
Introduction
Myeloid malignancies comprise three broad categories: acute myelogenous leukemia (AML) characterized by accumulation of immature myeloid cells in the bone marrow, myelodypastic syndromes (MDS) associated with ineffective hematopoiesis and chronic myeloproliferative disorders (MPDs), usually associated with an increased production of terminally differentiated myeloid cells. Despite this heterogeneity, all myeloid malignancies originate from progenitors that normally give rise to terminally differentiated cells of the myeloid series. Genetic and epigenetic events occurring early in hematopoietic stem cell maturation can lead to myeloid malignancies. Mutations can disrupt the normal course of hematopoiesis, resulting in a wide range of defects affecting the myeloid lineages [1].
Normal hematopoiesis involves a strict hierarchy of hematopoietic progenitor cells. The single primary source for the entire mammalian blood system is the hematopoietic stem cell (HSC), which is both multipotent and self-renewing in the absence of differentiation [2]. HSCs initially give rise to multipotent progenitors (MPPs), which have lost the ability to self-renew, but still maintain full-lineage differentiation potential [3]. MPPs can differentiate into common myeloid progenitors (CMPs), leading eventually to the generation of the myeloid lineage [2, 4-8].
Some myeloid malignancies are characterized by prominent and characteristic disease-causing mutations. For example, the vast majority of Polycythemia vera patients carry gain-of function mutations in Janus kinase 2 (JAK2 V617F) [9], while a hallmark of chronic myeloid leukemia (CML) is the generation of BCR-ABL fusion proteins [10]. On the other hand, two thirds of patients with myelodysplastic/myeloproliferative neoplasms (MDS/MPN) have a normal karyotype, but have several oncogenic mutations in a variety of genes encoding proteins involved in RNA splicing, chromatin modification, transcription or signaling [11]. A substantial fraction of MDS/MPN and their subtypes is characterized by the frequent occurrence of mutations in the gene encoding c-Cbl (Casitas B-lineage Lymphoma), a protein whose various enzymatic and non-enzymatic functions are now beginning to emerge.
Architecture and function of c-Cbl proteins
The attachment of ubiquitin to its client proteins regulates many different biological processes and occurs by a complex enzymatic cascade [12]. This process is critically dependent on ubiquitin activation by the E1 enzymes, followed by transfer of the activated ubiquitin to an ubiquitin-conjugating enzyme (E2) and finally, conjugation of ubiquitin to the target protein by an ubiquitin-protein ligase (E3). The E3 family of ubiquitin-protein ligases has a crucial role in substrate recognition and is of great importance in the (patho)physiology of intracellular signaling networks [13, 14]. Among E3 ligases, the small family of Cbl ubiquitin E3 ligases consists of the ubiquitously expressed proteins c-Cbl and Cbl-b as well as Cbl-c, which occur primarily in epithelial cells. All Cbl proteins share an N-terminal tyrosine-kinase-binding (TKB) domain that is followed by a RING (really interesting new gene) finger and a proline-rich domain [15]. The TKB domain is a modified SH2 (Src homology 2) domain that allows binding to phosphorylated tyrosines typically found in Cbl-interacting proteins. Cbl proteins are activated by phosphorylation at Y371 located in a linker region between the RING finger and the TKB domain [16]. This phosphorylation releases Cbl from its autoinhibited structure by triggering a conformational change that results in increased binding of the ubiquitin conjugating protein (or E2) to the RING finger of Cbl. This in turn leads to an enhanced transfer of ubiquitin from the E2 enzyme to the substrate proteins [17, 18]. The C-termini of Cbl-b and c-Cbl harbor extended proline-rich sequences and also a domain involved in ubiquitin binding and dimerization [19]. These domains allow for regulated protein/protein interactions via inducible association with SH2 and SH3 (Src homology 3) domain-containing proteins, as schematically displayed in Figure 1.
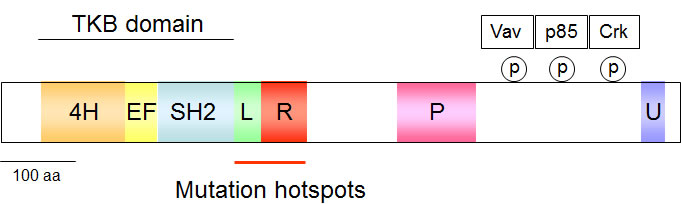
Figure 1: Schematic display of the c-Cbl structure. The various domains are given in colors, 4H: four helical bundle, EF: EF-hand calcium-binding domain, SH2: Src homology 2, L: linker helical region, P: proline-rich, U: Ubiquitin-binding domain. The phosphorylated tyrosines in the C-terminus allow docking of the indicated proteins and the frequently mutated region is highlighted.
Cbl proteins can exert multiple enzymatic functions. They can attach K48-branched polyubiquitin chains, resulting in proteasomal degradation of the decorated client proteins [20]. Cbl E3 ligases can also mediate the attachment of either K63-conjugated ubiquitin chains or single ubiquitin molecules to their substrate proteins to mediate non-proteolytic regulatory functions [21]. Recent evidence shows that c-Cbl also has the ability to mediate attachment of the ubiquitin-like protein NEDD8 (neural precursor cell expressed, developmentally down-regulated 8) to substrate proteins. It was shown that c-Cbl-mediated neddylation of the TGF-β type II receptor antagonizes its ubiquitination and degradation [22]. Currently, it is not clear what molecular events guide Cbl proteins to distinguish between these four distinct enzymatic functions.
In addition to these enzymatic activities, the various domains in Cbl proteins enable its function as an adapter by allowing constitutive or phososphorylation-dependent association with a plethora of different proteins [15, 23]. The dual function of c-Cbl as an enzyme and as an adapter protein is schematically displayed in Figure 2. Given the multiplicity of interaction partners and its various enzymatic functions it is not surprising that Cbl proteins affect signaling pathways involved in a wide range of processes, including the immune response, endocytic sorting, apoptosis and autophagy [24-26]. Single nucleotide polymorphism screens and sequencing showed that myeloproliferative neoplasms frequently show mutations in c-Cbl, but not in the other members of the Cbl family [27-29]. While mutations of c-Cbl are frequently discovered in myeloid neoplasms, mutations of the other Cbl family members, Cbl-b and Cbl-c, are hardly detected [30]. So far only five mutations within the RING finger domain of Cbl-b were reported, which likely disrupt its function as a ubiquitin E3 ligase [31, 32].
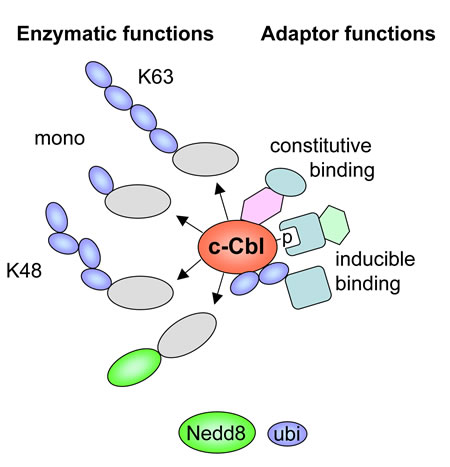
Figure 2: Schematic summary of c-Cbl functions as an adapter protein and as an enzyme. Inducible interactions by the ubiquitin binding domain and phosphotyrosine/SH2 interactions are shown. The various enzymatic functions mediating different modifications of the substrate proteins (grey) are displayed.
c-Cbl mutations in myelodysplastic and myeloproliferative neoplasms
Although approximately 10% of myeloid neoplasms show c-Cbl mutations, their frequency varies considerably between the subtypes. For example, while de novo AML has a relatively low frequency of such mutations at around 1-3%, juvenile myelomonocytic leukemia (JMML) and chronic myelomonocytic leukaemia (CMML) have the highest frequencies of c-Cbl mutations of around 13-15% [33-35]. Mutations in c-Cbl are typically nucleotide substitutions or small insertions/deletions that are heavily enriched in exons 8 and 9 and occur in the linker or RING finger domain of c-Cbl [27, 28, 36, 37]. Approximately 85% of the mutations are point mutations and approximately 15% are small deletions of the linker and RING finger regions. Approximately 10% of patients with MDS/MPN show c-Cbl mutations and frequently display clinical features such as splenomegaly, monocytosis and anemia [38]. Of the point mutations, about 70% are homozygous due to acquired uniparental disomy (aUPD), where both copies of a chromosome pair or parts of chromosomes have originated from one parent. Thus aUPD is a mechanism by which pathogenetic mutations in cancer may be reduced to homozygosity. Thus, an allelic conversion of the 11q arms leads to duplication of the mutated parental copy of 11q and loss of the remaining wild-type allele, resulting in homozygous c-Cbl mutations [36]. These mutations frequently result in the loss of ubiquitin E3 ligase activity [36], thus prohibiting lysosomal or ubiquitin/proteasome-mediated degradation of tyrosine kinases and thereby unleashing tyrosine kinase signaling. Therefore, although these mutations cause biochemical loss-of-function of c-Cbl, their functional consequences resemble classical gain-of-function mutations.
Mechanisms of c-Cbl mediated oncogenic transformation
The causal contribution of c-Cbl to the onset of myeloid malignancies was revealed in mouse models. c-Cbl-deficient mice are viable but show lymphoid hyperplasia, enhanced T-cell signaling and primary splenic extramedullary hemopoiesis [39]. In contrast, mice lacking Cbl-b do not share phenotypes with c-Cbl-null mice and are characterized by a reduced T-cell activation threshold. Mice with combined deletion of c-Cbl and Cbl-b in hematopoietic stem cells develop an early-onset lethal myeloproliferative disease within the first months after birth [40]. Mice engineered to express a c-Cbl RING domain point mutation (C379A) develop a myeloproliferative disease that progresses to leukemia [41], thus classifying c-Cbl mutations as driver mutations. This phenotype is even more severe than that of c-Cbl-deficient mice, suggesting that c-Cbl mutations act in a dominant negative manner and also affect the other endogenous members of the Cbl family. In addition to its function as a dominant negative protein mutant c-Cbl may also act as a positive signaling protein via its adaptor function. While a single c-Cbl point mutation can cause myeloid malignancies in mice, the situation is markedly different in humans. This difference is unlikely to be attributable to differences in expression levels or other experimental settings, as in humans over 10% of the tumors carry c-Cbl mutations together with 2-4 additional characteristic mutations [11, 37]. Furthermore, mutations at amino-acid residue C381 in human c-Cbl (equivalent to amino-acid residue C379 in mouse c-Cbl) amounts to ~0.8% of the mutations identified. The relative contribution of c-Cbl to development of MDS/MPN might be underestimated because some other genes known to be mutated or amplified in MDS/MPN, such as the duplicated gene encoding the SH3KBP1 (SH3-domain kinase binding protein) adapter protein, are part of the c-Cbl-regulated signaling network and may have the same effect as a c-Cbl mutation [42]. In addition, inactivation of c-Cbl may be mediated by events that do not affect the c-Cbl sequence. Phosphatases may keep c-Cbl in an inactivated state and increased auto-ubiquitination can limit the available protein amounts of the E3 ligase in tumor cells. Novel proteogenomic approaches for characterization of tumor cells will enable completely new insights in the relative protein abundance of c-Cbl and its client proteins [43]. In humans, c-Cbl mutations are not typical founding driver mutations allowing the first step in the leukemogenic process, but rather occur as subclonal secondary driver mutations, ensuring the further progress of the disease. Support for this model comes from the analysis of a patient with essential thrombocythemia, who had an intact c-Cbl gene at the early stage of disease, but acquired a c-Cbl R420Q mutation during disease progression to myelofibrosis [29]. The concept of c-Cbl mutations as secondary driver mutations is recapitulated in an animal model where transgenic mice expressing the oncogenic fusion protein NHD13 (NUP98-HOXD13) develop a myelodysplastic syndrome, which proceeds to acute leukemia. These animals pick up several secondary driver mutations in several genes including c-Cbl [44]. A deep sequencing study performed with a cell colony derived from a single cell detected ten mutations, compatible with the concept of a linear evolution of a dominant clone [34]. Recent advances in single cell genomics will now enable a detailed view into the stepwise acquisition of primary and secondary driver mutations [45]. It was suggested that c-Cbl mutations might indicate a poor clinical prognosis [38], but this is not consistently seen in other studies [46, 47]. It will be therefore important to consider also the prognostic value of these co-occurring secondary mutations in the future.
The importance of c-Cbl to myeloid differentiation and cancer is likely due to its involvement in numerous signaling pathways and biological functions. Mutation of c-Cbl can lead to aberrant activation of the JAK-STAT and PI3K-AKT pathways to transmit mitogenic and/or survival signals [48-50]. The mutant c-Cbl protein can also affect cytoskeleton organization via activation of Rac1 or Cdc42, and R-RAS [51] and affects integrin-mediated cell adhesion, spreading, and migration [52, 53]. Cbl proteins are important for the control of signaling thresholds in immune cells, and point mutations of c-Cbl consistently lead to enhanced granulocyte-macrophage colony-stimulating factor (GM-CSF)-triggered signaling via STAT5 and JAK2 [54, 55].
Cbl also contributes to myeloid differentiation by regulating the activity of colony stimulating factor-1 (CSF-1) receptor (CSF-1R). In the hematopoietic system, CSF-1 is believed to act specifically on myeloid progenitors, starting from the CMP stage, and to favour the differentiation of CMPs into the monocyte/macrophage lineage [56]. Indeed, transgenic mice engineered to secrete human CSF-1 show augmented frequencies and functions of human myeloid cells [57]. CSF-1 can directly induce the myeloid master regulator PU.1 and thus instruct myeloid cell-fate change in mouse HSCs both in vitro and in vivo [58]. Furthermore, intra-hepatic transfer of human fetal liver derived hematopoietic stem and progenitor cells (CD34+) in humanized CSF-1 newborn mice resulted in more efficient differentiation and enhanced frequencies of human monocytes/macrophages in the bone marrow, spleens, peripheral blood, lungs, liver and peritoneal cavity, pointing to its potential role in myeloid cell fate [57]. The magnitude and duration of signaling through activated CSF-1R is tightly regulated by its c-Cbl mediated ubiquitination-dependent down-regulation [59-61]. These c-Cbl-mediated effects on soluble factors such as CSF-1 raise the intriguing possibility that an oncogenic mutation in one tumor cell might affect neighbouring wildtype cells by paracrine mechanisms.
Future directions
Accumulating clinical evidence shows that progression mutations in c-Cbl and other key regulators occur during further clonal development of myeloid malignancies. Unlike the classical gain-of-function mutations exemplified by constitutive active JAK2 (V617F) or BCR-ABL [62], mutant c-Cbl has lost its enzymatic activity which renders it not an obvious drug target. This raises the need to identify druggable downstream components of the c-Cbl signaling pathways. Therefore, the downstream effectors of c-Cbl such as the JAK/STAT, PI3K, and ERK signaling pathways have been suggested as potential therapeutic targets. However, drugs acting on these signaling endpoints will not be specific for myeloid malignancies. As mutant c-Cbl proteins could display residual enzymatic activities as E3 ligases it may also be feasible to inhibit deubiquitinating enzymes that counteract c-Cbl. In order to develop drugs that are specifically tailored for the treatment of myeloid tumors with c-Cbl mutations we need a better understanding of the functional consequences of these mutations. The importance of this concept has been demonstrated by the use of mice with a c-Cbl RING finger mutation that develop a myeloproliferative disease progressing to leukemia. These mice exhibit augmented FLT3 (fms-related tyrosine kinase 3) signaling and inhibition of FLT3 kinase activity by quizartinib (AC220) effectively suppresses MPD development [41]. Deciphering how individual c-Cbl mutations affect its different enzymatic functions (neddylation, monoubiquitination, regulatory or proteolytic polyubiquitination) will provide therapeutic clues. As the activity of c-Cbl proteins is regulated by conformational changes [17, 18], it will be important to determine changes in the interactomes between wildtype and oncogenically mutated proteins. Also the intracellular localization of mutant c-Cbl and its posttranslational modifications need to be investigated, as phosphorylation of Y700 enables the interaction with further signal transmitting enzymes such as Vav1 [63], while phosphorylation at Y731 and Y774 allows binding of the p85 subunit of PI3K and the Crk-family of adapter proteins, respectively [48, 64]. Along this line, a comparative analysis of phosphoproteomes in cells expressing wildtype or mutant c-Cbl would help in the exploration of deregulated signaling pathways. New genetic tools such as inducibly expressed shRNAs or CRISPR-Cas9-mediated genome editing will enable synthetic lethality screens to identify druggable interactions between mutant c-Cbl and further components of the signaling network.
Acknowledgments
S.K. and M.L.S. are indebted to Dr. Susan Lewis for editing the manuscript.
Funding information
S.K. acknowledges the financial support by grants from the Israel Academy of Sciences, the Israel Cancer Research Foundation, the Israeli Cancer Association (ICA), with the generous assistance of the London friends of ICA in memory of the late Haim Yacobi, and the Hubert H. Humphrey Center for Experimental Medicine and Cancer Research. M.L.S. acknowledges the financial support by the Deutsche Forschungsgemeinschaft (projects DFG SCHM1417/9-1, SFB 1021/1, SFB/TRR81 and the Excellence Cluster Cardio-Pulmonary System ECCPS; EXC 147/2).
Conflicts of interest
None declared.
Abbreviations
AML: acute myelogenous leukemia; aUPD: acquired uniparental disomy; c-Cbl: Casitas B-lineage Lymphoma; CML: chronic myeloid leukemia; CMML: chronic myelomonocytic leukaemia; CMP: common myeloid progenitor; CSF-1R: colony stimulating factor-1 (CSF-1) receptor; FLT3: fms-related tyrosine kinase 3; GM-CSF: granulocyte-macrophage colony-stimulating factor; HSC: hematopoietic stem cell; JAK2: Janus kinase 2; JMML: juvenile myelomonocytic leukemia; MDS: myelodypastic syndromes; MDS/MPN: myelodysplastic/myeloproliferative neoplasms; MPDs:myeloproliferative disorders; MPPs:multipotent progenitors; NEDD8: neural precursor cell expressed, developmentally down-regulated 8; NHD13: NUP98-HOXD13; RING: really interesting new gene; SH2: Src homology 2; SH3: Src homology 3; SH3KBP1: SH3-domain kinase binding protein; TKB: tyrosine-kinase-binding.
References
1. Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, Harris NL, Le Beau MM, Hellstrom-Lindberg E, Tefferi A and Bloomfield CD. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009; 114: 937-951.
2. Chao MP, Seita J and Weissman IL. Establishment of a normal hematopoietic and leukemia stem cell hierarchy. Cold Spring Harb Symp Quant Biol. 2008; 73: 439-449.
3. Christensen JL and Weissman IL. Flk-2 is a marker in hematopoietic stem cell differentiation: A simple method to isolate long-term stem cells. Proceedings of the National Academy of Sciences of the United States of America. 2001; 98: 14541-14546.
4. Serwold T, Ehrlich LIR and Weissman IL. Reductive isolation from bone marrow and blood implicates common lymphoid progenitors as the major source of thymopoiesis. Blood. 2009; 113: 807-815.
5. Karsunky H, Inlay MA, Serwold T, Bhattacharya D and Weissman IL. Flk2(+) common lymphoid progenitors possess equivalent differentiation potential for the B and T lineages. Blood. 2008; 111: 5562-5570.
6. Akashi K, Traver D, Miyamoto T and Weissman IL. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature. 2000; 404:193-197.
7. Orkin SH and Zon LI. SnapShot: Hematopoiesis. Cell. 2008; 132: 712-U717.
8. Zandi S, Bryder D and Sigvardsson M. Load and lock: the molecular mechanisms of B-lymphocyte commitment. Immunological reviews. 2010; 238: 47-62.
9. Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, Vassiliou GS, Bench AJ, Boyd EM, Curtin N, Scott MA, Erber WN, Green AR and Cancer Genome P. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005; 365: 1054-1061.
10. Ben-Neriah Y, Daley GQ, Mes-Masson AM, Witte ON and Baltimore D. The chronic myelogenous leukemia-specific P210 protein is the product of the bcr/abl hybrid gene. Science. 1986; 233: 212-214.
11. Murati A, Brecqueville M, Devillier R, Mozziconacci MJ, Gelsi-Boyer V and Birnbaum D. Myeloid malignancies: mutations, models and management. BMC cancer. 2012; 12: 304.
12. Scheffner M, Nuber U and Huibregtse JM. Protein ubiquitination involving an E1-E2-E3 enzyme ubiquitin thioester cascade. Nature. 1995; 373: 81-83.
13. Lipkowitz S and Weissman AM. RINGs of good and evil: RING finger ubiquitin ligases at the crossroads of tumour suppression and oncogenesis. Nature reviews Cancer. 2011; 11: 629-643.
14. Metzger MB, Hristova VA and Weissman AM. HECT and RING finger families of E3 ubiquitin ligases at a glance. J Cell Sci. 2012; 125: 531-537.
15. Schmidt MHH and Dikic I. The Cbl interactome and its functions. Nat Rev Mol Cell Bio. 2005; 6: 907-918.
16. Ryan PE, Davies GC, Nau MM and Lipkowitz S. Regulating the regulator: negative regulation of Cbl ubiquitin ligases. Trends in biochemical sciences. 2006; 31: 79-88.
17. Dou H, Buetow L, Hock A, Sibbet GJ, Vousden KH and Huang DT. Structural basis for autoinhibition and phosphorylation-dependent activation of c-Cbl. Nature structural & molecular biology. 2012; 19: 184-192.
18. Kobashigawa Y, Tomitaka A, Kumeta H, Noda NN, Yamaguchi M and Inagaki F. Autoinhibition and phosphorylation-induced activation mechanisms of human cancer and autoimmune disease-related E3 protein Cbl-b. Proceedings of the National Academy of Sciences of the United States of America. 2011; 108: 20579-20584.
19. Davies GC, Ettenberg SA, Coats AO, Mussante M, Ravichandran S, Collins J, Nau MM and Lipkowitz S. Cbl-b interacts with ubiquitinated proteins; differential functions of the UBA domains of c-Cbl and Cbl-b. Oncogene. 2004; 23: 7104-7115.
20. Joazeiro CA, Wing SS, Huang H, Leverson JD, Hunter T and Liu YC. The tyrosine kinase negative regulator c-Cbl as a RING-type, E2-dependent ubiquitin-protein ligase. Science. 1999; 286: 309-312.
21. Mosesson Y, Shtiegman K, Katz M, Zwang Y, Vereb G, Szollosi J and Yarden Y. Endocytosis of receptor tyrosine kinases is driven by monoubiquitylation, not polyubiquitylation. The Journal of biological chemistry. 2003; 278: 21323-21326.
22. Zuo W, Huang F, Chiang YJ, Li M, Du J, Ding Y, Zhang T, Lee HW, Jeong LS, Chen Y, Deng H, Feng XH, Luo S, Gao C and Chen YG. c-Cbl-mediated neddylation antagonizes ubiquitination and degradation of the TGF-beta type II receptor. Mol Cell. 2013; 49: 499-510.
23. Tsygankov AY, Teckchandani AM, Feshchenko EA and Swaminathan G. Beyond the RING: CBL proteins as multivalent adapters. Oncogene. 2001; 20: 6382-6402.
24. Balagopalan L, Barr VA and Samelson LE. Endocytic events in TCR signaling: focus on adapters in microclusters. Immunological reviews. 2009; 232: 84-98.
25. Schmitz ML. Activation of T cells: releasing the brakes by proteolytic elimination of Cbl-b. Science signaling. 2009; 2: pe38.
26. Gay DL, Ramon H and Oliver PM. Cbl- and Nedd4-family ubiquitin ligases: balancing tolerance and immunity. Immunologic research. 2008; 42: 51-64.
27. Reindl C, Quentmeier H, Petropoulos K, Greif PA, Benthaus T, Argiropoulos B, Mellert G, Vempati S, Duyster J, Buske C, Bohlander SK, Humphries KR, Hiddemann W and Spiekermann K. CBL exon 8/9 mutants activate the FLT3 pathway and cluster in core binding factor/11q deletion acute myeloid leukemia/myelodysplastic syndrome subtypes. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009; 15: 2238-2247.
28. Dunbar AJ, Gondek LP, O’Keefe CL, Makishima H, Rataul MS, Szpurka H, Sekeres MA, Wang XF, McDevitt MA and Maciejewski JP. 250K single nucleotide polymorphism array karyotyping identifies acquired uniparental disomy and homozygous mutations, including novel missense substitutions of c-Cbl, in myeloid malignancies. Cancer research. 2008; 68: 10349-10357.
29. Grand FH, Hidalgo-Curtis CE, Ernst T, Zoi K, Zoi C, McGuire C, Kreil S, Jones A, Score J, Metzgeroth G, Oscier D, Hall A, Brandts C, Serve H, Reiter A, Chase AJ, et al. Frequent CBL mutations associated with 11q acquired uniparental disomy in myeloproliferative neoplasms. Blood. 2009; 113: 6182-6192.
30. Kales SC, Ryan PE, Nau MM and Lipkowitz S. Cbl and human myeloid neoplasms: the Cbl oncogene comes of age. Cancer research. 2010; 70: 4789-4794.
31. Caligiuri MA, Briesewitz R, Yu J, Wang L, Wei M, Arnoczky KJ, Marburger TB, Wen J, Perrotti D, Bloomfield CD and Whitman SP. Novel c-CBL and CBL-b ubiquitin ligase mutations in human acute myeloid leukemia. Blood. 2007; 110: 1022-1024.
32. Makishima H, Cazzolli H, Szpurka H, Dunbar A, Tiu R, Huh J, Muramatsu H, O’Keefe C, Hsi E, Paquette RL, Kojima S, List AF, Sekeres MA, McDevitt MA and Maciejewski JP. Mutations of e3 ubiquitin ligase cbl family members constitute a novel common pathogenic lesion in myeloid malignancies. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2009; 27: 6109-6116.
33. Klampfl T, Milosevic JD, Puda A, Schonegger A, Bagienski K, Berg T, Harutyunyan AS, Gisslinger B, Rumi E, Malcovati L, Pietra D, Elena C, Della Porta MG, Pieri L, Guglielmelli P, Bock C, et al. Complex patterns of chromosome 11 aberrations in myeloid malignancies target CBL, MLL, DDB1 and LMO2. PLoS One. 2013; 8: e77819.
34. Masuda S. Point mutations in myelodysplastic syndromes. The New England journal of medicine. 2011; 365: 1154; author reply 1155.
35. Murati A, Brecqueville M, Devillier R, Mozziconacci MJ, Gelsi-Boyer V and Birnbaum D. Myeloid malignancies: mutations, models and management. BMC cancer. 2012; 12: 304.
36. Sanada M, Suzuki T, Shih LY, Otsu M, Kato M, Yamazaki S, Tamura A, Honda H, Sakata-Yanagimoto M, Kumano K, Oda H, Yamagata T, Takita J, Gotoh N, Nakazaki K, Kawamata N, et al. Gain-of-function of mutated C-CBL tumour suppressor in myeloid neoplasms. Nature. 2009; 460: 904-908.
37. Naramura M, Nadeau S, Mohapatra B, Ahmad G, Mukhopadhyay C, Sattler M, Raja SM, Natarajan A, Band V and Band H. Mutant Cbl proteins as oncogenic drivers in myeloproliferative disorders. Oncotarget. 2011; 2: 245-250.
38. Schwaab J, Ernst T, Erben P, Rinke J, Schnittger S, Strobel P, Metzgeroth G, Mossner M, Haferlach T, Cross NC, Hochhaus A, Hofmann WK and Reiter A. Activating CBL mutations are associated with a distinct MDS/MPN phenotype. Annals of hematology. 2012; 91: 1713-1720.
39. Murphy MA, Schnall RG, Venter DJ, Barnett L, Bertoncello I, Thien CB, Langdon WY and Bowtell DD. Tissue hyperplasia and enhanced T-cell signalling via ZAP-70 in c-Cbl-deficient mice. Molecular and cellular biology. 1998; 18: 4872-4882.
40. Naramura M, Nandwani N, Gu H, Band V and Band H. Rapidly fatal myeloproliferative disorders in mice with deletion of Casitas B-cell lymphoma (Cbl) and Cbl-b in hematopoietic stem cells. Proceedings of the National Academy of Sciences of the United States of America. 2010; 107: 16274-16279.
41. Rathinam C, Thien CB, Flavell RA and Langdon WY. Myeloid leukemia development in c-Cbl RING finger mutant mice is dependent on FLT3 signaling. Cancer Cell. 2010; 18: 341-352.
42. Adelaide J, Gelsi-Boyer V, Rocquain J, Carbuccia N, Birnbaum DJ, Finetti P, Bertucci F, Mozziconacci MJ, Vey N, Birnbaum D and Chaffanet M. Gain of CBL-interacting protein, a possible alternative to CBL mutations in myeloid malignancies. Leukemia. 2010; 24: 1539-1541.
43. Zhang B, Wang J, Wang X, Zhu J, Liu Q, Shi Z, Chambers MC, Zimmerman LJ, Shaddox KF, Kim S, Davies SR, Wang S, Wang P, Kinsinger CR, Rivers RC, Rodriguez H, et al. Proteogenomic characterization of human colon and rectal cancer. Nature. 2014; 513:382-387.
44. Slape C, Liu LY, Beachy S and Aplan PD. Leukemic transformation in mice expressing a NUP98-HOXD13 transgene is accompanied by spontaneous mutations in Nras, Kras, and Cbl. Blood. 2008; 112: 2017-2019.
45. Macaulay IC and Voet T. Single cell genomics: advances and future perspectives. PLoS genetics. 2014; 10: e1004126.
46. Kao HW, Sanada M, Liang DC, Lai CL, Lee EH, Kuo MC, Lin TL, Shih YS, Wu JH, Huang CF, Ogawa S and Shih LY. A high occurrence of acquisition and/or expansion of C-CBL mutant clones in the progression of high-risk myelodysplastic syndrome to acute myeloid leukemia. Neoplasia. 2011; 13: 1035-1042.
47. Schnittger S, Bacher U, Alpermann T, Reiter A, Ulke M, Dicker F, Eder C, Kohlmann A, Grossmann V, Kowarsch A, Kern W, Haferlach C and Haferlach T. Use of CBL exon 8 and 9 mutations in diagnosis of myeloproliferative neoplasms and myelodysplastic/myeloproliferative disorders: an analysis of 636 cases. Haematologica. 2012; 97: 1890-1894.
48. Ueno H, Sasaki K, Honda H, Nakamoto T, Yamagata T, Miyagawa K, Mitani K, Yazaki Y and Hirai H. c-Cbl is tyrosine-phosphorylated by interleukin-4 and enhances mitogenic and survival signals of interleukin-4 receptor by linking with the phosphatidylinositol 3’-kinase pathway. Blood. 1998; 91: 46-53.
49. Grishin A, Sinha S, Roginskaya V, Boyer MJ, Gomez-Cambronero J, Zuo S, Kurosaki T, Romero G and Corey SJ. Involvement of Shc and Cbl-PI 3-kinase in Lyn-dependent proliferative signaling pathways for G-CSF. Oncogene. 2000; 19: 97-105.
50. Wang L, Rudert WA, Loutaev I, Roginskaya V and Corey SJ. Repression of c-Cbl leads to enhanced G-CSF Jak-STAT signaling without increased cell proliferation. Oncogene. 2002; 21: 5346-5355.
51. Gotoh T, Niino Y, Tokuda M, Hatase O, Nakamura S, Matsuda M and Hattori S. Activation of R-Ras by Ras-guanine nucleotide-releasing factor. The Journal of biological chemistry. 1997; 272: 18602-18607.
52. Meng F and Lowell CA. A beta 1 integrin signaling pathway involving Src-family kinases, Cbl and PI-3 kinase is required for macrophage spreading and migration. The EMBO journal. 1998; 17: 4391-4403.
53. Zell T, Warden CS, Chan AS, Cook ME, Dell CL, Hunt SW, 3rd and Shimizu Y. Regulation of beta 1-integrin-mediated cell adhesion by the Cbl adaptor protein. Current biology : CB. 1998; 8: 814-822.
54. Kotecha N, Flores NJ, Irish JM, Simonds EF, Sakai DS, Archambeault S, Diaz-Flores E, Coram M, Shannon KM, Nolan GP and Loh ML. Single-cell profiling identifies aberrant STAT5 activation in myeloid malignancies with specific clinical and biologic correlates. Cancer Cell. 2008; 14: 335-343.
55. Javadi M, Richmond TD, Huang K and Barber DL. CBL linker region and RING finger mutations lead to enhanced granulocyte-macrophage colony-stimulating factor (GM-CSF) signaling via elevated levels of JAK2 and LYN. The Journal of biological chemistry. 2013; 288: 19459-19470.
56. Sherr CJ, Rettenmier CW and Roussel MF. Macrophage colony-stimulating factor, CSF-1, and its proto-oncogene-encoded receptor. Cold Spring Harb Symp Quant Biol. 1988; 53 Pt 1: 521-530.
57. Rathinam C, Poueymirou WT, Rojas J, Murphy AJ, Valenzuela DM, Yancopoulos GD, Rongvaux A, Eynon EE, Manz MG and Flavell RA. Efficient differentiation and function of human macrophages in humanized CSF-1 mice. Blood. 2011; 118: 3119-3128.
58. Mossadegh-Keller N, Sarrazin S, Kandalla PK, Espinosa L, Stanley ER, Nutt SL, Moore J and Sieweke MH. M-CSF instructs myeloid lineage fate in single haematopoietic stem cells. Nature. 2013; 497: 239-243.
59. Xiong Y, Song D, Cai Y, Yu W, Yeung YG and Stanley ER. A CSF-1 receptor phosphotyrosine 559 signaling pathway regulates receptor ubiquitination and tyrosine phosphorylation. The Journal of biological chemistry. 2011; 286: 952-960.
60. Caveggion E, Continolo S, Pixley FJ, Stanley ER, Bowtell DD, Lowell CA and Berton G. Expression and tyrosine phosphorylation of Cbl regulates macrophage chemokinetic and chemotactic movement. Journal of cellular physiology. 2003; 195: 276-289.
61. Lee PS, Wang Y, Dominguez MG, Yeung YG, Murphy MA, Bowtell DD and Stanley ER. The Cbl protooncoprotein stimulates CSF-1 receptor multiubiquitination and endocytosis, and attenuates macrophage proliferation. The EMBO journal. 1999; 18: 3616-3628.
62. Quintas-Cardama A, Kantarjian H and Cortes J. Third-generation tyrosine kinase inhibitors and beyond. Seminars in hematology. 2010; 47: 371-380.
63. Marengere LE, Mirtsos C, Kozieradzki I, Veillette A, Mak TW and Penninger JM. Proto-oncoprotein Vav interacts with c-Cbl in activated thymocytes and peripheral T cells. J Immunol. 1997; 159: 70-76.
64. Hunter S, Burton EA, Wu SC and Anderson SM. Fyn associates with Cbl and phosphorylates tyrosine 731 in Cbl, a binding site for phosphatidylinositol 3-kinase. The Journal of biological chemistry. 1999; 274: 2097-2106.