance to imatinib in chronic myeloid leukaemia. The lancet oncology. 2007; 8:1018-1029.
103. Soverini S, Hochhaus A, Nicolini FE, Gruber F, Lange T, Saglio G, Pane F, Muller MC, Ernst T, Rosti G, Porkka K, Baccarani M, Cross NC, Martinelli G. BCR-ABL kinase domain mutation analysis in chronic myeloid leukemia patients treated with tyrosine kinase inhibitors: recommendations from an expert panel on behalf of European LeukemiaNet. Blood. 2011; 118:1208-1215.
104. Graham SM, Jorgensen HG, Allan E, Pearson C, Alcorn MJ, Richmond L, Holyoake TL. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood. 2002; 99:319-325.
105. Copland M, Hamilton A, Elrick LJ, Baird JW, Allan EK, Jordanides N, Barow M, Mountford JC, Holyoake TL. Dasatinib (BMS-354825) targets an earlier progenitor population than imatinib in primary CML but does not eliminate the quiescent fraction. Blood. 2006; 107:4532-4539.
106. Jiang X, Zhao Y, Smith C, Gasparetto M, Turhan A, Eaves A, Eaves C. Chronic myeloid leukemia stem cells possess multiple unique features of resistance to BCR-ABL targeted therapies. Leukemia. 2007; 21:926-935.
107. Jamieson CH, Ailles LE, Dylla SJ, Muijtjens M, Jones C, Zehnder JL, Gotlib J, Li K, Manz MG, Keating A, Sawyers CL, Weissman IL. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. The New England journal of medicine. 2004; 351:657-667.
108. Stuart SA, Minami Y, Wang JY. The CML stem cell: evolution of the progenitor. Cell Cycle. 2009; 8:1338-1343.
109. Peng C, Chen Y, Li D, Li S. Role of Pten in leukemia stem cells. Oncotarget. 2010; 1:156-160.
110. Sutherland HJ, Lansdorp PM, Henkelman DH, Eaves AC, Eaves CJ. Functional characterization of individual human hematopoietic stem cells cultured at limiting dilution on supportive marrow stromal layers. Proceedings of the National Academy of Sciences of the United States of America. 1990; 87:3584-3588.
111. Pettengell R, Luft T, Henschler R, Hows JM, Dexter TM, Ryder D, Testa NG. Direct comparison by limiting dilution analysis of long-term culture-initiating cells in human bone marrow, umbilical cord blood, and blood stem cells. Blood. 1994; 84:3653-3659.
112. Pereira C, Clarke E, Damen J. Hematopoietic colony-forming cell assays. Methods Mol Biol. 2007; 407:177-208.
113. Miyamoto N, Sugita K, Goi K, Inukai T, Lijima K, Tezuka T, Kojika S, Nakamura M, Kagami K, Nakazawa S. The JAK2 inhibitor AG490 predominantly abrogates the growth of human B-precursor leukemic cells with 11q23 translocation or Philadelphia chromosome. Leukemia. 2001; 15:1758-1768.
114. Xie S, Wang Y, Liu J, Sun T, Wilson MB, Smithgall TE, Arlinghaus RB. Involvement of Jak2 tyrosine phosphorylation in Bcr-Abl transformation. Oncogene. 2001; 20:6188-6195.
115. Samanta AK, Lin H, Sun T, Kantarjian H, Arlinghaus RB. Janus kinase 2: a critical target in chronic myelogenous leukemia. Cancer research. 2006; 66:6468-6472.
116. Samanta A, Perazzona B, Chakraborty S, Sun X, Modi H, Bhatia R, Priebe W, Arlinghaus R. Janus kinase 2 regulates Bcr-Abl signaling in chronic myeloid leukemia. Leukemia. 2011; 25:463-472.
117. Samanta AK, Chakraborty SN, Wang Y, Schlette E, Reddy EP, Arlinghaus RB. Destabilization of Bcr-Abl/Jak2 Network by a Jak2/Abl Kinase Inhibitor ON044580 Overcomes Drug Resistance in Blast Crisis Chronic Myelogenous Leukemia (CML). Genes & cancer. 2010; 1:346-359.
118. Parisi MA, Doherty D, Eckert ML, Shaw DW, Ozyurek H, Aysun S, Giray O, Al Swaid A, Al Shahwan S, Dohayan N, Bakhsh E, Indridason OS, Dobyns WB, Bennett CL, Chance PF, Glass IA. AHI1 mutations cause both retinal dystrophy and renal cystic disease in Joubert syndrome. Journal of medical genetics. 2006; 43:334-339.
119. Amann-Zalcenstein D, Avidan N, Kanyas K, Ebstein RP, Kohn Y, Hamdan A, Ben-Asher E, Karni O, Mujaheed M, Segman RH, Maier W, Macciardi F, Beckmann JS, Lancet D, Lerer B. AHI1, a pivotal neurodevelopmental gene, and C6orf217 are associated with susceptibility to schizophrenia. European journal of human genetics : EJHG. 2006; 14:1111-1119.
120. Ingason A, Sigmundsson T, Steinberg S, Sigurdsson E, Haraldsson M, Magnusdottir BB, Frigge ML, Kong A, Gulcher J, Thorsteinsdottir U, Stefansson K, Petursson H, Stefansson H. Support for involvement of the AHI1 locus in schizophrenia. European journal of human genetics : EJHG. 2007; 15:988-991.
121. Slonimsky A, Levy I, Kohn Y, Rigbi A, Ben-Asher E, Lancet D, Agam G, Lerer B. Lymphoblast and brain expression of AHI1 and the novel primate-specific gene, C6orf217, in schizophrenia and bipolar disorder. Schizophrenia research. 2010; 120:159-166.
122. Maria BL, Hoang KB, Tusa RJ, Mancuso AA, Hamed LM, Quisling RG, Hove MT, Fennell EB, Booth-Jones M, Ringdahl DM, Yachnis AT, Creel G, Frerking B. “Joubert syndrome” revisited: key ocular motor signs with magnetic resonance imaging correlation. Journal of child neurology. 1997; 12:423-430.
123. Joubert M, Eisenring JJ, Robb JP, Andermann F. Familial agenesis of the cerebellar vermis. A syndrome of episodic hyperpnea, abnormal eye movements, ataxia, and retardation. Neurology. 1969; 19:813-825.
124. Boltshauser E, Isler W. Joubert syndrome: episodic hyperpnea, abnormal eye movements, retardation and ataxia, associated with dysplasia of the cerebellar vermis. Neuropadiatrie. 1977; 8:57-66.
125. Maria BL, Boltshauser E, Palmer SC, Tran TX. Clinical features and revised diagnostic criteria in Joubert syndrome. Journal of child neurology. 1999; 14:583-590; discussion 590-581.
126. Gleeson JG, Keeler LC, Parisi MA, Marsh SE, Chance PF, Glass IA, Graham Jr JM, Maria BL, Barkovich AJ, Dobyns WB. Molar tooth sign of the midbrain-hindbrain junction: occurrence in multiple distinct syndromes. American journal of medical genetics Part A. 2004; 125A:125-134; discussion 117.
127. Valente EM, Salpietro DC, Brancati F, Bertini E, Galluccio T, Tortorella G, Briuglia S, Dallapiccola B. Description, nomenclature, and mapping of a novel cerebello-renal syndrome with the molar tooth malformation. American journal of human genetics. 2003; 73:663-670.
128. Torri F, Akelai A, Lupoli S, Sironi M, Amann-Zalcenstein D, Fumagalli M, Dal Fiume C, Ben-Asher E, Kanyas K, Cagliani R, Cozzi P, Trombetti G, Strik Lievers L, Salvi E, Orro A, Beckmann JS et al. Fine mapping of AHI1 as a schizophrenia susceptibility gene: from association to evolutionary evidence. The FASEB journal. 2010; 24:3066-3082.
129. Rivero O, Reif A, Sanjuan J, Molto MD, Kittel-Schneider S, Najera C, Topner T, Lesch KP. Impact of the AHI1 gene on the vulnerability to schizophrenia: a case-control association study. PloS one. 2010; 5:e12254.
130. Ozonoff S, Williams BJ, Gale S, Miller JN. Autism and autistic behavior in Joubert syndrome. Journal of child neurology. 1999; 14:636-641.
131. Holroyd S, Reiss AL, Bryan RN. Autistic features in Joubert syndrome: a genetic disorder with agenesis of the cerebellar vermis. Biological psychiatry. 1991; 29:287-294.
132. Chan EY, Nasir J, Gutekunst CA, Coleman S, Maclean A, Maas A, Metzler M, Gertsenstein M, Ross CA, Nagy A, Hayden MR. Targeted disruption of Huntingtin-associated protein-1 (Hap1) results in postnatal death due to depressed feeding behavior. Human molecular genetics. 2002; 11:945-959.
133. Li Y, Chin LS, Levey AI, Li L. Huntingtin-associated protein 1 interacts with hepatocyte growth factor-regulated tyrosine kinase substrate and functions in endosomal trafficking. The Journal of biological chemistry. 2002; 277:28212-28221.
134. Kittler JT, Thomas P, Tretter V, Bogdanov YD, Haucke V, Smart TG, Moss SJ. Huntingtin-associated protein 1 regulates inhibitory synaptic transmission by modulating gamma-aminobutyric acid type A receptor membrane trafficking. Proceedings of the National Academy of Sciences of the United States of America. 2004; 101:12736-12741.
135. Rong J, McGuire JR, Fang ZH, Sheng G, Shin JY, Li SH, Li XJ. Regulation of intracellular trafficking of huntingtin-associated protein-1 is critical for TrkA protein levels and neurite outgrowth. The Journal of neuroscience. 2006; 26:6019-6030.
136. Barbacid M. The Trk family of neurotrophin receptors. Journal of neurobiology. 1994; 25:1386-1403.
137. Chao MV. Neurotrophins and their receptors: a convergence point for many signalling pathways. Nature reviews Neuroscience. 2003; 4:299-309.
138. Louie CM, Caridi G, Lopes VS, Brancati F, Kispert A, Lancaster MA, Schlossman AM, Otto EA, Leitges M, Grone HJ, Lopez I, Gudiseva HV, O’Toole JF, Vallespin E, Ayyagari R, Ayuso C et al. AHI1 is required for photoreceptor outer segment development and is a modifier for retinal degeneration in nephronophthisis. Nature genetics. 2010; 42:175-180.
139. MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Developmental cell. 2009; 17:9-26.
140. Lai SL, Chien AJ, Moon RT. Wnt/Fz signaling and the cytoskeleton: potential roles in tumorigenesis. Cell research. 2009; 19:532-545.
141. Surendran K, Schiavi S, Hruska KA. Wnt-dependent beta-catenin signaling is activated after unilateral ureteral obstruction, and recombinant secreted frizzled-related protein 4 alters the progression of renal fibrosis. Journal of the American Society of Nephrology : JASN. 2005; 16:2373-2384.
142. Westfall JE, Hoyt C, Liu Q, Hsiao YC, Pierce EA, Page-McCaw PS, Ferland RJ. Retinal degeneration and failure of photoreceptor outer segment formation in mice with targeted deletion of the Joubert syndrome gene, Ahi1. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2010; 30:8759-8768.