
INTRODUCTION
Thyroid hormone as L-thyroxine (T4) is the major secretory product of the normal thyroid gland. It is converted to 3,5,3′-triiodo-L-thyronine (T3) by deiodination to effect genomic hormonal actions that require nuclear receptors for thyroid hormone (TRs) [1]. These actions in normal cells involve transcription of a large number of genes whose transcriptional products regulate a large number of metabolic processes essential to normal cell function. Nuclear receptors may reside in nonnuclear compartments, such as cytoplasm, and undergo nuclear importation that may be thyroid hormone-directed [2, 3]. Or, they may interact as a plasma membrane receptor with thyroid hormone [4, 5], to participate in nongenomic events that may not require nuclear uptake of TR.
In dividing endothelial cells, tumor cells, osteoclasts and certain other cells, the plasma membrane bears in large quantities a structural protein, integrin αvβ3, whose extracellular domain contains a specific receptor for thyroid hormone [6, 7]. This receptor site is not structurally related to the binding site for iodothyronines on TRs. The functions of this receptor on the integrin are distinct from those of the nuclear receptors. However, the hormone-binding site on αvβ3 may support nuclear uptake (trafficking) of cytoplasmic TR, phosphorylation of TRs and the formation of intranuclear complexes of coactivator proteins that are relevant to genomic hormonal actions involving TR-regulated genes [8]. At the integrin αvβ3 receptor for thyroid hormone, T4 and T3 initiate complex pro-angiogenic activities that may include transcription of vascular growth factor genes [9–12], nontranscriptional modulation of the function of vascular growth factor receptors adjacent to αvβ3 [13] and enhanced endothelial cell motility [14]. The thyroid hormone receptor on the integrin also has functions in neocortex expansion in developing brain [15], in determining lung smooth muscle phenotype [16] and in regulating macrophage function [17].
The thyroid hormone receptor site on αvβ3 on tumor cells and endothelial cells provides important support for tumor-related angiogenesis, a function that is obviously undesirable in the clinical setting. We have proposed that these pro-angiogenic activities of iodothyronines based at αvβ3 may limit the effectiveness of anti-angiogenic therapy in cancer patients [14, 18], and we have shown in vascularization models such as the chick chorioallantoic membrane (CAM) [18] that pharmacologic or immunologic inhibition of thyroid hormone action at αvβ3 profoundly reduces angiogenic activity of the hormone. The integrin is expressed or activated only limitedly in non-malignant cells and endothelial cells that are not undergoing cell division.
At the integrin thyroid hormone receptor, T4 is a proliferative factor as well for a number of human cancer cell lines that have been studied in vitro [19–22]. The role of T4 exceeds that of T3 in this regard, in that these hormones are equipotent stimulators of tumor cell proliferation, but the circulating levels of free T3 are lower than those of T4. Further, in a clinical experience involving pharmacologic induction of the euthyroid hypothyroxinemic state in advanced cancer patients, maintenance of normal circulating T3 levels and euthyroidism—with substantial reduction in circulating levels of T4—has been associated with stabilization of the tumor or reduction in tumor size [23]. On the other hand, differentiated and undifferentiated PC12 (pheochromocytoma) cells distinguish between T4 and T3 at αvβ3 [24], consistent with the existence of subspecialized hormone-binding domains of the receptor on the integrin [25].
Apoptosis is a regulated process of cell death in nonmalignant cells and is a conscientiously pursued therapeutic goal in oncology when the process can be limited to tumor cells. The tumor cell has a variety of anti-apoptotic transcriptional defenses and interacts with its microenvironment—malignant cell-cell adhesion, tumor cell-nonmalignant cell adhesion and tumor cell-extracellular matrix (ECM) interaction—to erect defenses against radiotherapy and chemotherapeutic measures that induce apoptosis [26]. The intrinsic apoptosis pathway involves induction by chemotherapy or radiation therapy of DNA damage and/or free radical generation that can initiate mitochondrial death signaling. Activated nuclear or mitochondrial p53 is an important mediator of such signaling. The extrinsic apoptosis pathway is activated by factors at the cell surface, for example, tumor necrosis factor-α (TNF-α) and Fas ligand [27]. The extrinsic and intrinsic pathways (Figure 1) converge at the mitochondrial membrane and control its porosity, but other events in apoptosis include loss of cytoskeletal structure, breakdown of the nuclear membrane and fragmentation of DNA. Thus, the latter may be a primary (inducing) event in response to radiation in the intrinsic pathway or a secondary event in the extrinsic pathway when DNases are activated. Thyroid hormone has anti-apoptotic actions on both the extrinsic and intrinsic pathways. The hormone also stabilizes the actin cytoskeleton via conversion of soluble actin to fibrous action (F-actin) [28] and reduces DNA breakdown by reducing caspase-activated DNase activity [29].
Against this background, it is not surprising that thyroid hormone is anti-apoptotic in tumor cells and in certain non-tumor cells. Anti-apoptosis may be desirable in nondividing neurons or endothelial cells, but is an obvious disadvantage in the setting of cancer management. Indeed, the anti-apoptotic activity of host thyroid hormone could contribute to chemotherapeutic resistance [30]. In this brief analysis, we examine the evidence that thyroid hormone is anti-apoptotic in cancer cells and the mechanisms that are involved. For example, thyroid hormone and hormone analogues affect the function of pro-apoptotic p53 [21]; that is, T4 blocks p53-dependent apoptosis and the T4 antagonist, and tetraiodothyroacetic acid (tetrac) and its formulations, facilitate apoptosis. Thyroid hormone and certain of its analogues also affect transcription of a variety of genes relevant to apoptosis [31–33]. This effect includes downregulation of expression of certain caspase genes [31]. Activated caspases are proteases essential to apoptosis by assuring structural disorganization of the nucleus and activation of DNAses, as well as contributing to degradation of cytoplasmic proteins. Tetrac is pro-apoptotic and acts at integrin αvβ3 to upregulate caspase gene transcription [31, 34]. The hypoxia-inducible factor-1α (HIF1α) gene in cancer cells is also upregulated by T4 [25]; the gene product has a number of defensive actions in tumor cells, including support of anti-apoptosis [35, 36]. Other examples of the actions of hormone analogues on apoptosis-modulating genes are discussed below.
We would also point out that thyroid hormone deprivation is associated, not unexpectedly, with increased apoptosis in breast cancer in the intact animal [37] and in hepatoma cells [38]. This may relate to increased expression of apoptosis-promoting p53 described in tumors in hypothyroid animals [39].
Anti-apoptotic actions of thyroid hormone on p53-related apoptosis
Resveratrol has been shown by a number of research groups to be pro-apoptotic. The cellular uptake and metabolism of resveratrol are rapid, underlying the abbreviated bioavailability of the systemically administered stilbene. A specific receptor for resveratrol exists on the extracellular domain integrin αvβ3 [40, 41] and is distinct from the receptor for thyroid hormone. The existence of the cell surface receptor for resveratrol explains why relatively brief exposure of tumor cells to the drug nonetheless results in apoptosis [42]. We have shown that apoptosis induced by resveratrol is p53-dependent, requires phosphorylation of Ser-15 of p53 by mitogen-activated protein kinase (MAPK, ERK1/2) and, interestingly, can involve nuclear accumulation of an inducible pool of cyclooxygenase-2 (COX-2) [43]. Constitutive expression of COX-2 is a biomarker of tumor cell aggressiveness [44], but the inducible nuclear pool of the enzyme has a wholly different function in resveratrol-exposed cells [21, 43, 45, 46] in which it forms complexes with pERK1/2, p53 and SUMO-1 to act as a co-activator for p53-responsive genes [47].
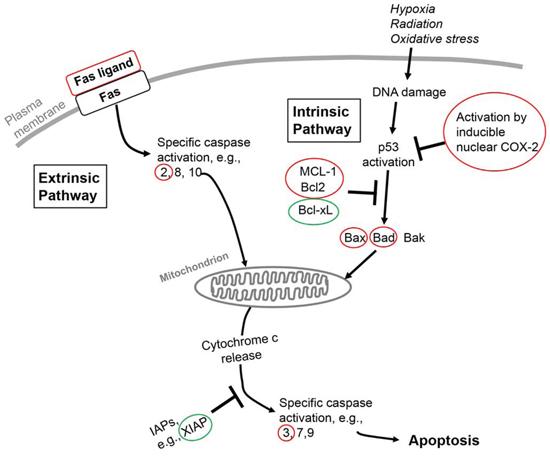
Figure 1: Schematic overview of extrinsic and intrinsic apoptosis pathways in the cell and points at which thyroid hormone in these pathways is anti-apoptotic. The pathways converge at the mitochondrion and cause its permeabilization, with release of cytochrome c and consequent apoptosis. Genes or proteins circled in red or green identify loci of differential actions of thyroid hormone on these multiple factors in apoptosis that are discussed in the current review. Red identifies downregulation of the factor and green identifies upregulation. The Fas receptor is an activator of the extrinsic pathway by its interaction with Fas ligand. An activator of the intrinsic pathway is DNA damage resulting from factors such as radiation or chemotherapy. Bcl-2, B cell lymphoma-2; Bcl-xL, Bcl-2-related gene, long form; Bad, Bcl-2/Bcl-xL-associated death domain protein; Bak, Bcl-2 homologus antagonist killer protein; Bax, Bcl-2-associated X protein; IAPs, inhibitors of apoptosis; MCL-1, myeloid leukemia cell-1; XIAP, X-linked inhibitor of apoptosis.
In the model of resveratrol-induced apoptosis, physiological concentrations of T4 prevent serine phosphorylation of p53 and block stilbene-induced cancer cell apoptosis. The nuclear accumulation of COX-2 is also inhibited [21]. These actions on resveratrol action are rapidly initiated at the binding site for thyroid hormone on αvβ3 [42]. The limited clinical success of resveratrol as a cancer chemotherapeutic agent [48] has been attributed to the limited bioavailability of the stilbene. We have speculated instead that endogenous (host) circulating thyroid hormone is the principal cause of ineffectiveness of resveratrol in the clinical setting [18]. As noted in the next sections, there are a number of anti-apoptotic mechanisms of thyroid hormone beyond p53 activation in tumor cells.
TNF-α and Fas and thyroid hormone
The extrinsic apoptosis pathway in cancer cells and nonmalignant cells is initiated by interaction of so-called death-related ligands with specific receptors on the cell surface. TNF-α and Fas ligand are such factors. Thyroid hormone has been shown to suppress apoptosis in trophoblast cells by decreasing expression of TNF-α and FAS and Fas ligand genes [49]. In the same cells, the hormone decreased the activity of caspase-3, another anti-apoptotic action. In mouse hepatocytes, Sukocheva and Carpenter [27] have shown that T3 is anti-apoptotic via interference with TNF-α\Fas mechanisms. The hormone prevented specific caspase activation and DNA fragmentation and blocked TNF-induced cell acidification. That acidification is an important component of the thyroid hormone-TNF-α apoptotic pathway was shown in these studies by pharmacologic inhibition of the Na+/H+ exchanger (NHE1) that eliminated the anti-apoptotic effect of T3. The exchanger is activated by T3 [50]. The intracellular alkalinization that can be induced by thyroid hormone may also be relevant to the function of P-glycoprotein (P-gp; MDR1), a plasma membrane pump that exports a number of cancer chemotherapeutic agents (see below).
Thyroid hormone action via αvβ3 on transcription of apoptosis-relevant genes
We know that the plasma membrane thyroid hormone/hormone analogue receptor site on the integrin controls transcription of apoptosis-promoting genes, such as CASP2 and BCL2L14 (Bcl-2-like protein 14), and anti-apoptosis genes, such as X-linked inhibitor of apoptosis (XIAP) and myeloid cell leukemia 1 (MCL1) [29, 34]. Tetrac blocks binding of both T4 and T3 to integrin αvβ3 [7, 25]. Tetrac and a nanoparticulate tetrac formulation (Nanotetrac) that is excluded from the cell interior and acts exclusively at the integrin cause suppression of XIAP and MCL1 gene expression and induce transcription of CASP2 and BCL2L14 and other pro-apoptotic genes [7, 51]. Such pro-apoptotic actions of the T4 inhibitor (tetrac/Nanotetrac) are consistent with the demonstrated anti-apoptotic activity of T4, and direct examination of the actions of T4 or T3 on apoptosis-relevant genes has indeed shown this to be the case. For example, T3 decreases transcription of pro-apoptotic BAD (Bcl-2-associated death promoter) and upregulates anti-apoptotic BCL2 in nonmalignant cells [52] without affecting pro-apoptotic BAX (Bcl-2-associated X protein) or AIF (apoptosis inducing factor). Thyroid hormone also decreases cellular abundance of caspases, e.g., caspase-3, and of BAX [32, 39, 53], and increased the expression of XIAP [54]. As mentioned above, the hormone also increases expression of HIF1α, the gene product of which has complex anti-apoptotic functions [35, 55, 56]. The tumor growth arrest that is achieved with imposition clinically of the euthyroid hypothyroxinemic state mentioned above [23] is also consistent with removal of the anti-apoptotic influence of T4.
Apoptosis-relevant action of thyroid hormone on mitochondria
The recognition of the existence of anti-apoptotic activity of the hormone and of known effects of the hormone on mitochondrial activity [1] raised the possibility that mitochondria-directed (intrinsic pathway) apoptosis might be affected by iodothyronines. Mukherjee and co-workers [57] have recently shown that T3 administration rescues hypothyroid rat liver cells from apoptosis induced by oxidative stress that causes inner mitochondrial membrane damage. Thus, thyroid hormone is anti-apoptotic in this model system.
Activation of caspases that begins in the extrinsic apoptosis pathway converges with the intrinsic pathway at mitochondria, with disruption of the mitochondrial membrane and freeing of cytochrome c, a hallmark event in apoptosis. Cytochrome c release may in turn further activate caspases. Support by thyroid hormone of expression of the BCL2 family of genes [58, 59] also has anti-apoptotic consequences. A principal Bcl-2-related protein, MCL1, resides in the outer mitochondrial membrane and binds Bak (Bcl-2 homologous antagonist/killer) and Bax proteins, thus preventing their destabilization of the membrane. Thyroid hormone activates expression of MCL1 [59], preventing mitochondrial membrane destabilization and the formation of channels by which mitochondrial cytochrome c is released and apoptosis induced. T4 downregulates expression of the BAX gene [58], as noted above, whose gene product is pro-apoptotic at the mitochondrion.
Thyroid hormone and residence time in cancer cells of anti-apoptotic chemotherapeutic agents
Acting at the thyroid hormone receptor on integrin αvβ3, the hormone receptor antagonist, tetrac, has been shown to increase residence time in tumor cells of a variety of cancer chemotherapeutic agents [60]. Among these agents are agents with pro-apoptotic properties, such as paclitaxel, etoposide and doxorubicin. These drugs are subject to export from cells by the P-glycoprotein (P-gp; MDR1, ABCB1) membrane pump and expression of the MDR1 gene and pump function are both known to be increased by thyroid hormone [61, 62]. Thus, an ‘anti-apoptotic’ action of the hormone may involve shortening of intracellular residence time and effectiveness of pro-apoptotic chemotherapeutic agents. We attribute the effect of tetrac to prolong residence time in cancer cells of doxorubicin and paclitaxel—increasing drug efficacy—to blockade of the thyroid hormone effect on MDR1. Hypoxia-induced multidrug resistance may also be supported by thyroid hormone via its action on HIF-1α [63].
That the hormonal effect on intracellular concentrations of pro-apoptotic chemotherapeutic agents is more complicated than regulation of MDR1 pump activity is emphasized by the example of cisplatin. Cisplatin is not a ligand of MDR1, but its intracellular concentration is lower in thyroid hormone-treated cells than in control cells [60]. This effect of the hormone may involve increased activity of the organic cation transporter that imports cisplatin into cancer cells, but this possibility has not been examined experimentally.
Involvement of TRs in anti-apoptotic action of thyroid hormone
The foregoing review has dealt with anti-apoptotic activity of thyroid hormone that is initiated by nongenomic mechanisms. However, in human hepatoma cells that overexpress TRα, exposure to T3 has been shown to induce resistance to apoptosis [38]. This effect may depend at least in part upon increased expression of Bcl-xL. Interestingly, these cells were resistant to apoptosis despite increased transcription of tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) and, in fact, the enhanced production of TRAIL was associated with increased, rather than decreased, metastatic potential of the cells. Another TR-related mechanism by which thyroid hormone is anti-apoptotic involves mitochondria studied in nonmalignant cells by Saelim and co-workers [64]. Here, at 10−7 M in Xenopus oocytes, T3 targeted to mitochondria by a short form of TRβ1 decreased apoptosis dependent upon cytochrome c release. Similar results were obtained in primate kidney (CV-1) cells. Thus, several TR-dependent mechanisms exist by which T3 may be anti-apoptotic, but demonstration of the existence of these mechanisms required supraphysiologic concentrations of T3.
In contrast, the liganded wild-type TRβ has thyroid cancer suppressor activity, as shown by Cheng and associates [65], and the receptor may induce apoptosis [66]. Multiple mechanisms are involved in tumor suppressor/pro-apoptosis effects, including downregulation of β-catenin expression and rescue by a nongenomic mechanism of p53 from binding to/inactivation by SV40 large T antigen (SV40Tag). The cellular Src kinase (cSrc) phosphorylation site on TRβ that confers tumor suppressor behavior has been identified [67], as have specific miRNAs that contribute to TRβ-directed decreases in cancer cell proliferation and invasiveness [68]. Mutated TRβ loses tumor suppressor activity and may increase cell proliferation and inhibit apoptosis [69]. Others have shown that T3 at very high concentrations (10−6 M) induces apoptosis in human breast cancer cells [70], apparently via TRβ-dependent downregulation of expression in tumor cells of anti-apoptotic senescence marker protein-30 gene (SMP30). Thus, the anti-apoptotic properties of thyroid hormone expressed by thyroid hormone by a variety of nongenomic mechanisms—most of which originate at integrin αvβ3—are well-substantiated, but the contributions of nuclear receptors TRβ and TRα to regulation of apoptosis are varied in result and appear to depend in part upon the model systems used.
DISCUSSION
Tightly regulated apoptosis is essential to normal tissue development, growth and maintenance of mass. In contrast, the state of anti-apoptosis in cancer cells permits tumor growth, and a number of pro-apoptotic chemotherapeutic agents have been developed to oppose cancer cells’ anti-apoptotic defense mechanisms. Thyroid hormone is an essential factor in the regulation of energy and protein metabolism in nonmalignant cells and normal tissues [1]. In the setting of cancer, however, thyroid hormone, particularly T4, acts via its cell surface receptor on integrin αvβ3 as a proliferative factor and is pro-angiogenic [1, 13]. Results of in vitro and of human xenograft studies support these roles of the hormone [19, 22, 25, 71]. Clinical support for such observations comes from medical interventions to reduce circulating levels of thyroid hormone [23, 72], from effects of hypothyroidism secondary to tyrosine kinase inhibitor use in patients with renal cell carcinoma [73, 74] and from the effects of spontaneous hypothyroidism on the clinical behavior of breast cancer [75].
That thyroid hormone is desirably anti-apoptotic in nonmalignant cells has been emphasized by other authors [58, 76, 77]. We have previously reported the presence of anti-apoptotic activity of thyroid hormone in cancer cells, a finding that serves to engage the hormone in cancer cell defense [20, 21, 41]. This action of the hormone was initiated at a cell surface receptor for thyroid hormone integrin αvβ3 and was inhibited by tetrac or tetrac formulations. In the present review, we have examined the molecular mechanisms by which the hormone can be anti-apoptotic and by which tetrac or its nanoparticulate formulation can be pro-apoptotic. What is clear is that multiple components of the extrinsic and intrinsic apoptosis pathways are largely modulated from αvβ3 by thyroid hormone—chiefly, T4—at physiological concentrations. These components include expression of genes for Fas ligand, caspases, multiple members of the Bcl-2 family and XIAP (Figure 1). Also affected is phosphorylation of p53. As a result of the congruence of these factors, cytochrome c release by mitochondria is decreased. Thus, a panel of proteins is affected with roles in apoptosis that affect the integrity of the mitochondrion. Finally, the hormone may limit chemotherapy-induced apoptosis by shortening intracellular residence time of anticancer drugs that are pro-apoptotic. All of these factors are possible clinical contributors to chemotherapy resistance, such as that which has been modeled in vitro in myeloma cells exposed to bortezomib and thyroid hormone [78].
How broadly the anti-apoptotic activity of thyroid hormone may affect chemotherapy is not yet known. It is possible to begin to examine the extent of anti-apoptosis with reduction in circulating levels of host cancer patient T4 while maintaining normal blood levels of T3 [23]. It may also be possible in the future to block the anti-apoptotic and other cancer support actions of T4 at integrin αvβ3 with pharmaceutical agents [7, 21], and monitor effectiveness of existing cancer chemotherapeutic drugs with pro-apoptotic properties.
REFERENCES
1. Cheng SY, Leonard JL, Davis PJ. Molecular aspects of thyroid hormone actions. Endocr Rev. 2010; 31:139–170.
2. Lin HY, Zhang S, West BL, Tang HY, Passaretti T, Davis FB, Davis PJ. Identification of the putative MAP kinase docking site in the thyroid hormone receptor-beta1 DNA-binding domain: functional consequences of mutations at the docking site. Biochemistry. 2003; 42:7571–7579.
3. Cao HJ, Lin HY, Luidens MK, Davis FB, Davis PJ. Cytoplasm-to-nucleus shuttling of thyroid hormone receptor-beta1 (Trbeta1) is directed from a plasma membrane integrin receptor by thyroid hormone. Endocr Res. 2009; 34:31–42.
4. Kalyanaraman H, Schwappacher R, Joshua J, Zhuang S, Scott BT, Klos M, Casteel DE, Frangos JA, Dillmann W, Boss GR, Pilz RB. Nongenomic thyroid hormone signaling occurs through a plasma membrane-localized receptor. Science Signal. 2014; 7:ra48.
5. Moeller LC, Broecker-Preuss M. Transcriptional regulation by nonclassical action of thyroid hormone. Thyroid Res. 2011; 4:S6.
6. Bergh JJ, Lin HY, Lansing L, Mohamed SN, Davis FB, Mousa S, Davis PJ. Integrin alphaVbeta3 contains a cell surface receptor site for thyroid hormone that is linked to activation of mitogen-activated protein kinase and induction of angiogenesis. Endocrinology. 2005; 146:2864–2871.
7. Davis PJ, Davis FB, Mousa SA, Luidens MK, Lin HY. Membrane receptor for thyroid hormone: physiologic and pharmacologic implications. Annu Rev Pharmacol Toxicol. 2011; 51:99–115.
8. Davis PJ, Lin HY, Tang HY, Davis FB, Mousa SA. Adjunctive input to the nuclear thyroid hormone receptor from the cell surface receptor for the hormone. Thyroid. 2013; 23:1503–1509.
9. Luidens MK, Mousa SA, Davis FB, Lin HY, Davis PJ. Thyroid hormone and angiogenesis. Vasc Pharmacol. 2010; 52:142–145.
10. Cayrol F, Diaz Flaque MC, Fernando T, Yang SN, Sterle HA, Bolontrade M, Amoros M, Isse B, Farias RN, Ahn H, Tian YF, Tabbo F, Singh A, et al. Integrin alphavbeta3 acting as membrane receptor for thyroid hormones mediates angiogenesis in malignant T cells. Blood. 2015; 125:841–851.
11. Liu X, Zheng N, Shi YN, Yuan J, Li L. Thyroid hormone induced angiogenesis through the integrin alphavbeta3/protein kinase D/histone deacetylase 5 signaling pathway. J Mol Endocrinol. 2014; 52:245–254.
12. Yoshida T, Gong J, Xu Z, Wei Y, Duh EJ. Inhibition of pathological retinal angiogenesis by the integrin alphavbeta3 antagonist tetraiodothyroacetic acid (tetrac). Exp Eye Res. 2012; 94:41–48.
13. Mousa SA, Bergh JJ, Dier E, Rebbaa A, O’Connor LJ, Yalcin M, Aljada A, Dyskin E, Davis FB, Lin HY, Davis PJ. Tetraiodothyroacetic acid, a small molecule integrin ligand, blocks angiogenesis induced by vascular endothelial growth factor and basic fibroblast growth factor. Angiogenesis. 2008; 11:183–190.
14. Mousa SA, Lin HY, Tang HY, Hercbergs A, Luidens MK, Davis PJ. Modulation of angiogenesis by thyroid hormone and hormone analogues: implications for cancer management. Angiogenesis. 2014; 17:463–469.
15. Stenzel D, Wilsch-Brauninger M, Wong FK, Heuer H, Huttner WB. Integrin alphavbeta3 and thyroid hormones promote expansion of progenitors in embryonic neocortex. Development. 2014; 141:795–806.
16. Dekkers BG, Naeimi S, Bos IS, Menzen MH, Halayko AJ, Hashjin GS, Meurs H. L-thyroxine promotes a proliferative airway smooth muscle phenotype in the presence of TGF-beta1. Am J Physiol Lung Cell Mol Physiol. 2015; 308:L301–306.
17. Chen Y, Sjolinder M, Wang X, Altenbacher G, Hagner M, Berglund P, Gao Y, Lu T, Jonsson AB, Sjolinder H. Thyroid hormone enhances nitric oxide-mediated bacterial clearance and promotes survival after meningococcal infection. PLoS One. 2012; 7:e41445.
18. Davis PJ, Yalcin M, Lin HY, Tang HY, Hercbergs A, Leith JT, Davis FB, Luidens MK, SA M. Incomplete success of angioinhibitor therapy in cancer: Estimation of contribution of pro-angiogenic activity of patient thyroid hormone. J Cancer Sci Ther. 2013; 5:441–445.
19. Davis FB, Tang HY, Shih A, Keating T, Lansing L, Hercbergs A, Fenstermaker RA, Mousa A, Mousa SA, Davis PJ, Lin HY. Acting via a cell surface receptor, thyroid hormone is a growth factor for glioma cells. Cancer Res. 2006; 66:7270–7275.
20. Lin HY, Tang HY, Shih A, Keating T, Cao G, Davis PJ, Davis FB. Thyroid hormone is a MAPK-dependent growth factor for thyroid cancer cells and is anti-apoptotic. Steroids. 2007; 72:180–187.
21. Lin HY, Tang HY, Keating T, Wu YH, Shih A, Hammond D, Sun M, Hercbergs A, Davis FB, Davis PJ. Resveratrol is pro-apoptotic and thyroid hormone is anti-apoptotic in glioma cells: both actions are integrin and ERK mediated. Carcinogenesis. 2008; 29:62–69.
22. Tang HY, Lin HY, Zhang S, Davis FB, Davis PJ. Thyroid hormone causes mitogen-activated protein kinase-dependent phosphorylation of the nuclear estrogen receptor. Endocrinology. 2004; 145:3265–3272.
23. Hercbergs A, Johnson RE, Ashur-Fabian O, Garfield DH, Davis PJ. Medically induced euthyroid hypothyroxinemia may extend survival in compassionate need cancer patients: an observational study. Oncologist. 2015; 20:72–76.
24. Barbakadze T, Natsvlishvili N, Mikeladze D. Thyroid hormones differentially regulate phosphorylation of ERK and Akt via integrin alphavbeta3 receptor in undifferentiated and differentiated PC-12 cells. Cell Biochem Funct. 2014; 32:282–286.
25. Lin HY, Sun M, Tang HY, Lin C, Luidens MK, Mousa SA, Incerpi S, Drusano GL, Davis FB, Davis PJ. L-Thyroxine vs. 3,5,3′-triiodo-L-thyronine and cell proliferation: activation of mitogen-activated protein kinase and phosphatidylinositol 3-kinase. Am J Physiol Cell Physiol. 2009; 296:C980–991.
26. Westhoff MA, Zhou S, Bachem MG, Debatin KM, Fulda S. Identification of a novel switch in the dominant forms of cell adhesion-mediated drug resistance in glioblastoma cells. Oncogene. 2008; 27:5169–5181.
27. Sukocheva OA, Carpenter DO. Anti-apoptotic effects of 3,5,3′-tri-iodothyronine in mouse hepatocytes. J Endocrinol. 2006; 191:447–458.
28. Davis PJ, Leonard JL, Davis FB. Mechanisms of nongenomic actions of thyroid hormone. Front Neuroendocrinol. 2008; 29:211–218.
29. Farwell AP, Dubord-Tomasetti SA, Pietrzykowski AZ, Stachelek SJ, Leonard JL. Regulation of cerebellar neuronal migration and neurite outgrowth by thyroxine and 3,3′,5′-triiodothyronine. Brain Res Dev Brain Res. 2005; 154:121–135.
30. Davis PJ, Lin HY, Sudha T, Yalcin M, Tang HY, Hercbergs A, Leith JT, Luidens MK, Ashur-Fabian O, Incerpi S, Mousa SA. Nanotetrac targets integrin alphavbeta3 on tumor cells to disorder cell defense pathways and block angiogenesis. OncoTargets Ther. 2014; 7:1619–1624.
31. Glinskii AB, Glinsky GV, Lin HY, Tang HY, Sun M, Davis FB, Luidens MK, Mousa SA, Hercbergs AH, Davis PJ. Modification of survival pathway gene expression in human breast cancer cells by tetraiodothyroacetic acid (tetrac). Cell Cycle. 2009; 8:3562–3570.
32. Verga Falzacappa C, Timperi E, Bucci B, Amendola D, Piergrossi P, D’Amico D, Santaguida MG, Centanni M, Misiti S. T(3) preserves ovarian granulosa cells from chemotherapy-induced apoptosis. J Endocrinol. 2012; 215:281–289.
33. Lin HY, Su YF, Hsieh MT, Lin S, Meng R, London D, Lin C, Tang HY, Hwang J, Davis FB, Mousa SA, Davis PJ. Nuclear monomeric integrin alphav in cancer cells is a coactivator regulated by thyroid hormone. FASEB J. 2013; 27:3209–3216.
34. Yalcin M, Dyskin E, Lansing L, Bharali DJ, Mousa SS, Bridoux A, Hercbergs AH, Lin HY, Davis FB, Glinsky GV, Glinskii A, Ma J, Davis PJ, et al. Tetraiodothyroacetic acid (tetrac) and nanoparticulate tetrac arrest growth of medullary carcinoma of the thyroid. J Clin Endocrinol Metab. 2010; 95:1972–1980.
35. Liao HY, Wang GP, Huang SH, Li Y, Cai SW, Zhang J, Chen HG, Wu WB. HIF-1alpha silencing suppresses growth of lung adenocarcinoma A549 cells through induction of apoptosis. Molec Med Rep. 2014; 9:911–915.
36. Zhang H, Chen GG, Zhang Z, Chun S, Leung BC, Lai PB. Induction of autophagy in hepatocellular carcinoma cells by SB203580 requires activation of AMPK and DAPK but not p38 MAPK. Apoptosis. 2012; 17:325–334.
37. Lopez-Fontana CM, Sasso CV, Maselli ME, Santiano FE, Semino SN, Cuello Carrion FD, Jahn GA, Caron RW. Experimental hypothyroidism increases apoptosis in dimethylbenzanthracene-induced mammary tumors. Oncol Rep. 2013; 30:1651–1660.
38. Chi HC, Chen SL, Liao CJ, Liao CH, Tsai MM, Lin YH, Huang YH, Yeh CT, Wu SM, Tseng YH, Chen CY, Tsai CY, Chung IH, et al. Thyroid hormone receptors promote metastasis of human hepatoma cells via regulation of TRAIL. Cell Death Differ. 2012; 19:1802–1814.
39. Sterle HA, Valli E, Cayrol F, Paulazo MA, Martinel Lamas DJ, Diaz Flaque MC, Klecha AJ, Colombo L, Medina VA, Cremaschi GA, Barreiro Arcos ML. Thyroid status modulates T lymphoma growth via cell cycle regulatory proteins and angiogenesis. J Endocrinol. 2014; 222:243–255.
40. Lin HY, Lansing L, Merillon JM, Davis FB, Tang HY, Shih A, Vitrac X, Krisa S, Keating T, Cao HJ, Bergh J, Quackenbush S, Davis PJ. Integrin alphaVbeta3 contains a receptor site for resveratrol. FASEB J. 2006; 20:1742–1744.
41. Lin HY, Davis PJ, Tang HY, Mousa SA, Luidens MK, Hercbergs AH, Davis FB. The pro-apoptotic action of stilbene-induced COX-2 in cancer cells: convergence with the anti-apoptotic effect of thyroid hormone. Cell Cycle. 2009; 8:1877–1882.
42. Chin YT, Hsieh MT, Yang SH, Tsai PW, Wang SH, Wang CC, Lee YS, Cheng GY, HuangFu WC, London D, Tang HY, Fu E, Yen Y, et al. Anti-proliferative and gene expression actions of resveratrol in breast cancer cells in vitro. Oncotarget. 2014; 5:12891–12907.
43. Tang HY, Shih A, Cao HJ, Davis FB, Davis PJ, Lin HY. Resveratrol-induced cyclooxygenase-2 facilitates p53-dependent apoptosis in human breast cancer cells. Mol Cancer Ther. 2006; 5:2034–2042.
44. Misra S, Sharma K. COX-2 signaling and cancer: new players in old arena. Curr Drug Targets. 2014; 15:347–359.
45. Lin HY, Sun M, Tang HY, Simone TM, Wu YH, Grandis JR, Cao HJ, Davis PJ, Davis FB. Resveratrol causes COX-2- and p53-dependent apoptosis in head and neck squamous cell cancer cells. J Cell Biochem. 2008; 104:2131–2142.
46. Lin HY, Delmas D, Vang O, Hsieh TC, Lin S, Cheng GY, Chiang HL, Chen CE, Tang HY, Crawford DR, Whang-Peng J, Hwang J, Liu LF, et al. Mechanisms of ceramide-induced COX-2-dependent apoptosis in human ovarian cancer OVCAR-3 cells partially overlapped with resveratrol. J Cell Biochem. 2013; 114:1940–1954.
47. Lin HY, Tang HY, Davis FB, Davis PJ. Resveratrol and apoptosis. Ann NY Acad Sci. 2011; 1215:79–88.
48. Carter LG, D’Orazio JA, Pearson KJ. Resveratrol and cancer: focus on in vivo evidence. Endocr-Relat Cancer. 2014; 21:R209–225.
49. Laoag-Fernandez JB, Matsuo H, Murakoshi H, Hamada AL, Tsang BK, Maruo T. 3,5,3′-Triiodothyronine down-regulates Fas and Fas ligand expression and suppresses caspase-3 and poly (adenosine 5′-diphosphate-ribose) polymerase cleavage and apoptosis in early placental extravillous trophoblasts in vitro. J Clin Endocrinol Metab. 2004; 89:4069–4077.
50. D’Arezzo S, Incerpi S, Davis FB, Acconcia F, Marino M, Farias RN, Davis PJ. Rapid nongenomic effects of 3,5,3′-triiodo-L-thyronine on the intracellular pH of L-6 myoblasts are mediated by intracellular calcium mobilization and kinase pathways. Endocrinology. 2004; 145:5694–5703.
51. Davis PJ, Glinsky GV, Lin HY, Leith JT, Hercbergs A, Tang HY, Ashur-Fabian O, Incerpi S, Mousa SA. Cancer cell gene expression Modulated from Plasma Membrane Integrin alphavbeta3 by Thyroid Hormone and Nanoparticulate Tetrac. Front Endocrinol. 2014; 5:240.
52. Zhang L, Cooper-Kuhn CM, Nannmark U, Blomgren K, Kuhn HG. Stimulatory effects of thyroid hormone on brain angiogenesis in vivo and in vitro. J Cerebr Blood Flow Metab. 2010; 30:323–335.
53. Pantos C, Mourouzis I, Saranteas T, Clave G, Ligeret H, Noack-Fraissignes P, Renard PY, Massonneau M, Perimenis P, Spanou D, Kostopanagiotou G, Cokkinos DV. Thyroid hormone improves postischaemic recovery of function while limiting apoptosis: a new therapeutic approach to support hemodynamics in the setting of ischaemia-reperfusion? Basic Res Cardiol. 2009; 104:69–77.
54. Zhang C, Xia G, Tsang BK. Interactions of thyroid hormone and FSH in the regulation of rat granulosa cell apoptosis. Front Biosci. 2011; 3:1401–1413.
55. Yang B, He K, Zheng F, Wan L, Yu X, Wang X, Zhao D, Bai Y, Chu W, Sun Y, Lu Y. Over-expression of hypoxia-inducible factor-1 alpha in vitro protects the cardiac fibroblasts from hypoxia-induced apoptosis. J Cardiovasc Med. 2014; 15:579–586.
56. Lambert CM, Roy M, Robitaille GA, Richard DE, Bonnet S. HIF-1 inhibition decreases systemic vascular remodelling diseases by promoting apoptosis through a hexokinase 2-dependent mechanism. Cardiovasc Res. 2010; 88:196–204.
57. Mukherjee S, Samanta L, Roy A, Bhanja S, Chainy GB. Supplementation of T3 recovers hypothyroid rat liver cells from oxidatively damaged inner mitochondrial membrane leading to apoptosis. BioMed Res Int. 2014; 2014:590897.
58. Genovese T, Impellizzeri D, Ahmad A, Cornelius C, Campolo M, Cuzzocrea S, Esposito E. Post-ischaemic thyroid hormone treatment in a rat model of acute stroke. Brain Res. 2013; 1513:92–102.
59. Pietrzak M, Puzianowska-Kuznicka M. Triiodothyronine utilizes phosphatidylinositol 3-kinase pathway to activate anti-apoptotic myeloid cell leukemia-1. J Molec Endocrinol. 2008; 41:177–186.
60. Rebbaa A, Chu F, Davis FB, Davis PJ, Mousa SA. Novel function of the thyroid hormone analog tetraiodothyroacetic acid: a cancer chemosensitizing and anti-cancer agent. Angiogenesis. 2008; 11:269–276.
61. Nishio N, Katsura T, Inui K. Thyroid hormone regulates the expression and function of P-glycoprotein in Caco-2 cells. Pharmaceut Res. 2008; 25:1037–1042.
62. Burk O, Brenner SS, Hofmann U, Tegude H, Igel S, Schwab M, Eichelbaum M, Alscher MD. The impact of thyroid disease on the regulation, expression, and function of ABCB1 (MDR1/P glycoprotein) and consequences for the disposition of digoxin. Clin Pharm Ther. 2010; 88:685–694.
63. Li DW, Dong P, Wang F, Chen XW, Xu CZ, Zhou L. Hypoxia induced multidrug resistance of laryngeal cancer cells via hypoxia-inducible factor-1alpha. Asian Pacific J Cancer Prev. 2013; 14:4853–4858.
64. Saelim N, Holstein D, Chocron ES, Camacho P, Lechleiter JD. Inhibition of apoptotic potency by ligand stimulated thyroid hormone receptors located in mitochondria. Apoptosis. 2007; 12:1781–1794.
65. Guigon CJ, Kim DW, Zhu X, Zhao L, Cheng SY. Tumor suppressor action of liganded thyroid hormone receptor beta by direct repression of beta-catenin gene expression. Endocrinology. 2010; 151:5528–5536.
66. Kim DW, Zhao L, Hanover J, Willingham M, Cheng SY. Thyroid hormone receptor beta suppresses SV40-mediated tumorigenesis via novel nongenomic actions. Am J Cancer Res. 2012; 2:606–619.
67. Park JW, Zhao L, Webb P, Cheng SY. Src-dependent phosphorylation at Y406 on the thyroid hormone receptor β confers the tumor suppressor activity. Oncotarget. 2014; 5:10002–10016.
68. Ruiz-Llorente L, Ardila-Gonzalez S, Fanjul LF, Martinez-Iglesias O, Aranda A. MicroRNAs 424 and 503 are mediators of the anti-proliferative and anti-invasvie action of the thyroid hormone receptor beta. Oncotarget. 2014; 5:2918–2933.
69. Park JW, Zhao L, Willingham M, Cheng SY. Oncogenic mutations of thyroid hormone receptor β. Oncotarget. 2015; 6:8115–831.
70. Sar P, Peter R, Rath B, Das Mohapatra A, Mishra SK. 3,3′5 Triiodo L thyronine induces apoptosis in human breast cancer MCF-7 cells, repressing SMP30 expression through negative thyroid response elements. PLoS One. 2011; 6:e20861.
71. Fabian ID, Rosner M, Fabian I, Vishnevskia-Dai V, Zioto O, Maman ES, Cohen K, Ellis M, Lin HY, Hercbergs A, Davis PJ, Ashur-Fabian O. Low thyroid hormone levels improve survival in murine model for ocular melanoma. Oncotarget. 2015; Mar 4 [Epub ahead of print].
72. Hercbergs AA, Goyal LK, Suh JH, Lee S, Reddy CA, Cohen BH, Stevens GH, Reddy SK, Peereboom DM, Elson PJ, Gupta MK, Barnett GH. Propylthiouracil-induced chemical hypothyroidism with high-dose tamoxifen prolongs survival in recurrent high grade glioma: a phase I/II study. Anticancer Res. 2003; 23:617–626.
73. Sella A, Hercbergs AH, Hanovich E, Kovel S. Does sunitinib-induced hypothyroidism play a role in the activity of sunitinib in metastatic renal cell carcinoma? Chemotherapy. 2012; 58:200–205.
74. Schmidinger M, Vogl UM, Bojic M, Lamm W, Heinzl H, Haitel A, Clodi M, Kramer G, Zielinski CC. Hypothyroidism in patients with renal cell carcinoma: blessing or curse? Cancer. 2011; 117:534–544.
75. Cristofanilli M, Yamamura Y, Kau SW, Bevers T, Strom S, Patangan M, Hsu L, Krishnamurthy S, Theriault RL, Hortobagyi GN. Thyroid hormone and breast carcinoma. Primary hypothyroidism is associated with a reduced incidence of primary breast carcinoma. Cancer. 2005; 103:1122–1128.
76. Cao X, Kambe F, Yamauchi M, Seo H. Thyroid-hormone-dependent activation of the phosphoinositide 3-kinase/Akt cascade requires Src and enhances neuronal survival. Biochem J. 2009; 424:201–209.
77. Crupi R, Paterniti I, Campolo M, Di Paola R, Cuzzocrea S, Esposito E. Exogenous T3 administration provides neuroprotection in a murine model of traumatic brain injury. Pharmacol Res. 2013; 70:80–89.
78. Cohen K, Ellis M, Shinderman E, Khoury S, Davis PJ, Hercbergs A, Ashur-Fabian O. Relevance of the thyroid hormones-alphavbeta3 pathway in primary myeloma bone marrow cells and to bortezomib action. Leuk Lymphoma. 2014; 1–8.