
Introduction
Meningiomas are one of the most frequent primary brain neoplasias –30-35% of all cerebral nervous system (CNS) tumors–, which originate from the meningeal coverings of the brain and the spinal cord [1]. Despite most meningiomas correspond to histologically benign (i.e. WHO grade I) slow growing tumors [2], as a whole they display a broad spectrum of clinical, histological and cytogenetic features, even within the same WHO grade [3].
In recent years, important advances have been achieved in the identification and characterization of the genetic and molecular alterations of meningiomas and their association with the behavior of the disease. Consequently several chromosomal regions and candidate targeted genes have been identified. Monosomy 22/del(22q) in association or not with various mutations of the NF2 gene, is by far the most frequent cytogenetic event, potentially occurring at the early stages of the disease; however, other isolated chromosomal alterations and gene mutations, together with more complex karyotypes, have also been reported in meningiomas at relatively high frequencies, usually in association with a more aggressive tumor behavior [4].
Here we review the most relevant genetic and molecular alterations described so far in meningiomas, particularly focusing on the most frequently altered chromosomes, genes and signaling pathways.
Genetic alterations of chromosome 22 in meningiomas
Monosomy 22 in association or not with mutation of the NF2 gene, is by far the most frequent chromosomal alteration in meningiomas although other genes in this chromosome, as well as other chromosomes have also been found to be recurrent altered in these tumors (Table 1).
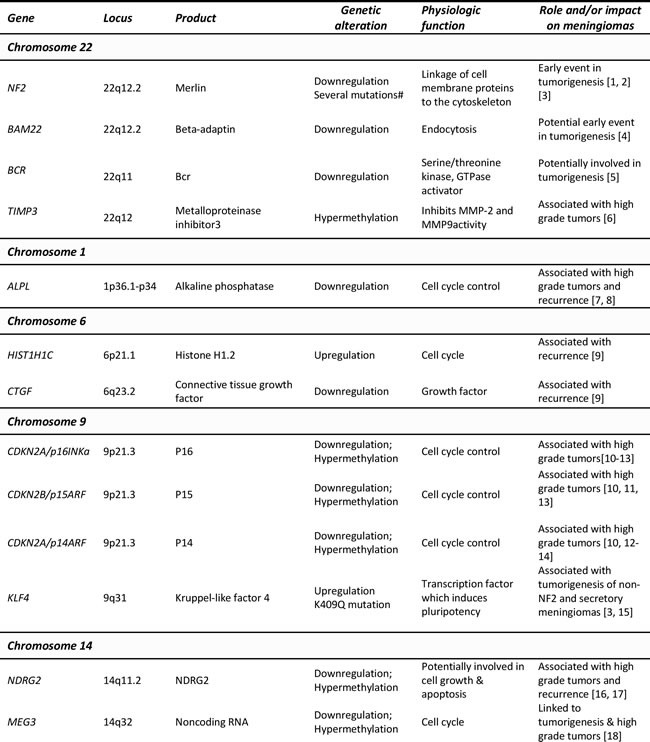
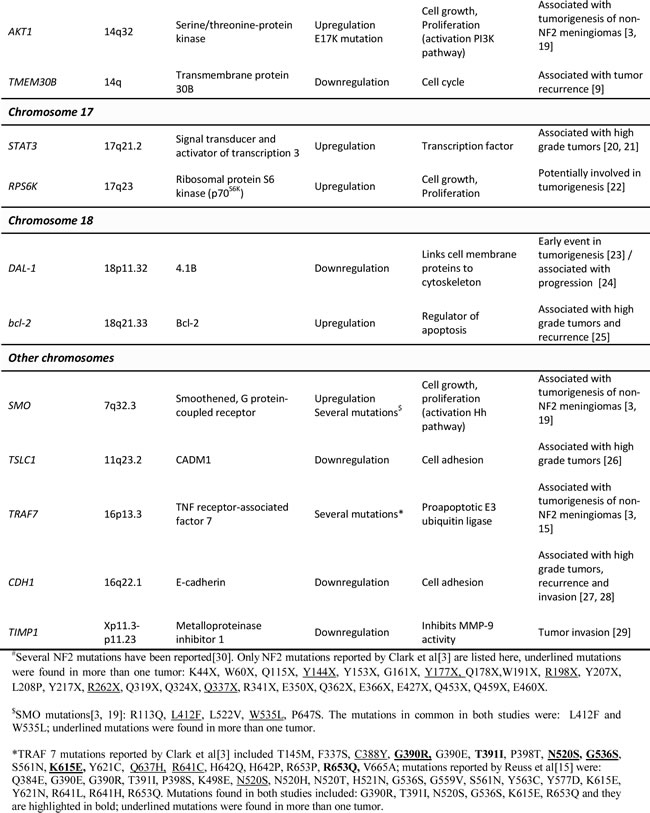
Table 1: Most relevant genes involved in the pathogenesis of meningiomas and coded by those chromosomes more frequently altered in these tumors: chromosomal localization, type of genetic alteration and function.
The NF2 gene and the merlin protein
A high proportion of meningiomas display recurrent genetic alterations of chromosome 22 and the NF2 tumor suppressor gene coded in the long arm of chromosome 22 (22q). Accordingly, monosomy 22 is found in around half of meningioma cases, the great majority of NF2-associated meningiomas, as well as between 40-70% of sporadic meningiomas, displaying allelic losses (loss of heterozygosity, LOH) at the 22q12.2 chromosomal region, where the NF2 gene is encoded [1, 3]. Additionally, up to 60% of these meningiomas carry inactivating mutations in the remainder NF2 allele [1, 5], consistent with the classical two-hit hypothesis of tumor suppressor gene inactivation. So far, a range of NF2 mutations have been reported in meningiomas most of which consist of small insertions, deletions, or nonsense mutations affecting the splicing sites [1, 6]. As the frequency of NF2 mutation is roughly equal among the different WHO grades, it has been considered a relevant genetic alteration in tumor initiation rather than in malignant progression [1, 3]. Despite Lomas et al. [7] have reported that the NF2 gene may be alternatively inactivated in meningiomas by aberrant promoter methylation, later studies indicated that methylation of the NF2 promoter does not play a major role in meningioma development [8, 9].
The merlin protein (also known as schwannomin) is the product of the NF2 gene. Merlin is a member of the 4.1 family of proteins which link integral membrane proteins to the cytoskeleton, and that are involved in the regulation of cell growth, proliferation and motility. Meningioma-associated NF2 mutations commonly result in a truncated, non-functional protein, which may lead to abnormal cell growth and motility through destabilization of adherens junctions. The main characteristic of cells lacking the NF2 protein is the loss of contact-mediated inhibition of cell proliferation [5]. Additionally, loss of merlin activity has been associated with increased levels of ErbB receptors in primary Schwann cells, which regulate downstream mitogenic signaling pathways (e.g. Ras/Raf/MEK/ERK and PI3K/AKT); altogether, these findings support a relevant role of merlin in tumorigenesis in meningiomas [10]. In line with this hypothesis, mice which are heterozygous for NF2 mutations more frequently develop metastatic tumors, and both in vivo and in vitro re-expression of wild type merlin leads to reduced tumor growth and decreased cell motility [3, 11].
Since the merlin functions include linking membrane proteins to the cytoskeleton, it has been hypothesized that alterations in merlin may substantially affect cell shape and might favor the appearance of a more mesenchymal-like phenotype rather than the epithelioid one, which is more commonly observed in wild type NF2 meningiomas [1]. Of note, several studies have reported different frequencies of NF2 mutation in meningiomas displaying distinct histopathological features; thus, alterations (e.g. monosomy) of chromosome 22q are more frequently observed in transitional and fibrous meningiomas than in the meningothelial variant [12, 13]. In addition, an association between the NF2 gene and tumor localization has also been reported; Kros et al. [13] demonstrated that tumors of the convexity are more prone to have NF2 mutations than anterior cranial-based tumors, and Clark et al. [14] recently correlated meningiomas with mutant NF2 and/or chromosome 22 loss with tumor localization in the cerebral and cerebellar hemispheres. In line with the unique features associated with NF2 mutation in meningiomas, an association with postmenopausal women tumors carrying monosomy 22 has also been reported recently [6].
Other candidate genes coded in chromosome 22
Although NF2 is the most frequently altered gene in chromosome 22, the frequency of deletions at this chromosomal region exceeds by far that of NF2 mutations in meningioma, suggesting that other genes encoded at chromosome 22 may also be involved in meningioma tumorigenesis. In this regard, an early report found BAM22 –a gene from the β-adaptin family coded at chromosome 22q12– to be inactivated in 9/71 meningiomas [15], and another more recent study found reduced expression of the BCR (breakpoint cluster region) gene coded at chromosome 22q11 in meningiomas with 22q LOH [16], further supporting the existence of candidate genes other than NF2 in the pathogenesis of meningiomas (Table 1).
Tissue inhibitors of metalloproteinases (TIMP) are proteins that regulate matrix metalloproteinases (MMP) and thereby also cell proliferation, apoptosis, and angiogenesis. The TIMP3 gene is coded at chromosome 22q12, is currently the best understood tumor suppressor gene being altered by epigenetic mechanisms in meningiomas [9]; of note epigenetic inactivation of TIMP3 has been recently associated with a more aggressive and higher-grade (grade II-III) meningioma phenotype [9, 17, 18].
Other relevant chromosomes and genes in meningiomas
In addition to monosomy 22/del(22q), other isolated and combined chromosomal alterations have also been identified in meningiomas. Thus, losses of chromosomes 1p, 10, 14/14q, and less frequently also of chromosomes 6q, 9p, 18q and the sex chromosomes, together with gains of chromosomes 1q, 9q, 12q, 15q, 17q, and 20q, have all been recurrently detected in a variable proportion of cases that overall account for around 30% of all meningiomas [1, 4, 19-22]. Of note, several genes coded in these chromosome have been recently identified by next generation sequencing as recurrently altered in meningiomas; among others, these include the AKT1 (E17K mutation), SMO (L412F and W535L mutations), KLF4 (K409Q mutation) and TRAF7 (several mutations mapped at the WD40 domains) genes [14, 23, 24]. Most interestingly, such mutations were shown to correlate with specific clinico-histopathological characteristics (e.g. tumor localization and histopathological subgroups) as well as with a subset of meningiomas lacking NF2 mutation, bringing new insight into NF2 non-mutated tumors [14, 23, 24].
Despite several other candidate genes have been proposed to be targeted by these chromosomal alterations, the specific relevant genes involved in many of them, still remain most frequently unknown.
Genetic alterations of chromosome 1
Chromosome 1p deletions comprise the second most common chromosomal alteration in meningiomas [1, 3]. Del(1p) is typically associated with more aggressive meningiomas, its frequency increasing from grade I (13-26%) to grade II (40-76%) and grade III (70-100%) tumors [3, 25]; from the clinical point of view, loss of chromosome 1p is also associated with higher tumor recurrence rates [26, 27] (Table 1). The most frequently targeted regions of chromosome 1p involve the 1p33-34 and 1p36 cytobands [4], where several candidate genes have been identified, e.g. TP73, CDKN2C, EPB41, GADD45A, and ALPL [3]. However, current results do not support a key role in tumorigenesis and/or malignant progression for most of these genes as meningioma-specific tumor suppressors namely CDKN2C, EPB41 and GADD45A, since they all failed to show consistent structural alterations. In contrast, although mutation of the TP73 gene has not been found, methylation-mediated inactivation of TP73 has been recurrently reported as frequent in meningiomas [17, 28]. Similarly, evidences exist about the potential role of the ALPL gene that encodes for the alkaline phosphatase enzyme at 1p36.1-34, as a tumor suppressor gene, because loss of chromosome 1p in meningiomas is strongly associated with loss of alkaline phosphatase activity [20], a predictor of meningioma recurrence [29]. Despite this, mutational analysis of ALPL has not been reported so far in the literature.
In turn, gains of chromosome 1q mainly involving two chromosomal regions (1q25.1 and 1q25.3 to 1q32.1), have been reported in around 60% of atypical meningiomas [30]. From the prognostic point of view, gains of chromosome 1q have been recurrently associated with a shorter progression-free survival [30]. However, detailed examination and identification of those genes with oncogenic potential which may be coded in this chromosome arm, still deserves further investigation.
Genetic alterations of chromosome 6
Genetic losses involving the long arm of chromosome 6 are a relatively common finding in meningiomas, particularly among high grade tumors; reported frequencies range from 9% of grade I to 25-33% of grade II and 50-63% of grade III meningiomas [4, 21, 25] (Table 1). A common deleted segment at chromosome 6q includes the 6q24.1-qter region, where the ESR1, IGF2R [4], DLL1, and CTGF [31] cancer-associated genes are encoded. However, the functional role of these genes and the impact of their alterations in meningiomas are still far from being fully understood. In addition to del(6q), Pérez-Magán et al. [31] also reported overexpression of the histone cluster 1 genes coded at chromosome 6p (e.g. the HIST1H1c gene) in 27% and 89% of primary and recurrent meningiomas, respectively; recent results suggest that physical interaction of the H1.2 protein could be involved in epigenetic regulation of gene expression by maintaining specific DNA methylation patterns, but its functional role in meningiomas still remains to be elucidated.
Genetic alterations of chromosome 9
Genetic losses at chromosome 9p have been reported in 5-17% of grade I, 18-52% of grade II, and 38-74% of grade III meningiomas [3] (Table 1). In contrast with other chromosomal alterations, the role of chromosome 9 in the development of malignant meningiomas is better defined, since it has been mostly associated with three tumor suppressor genes coded at chromosome 9p21: CDKN2A/p16INKa, CDKN2A/p14ARF and CDKN2B/p15ARF. Proteins coded by these three genes are all well known to play an important role in cell cycle regulation and apoptosis; thus p16INKa and p15ARF regulate cell cycle progression at the G1/S-phase checkpoint by inhibiting cyclin-cdk complexes, whereas p14ARF regulates apoptosis through a blockade of MDM2-mediated degradation of p53 [3]. In addition, homozygous deletion and somatic mutation of these genes have been reported in anaplastic meningiomas, supporting the notion that inactivation of cell cycle regulation is an important feature of malignant progression [32]. For example, Boström et al. [33] found homozygous deletions of CDKN2A/p16INKa, CDKN2B/p15ARF and CDKN2A/p14ARF in 46% of anaplastic vs. 3% of atypical meningiomas. In a similar way, Goutagny et al. [34] have recently shown by SNP-arrays that the most frequent genomic alteration of meningiomas upon progression to grade III was loss of CDKN2A/CDKN2B. Additionally, inactivation through hypermethylation of CpG islands has also been reported in a smaller proportion of meningiomas, including hypermethylation of CDKN2A/ p16INKa in 8-17% of cases, CDKN2A/p14ARF in 4-13%, and CDKN2B/p15ARF in 4% of these tumors [17, 35]. All such correlations also appear to have a prognostic impact since meningiomas with 9p21 losses have a significantly shorter survival and a worse clinical outcome than cases showing no alterations of chromosome 9p21 [36]. In addition to the CDKN2A/CDKN2B genes, the KLF4 (Kruppel like factor 4) gene has also been recently reported to be mutated in meningiomas (particularly among secretory tumors) in association with lack of NF2 mutation.
Genetic alterations of chromosome 10
Losses of part or the whole chromosome 10 are present in a significant proportion of meningiomas, their frequency increasing from grade I (5-12%) to grade II (29-40%), and grade III (40-58%) tumors [3, 25, 37]. In addition, LOH at specific regions of chromosome 10 have been associated with both higher recurrence rates and a poorer survival [37]. Some of the potential candidate genes encoded in such chromosomal regions (e.g. 10q23-q25) include the PTEN, MXI1 and DMBT1 genes, but studies have failed to identify frequent/recurrent mutations of these genes in meningiomas [38].
More recently, Dobbins et al. [39] identified a new susceptibility locus for meningioma at chromosome 10p12.31, which encompasses the MLLT10 (myeloid/lymphoid or mixed-lineage leukemia translocated to 10) gene; this gene is involved in chromatin remodeling and modulation of transcription. However, further investigations about the potential role of this gene in meningiomas are still needed.
Genetic alterations of chromosome 14
Overall, monosomy 14/del(14q) represents the third most common chromosomal alteration in meningiomas, being present in up to 31% of grade I, 40–70% grade II, and up to 100% of grade III meningiomas [40]. From the prognostic point of view, the chromosome 14q status has been identified as a prognostic indicator for tumor recurrence [40, 41]. Because of this, among other chromosome 14 regions, the 14q32 region has been suggested to be potentially relevant for meningioma progression. In this regard, Zang et al. [42] have identified the maternally expressed gene 3 (MEG3) coded in this chromosomal region, as a candidate tumor suppressor gene with antiproliferative activity. MEG3 is an imprinting gene that encodes for a non-coding RNA. In meningiomas, loss of MEG3 expression, its deletion at the genomic DNA and the degree of methylation of its promoter, have all been associated with higher tumor growth [43] (Table 1). In turn, functional studies have demonstrated that MEG3 mediates its anti-tumoral effect through inhibition of DNA synthesis, colony formation and proliferation of meningioma cell lines, being also found to transactivate p53 [42]. Altogether, these findings suggest that MEG3 may have an important role as a novel long non-coding RNA tumor suppressor in meningiomas. Recently, the AKT1 gene (coded at chromosome 14q32) has been reported to be mutated in meningiomas lacking NF2 mutation [14, 23], the reported mutation (E17K) resulting in constitutive AKT1 activation [23]; therefore, the AKT1 gene has become one of the most attractive target genes for cases with monosomy 14/del(14q).
Of note, the N-myc downstream-regulated gene 2 (NDRG2) has also been identified as a potential tumor suppressor gene coded at chromosome 14q11.2. Accordingly, NDRG2 was found to be frequently downregulated at both the transcript and the protein levels in anaplastic meningiomas and in a subset of lower-grade and atypical cases with an aggressive clinical behavior [44], as well as in recurrent meningiomas [45]. Reduced expression of NDRG2 appears to be closely associated with hypermethylation of its promoter [44]. Despite the precise mechanism of action of NDRG2 remains largely unknown, this gene has been involved in the regulation of cell growth, differentiation and apoptosis [9].
Genetic alterations of chromosome 17
Chromosome 17 gains and/or amplification of the 17q21-qter chromosomal region have been recurrently reported, mostly in malignant meningiomas [4]. RPS6K (ribosomal protein S6 kinase; p70S6K) is a proto-oncogene coded at chromosome 17q23, which has been found to be overexpressed at the protein level [46]; however, amplification of this gene appears to occur only in a small subset of higher grade meningiomas, even when amplification of the loci adjacent to this gene is present [47], suggesting that other genes coded in the vicinity of RPS6K may be the main targets for 17q amplification. In this regard, recent studies have also investigated the potential role of STAT3 (coded at 17q21.2), showing a higher frequency of enhanced expression of STAT3 with increasing tumor grade [48] (Table 1). Zhang et al. [48] further reported that constitutively active STAT3 was significantly associated with expression of the vascular endothelial growth factor (VEGF), a major inducer of tumor angiogenesis, and Johnson et al. [49] suggested that the cerebrospinal fluid itself may act as a stimulus for STAT3 phosphorylation/activation.
Genetic alterations of chromosome 18
Losses at chromosome 18q have been recurrently reported in meningiomas [3, 4, 50]; however, the specific targeted genes still remain to be identified. The expression of the bcl-2 oncoprotein coded at 18q21.3, has been associated with both a higher tumor grade and recurrence rate [51, 52]. In parallel, due to the role of the merlin protein in meningioma tumorigenesis, several studies have further investigated other members of the 4.1 family of membrane-associated proteins coded at chromosome 18q. These include the DAL-1 (differentially expressed in adenocarcinoma of the lung) gene which encodes for the 4.1B protein and has been claimed to act as a potential tumor suppressor gene in meningiomas [53]. Thus, loss of heterozygosity of DAL-1 at chromosome 18p11.32 was initially reported to occur in 60-76% of sporadic meningiomas [54], independently of the histological grade, suggesting that it could represent an early event in the pathogenesis of the disease (Table 1). However, more recent studies showed conflicting results. Thus, Yi et al. [55] reported that transgenic mice lacking DAL-1 do not develop tumors, and Nunes et al. [56] reported that only 12/62 (19%) meningiomas had LOH of DAL-1, 11 of such 12 cases also showing LOH of the NF2 gene. Altogether, these results suggest that DAL-1 may be involved in progression rather than initiation of meningiomas, as supported by the presence of monosomy 18 and/or del(18p) in 3/4 WHO grade II tumors vs. 2/13 WHO grade I meningiomas [56]. Other recent reports in meningiomas found no losses involving the genomic regions which contain the DAL-1 gene [8, 23], and another study found this gene to be mutated at a very low frequency [57].
Altered signaling pathways in meningiomas
At present, it is well known that most of the above described genetic alterations have an impact on one or more signaling pathways which are recurrently involved in cancer. The most relevant and frequently altered signaling pathways in meningioma are reviewed in this next section and illustrated in Figure 1.
The RB/p53 pathways and its impact on cell cycle dysregulation
RB has a central role in the inhibition of cell cycle progression at the G1/S-phase checkpoint. Briefly, RB binds (and inhibits) to the E2F transcription factor. Once cyclin D expression is upregulated (e.g. under mitogenic stimuli) it binds to either Cdk4 or Cdk6, and phosphorylates RB; RB phosphorylation induces release of the active E2F factor, leading to the transcription of genes which are critical for the transition from the G1 to the S-phase. p16INK4a and p15INK4b prevent S-phase entry by inhibiting the Cdk4/cyclin D complex [3]. In turn, the p53 pathway acts as a feedback inhibitor of the RB pathway, by inducing cell cycle arrest, DNA repair and apoptosis in case of aberrant RB pathway activation (Figure 1). The RB and p53 pathways are connected via p14ARF. Release of the E2F transcription factor following RB phosphorylation, also induces transcription of p14ARF, which promotes p53 activity through negative regulation of the MDM2 (murine double minute 2 protein) proto-oncogene. Dysregulation of these two pathways in higher-grade meningiomas is frequently associated with loss of p16INK4a, p15INK4b and p14ARF, increased cell proliferation and tumor progression [33, 34]. In addition, accumulating evidences indicate that hypermethylation- associated loss of function of RB [8, 58], overexpression of the MDM2 gene/protein [25, 26] and loss of expression of MEG3 (an anti-proliferative tumor suppressor that induces activation of p53 by a transcriptional effect) [42] in higher grade meningiomas, might further contribute to dysregulation of both cell cycle-associated pathways during meningioma progression.
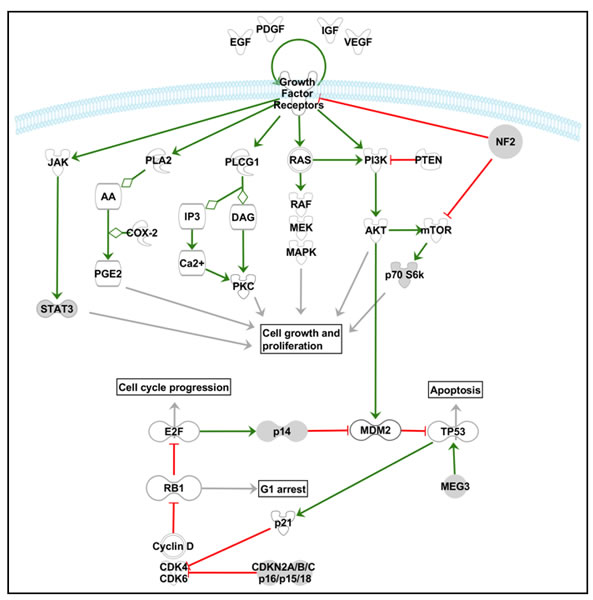
Figure 1: Schematic diagram illustrating the key elements of some of the most relevant signaling pathways involved in the pathogenesis of meningiomas. The Ras-Raf-MEK-MAPK/ERK, PI3K-Akt/PKB, PLCγ1-PKC, PLA2-COX, JAK-STAT3 and mTOR signaling pathways are represented in the upper part of the scheme, while the relationships between the pRb and p53 cell cycle associated signaling pathways are illustrated in the lower part of the scheme. The pathway scheme displayed was generated with the Ingenuity Pathway Analysis (IPA) software (Ingenuity Systems Inc., Redwood City, CA, USA).
Growth factors and autocrine loops
Multiple studies have demonstrated enhanced expression of several growth factors, and activation of autocrine loops, which act as extra- and intracellular signals that induce tumor growth, cell migration, and angiogenesis, mostly via the MAPK and PI3K/Akt signaling pathways (Figure 1). Among others, the platelet-derived growth factor BB (PDGF-BB) and its PDGFR-β receptor have been found to be frequently overexpressed in meningiomas (typically at greater levels in high vs. low grade tumors), leading to meningioma cell proliferation via an autocrine and/or paracrine loop. Similarly, the epidermal growth factor receptor (EGFR), and both their EGF and transforming growth factor-alpha (TGFα) ligands, as well as some members of the insulin-like growth factor (IGF) system (e.g. IGF2) [59], have all been associated with meningioma cell proliferation and tumor progression. Interestingly, Lallemand et al. [10] reported that merlin regulates cell contact-mediated inhibition of proliferation by limiting the delivery of several growth factor receptors (e.g. ErbB2, ErbB3, IGF1R and PDGFR) at the plasma membrane of primary Schwann cells, and thereby inhibit the activity of the downstream mitogenic signaling pathways.
VEGFA and its VEGFR-1 receptor have been associated with regulation of the development of new blood vessels and peritumoral edema in brain tumors, a common feature in meningioma patients [3]. Of note, meningiomas express both VEGF and VEGFR, and VEGF expression correlates with the severity of peritumoral edema [60, 61] and tumor vascularization [61]. Despite this, the precise mechanisms that regulate VEGF expression in meningiomas remain unknown. In human cells, VEGF is mainly regulated by the hypoxia inducible factor-1 (HIF-1) transcription factor and in meningiomas, HIF-1 expression correlates with VEGF expression and the degree of peritumoral edema. In addition, upregulation of VEGF expression can also be induced by other growth factors such as EGF and PDGF, suggesting that both growth factors and hypoxia stimulation may all contribute to control VEGF expression in these tumors.
Other growth factors that have been associated with the pathogenesis of meningiomas include: 1) the stromal cell-derived factor 1 (SDF1) CXC chemokine and its CXCR4 receptor, which might exert its mitogenic effects through the MAPK pathway [62]; 2) the bone morphogenic proteins (BMPs) and their receptors (BMPR), which are associated with Smad 1 signaling, and; 3) the fibroblast growth factor (FGF) and its FGFR3 receptor, which are activated by the PI3K/Akt pathway [63]. In contrast, TGF-β and its receptors (TGF-βRI and TGF-βRII) may act as potential inhibitors of meningioma growth/proliferation through the Smad 2/3 apoptotic pathway [64], although the role of TGF-β in meningioma tumorigenesis remains to be fully established.
The MAPK and PI3K/Akt signaling pathways
The mitogen-activated protein kinase (MAPK) pathway and the phosphatidylinositol 3-kinase (PI3K)/Akt pathway are both involved in multiple cellular processes (e.g. differentiation, growth, and apoptosis) associated with the pathogenesis of meningiomas, particularly with those tumors showing deregulated cell proliferation [3]. MAPKs are intracellular serine/threonine-specific protein kinases which are activated by extracellular stimuli (e.g., mitogen signals), leading to sequential activation of a kinase cascade triggered by the Ras/Raf-1/MEK-1/MAPK/ERK pathway and that ultimately, leads to phosphorylation/activation of transcription factors in the nucleus [64]. PI3Ks are a family of intracellular signal transducer enzymes that phosphorylate inositol phospholipids. Activation of PI3K results in phosphorylation/activation of PKB/Akt and subsequently p70S6K, which are key elements of the cell growth-promoting effects of this pathway; alternatively, activating mutations of AKT have also been recently reported in a subset of meningiomas [64]. In line with this, Johnson et al. [63] found evidences for the activation of both the MAPK and the Akt/PKB pathways in meningiomas, upon growth factor receptor signaling via e.g. PDGF-BB and PDGFβ; furthermore, these authors showed that administration of MAPK or PI3K inhibitors induces progressive growth inhibition of meningioma cells in association with reduced phosphorylation of MAPK or Akt and p70S6K, respectively. Interestingly, Mawrin et al. [65] have also found higher levels of phospho-Akt/PKB in association with lower levels of activation of MAPK in anaplastic and atypical vs. benign meningiomas; moreover, in vitro studies revealed decreased meningioma cell growth in the absence of apoptosis induced by a PI3K blocker, whereas inhibition of MAPK resulted in cell death through apoptosis [65]; based on these findings, the authors hypothesized that PI3K/Akt activation is associated with uncontrolled growth in malignant meningiomas, whereas MAPK activation appears to be involved in both meningioma cell proliferation and apoptosis [65].
The PLCγ/PKC-calcium signaling pathway
Tyrosine kinase receptors also activate (e.g. phosphorylate) phospholipase C-γ1 (PLC-γ1), leading to hydrolysis of PIP2 (phosphatidylinositol 4,5-biphosphate) into two intracellular active second messengers: IP3 (inositol 1,4,5-triphosphate) and 1,2-DAG (1,2-diacylglycerol) (Figure 1). DAG activates protein kinase C (PKC), which enters the nucleus and activates transcription factors, resulting in cell proliferation and inhibition of apoptosis [64]. The activation of the EGFR kinase in meningioma cells further activates PLC-γ1 and increases its catalytic activity, leading to another mechanism that promotes meningioma cell growth; additional evidences indicate that PLCγ expression does not differ significantly between meningiomas of different histopathological grades [65]. In turn, IP3 mediates calcium release from intracellular stores resulting in increased free cytosolic calcium. Interestingly, calcium channel antagonists can block in vitro primary meningioma cell growth after stimulation with EGF and PDGF, as well as in vivo meningioma growth in a subcutaneous meningioma mouse model [66]; the specific mechanisms involved in such blockade of IP3-mediated intracellular calcium signaling pathways in meningiomas, still deserves further investigation.
The cyclooxygenase-2 signaling pathway
The phospholipase A2 (PLA2)-cyclooxygenase (COX) signaling pathway has also been recently investigated in meningiomas [64]. COX-2 is an enzyme that serves as the rate-limiting step for the synthesis of prostaglandins from arachidonic acid. Prostaglandins (e.g. PGE2) are mediators of several critical cellular processes such as cell growth, proliferation, angiogenesis, suppression of apoptosis, and inflammation [3]. Normally, the cytoplasmic levels of arachidonic acid are relatively low, which limits the production of prostaglandins. However, altered levels of arachidonic acid and COX-2 overexpression have been associated with cancer growth and progression, possibly driven by signaling pathways such as those involving growth factors and the MAPK pathway [64]. Of note, high levels of arachidonic acid, increased prostaglandin production, as well as COX-2 overexpression [26, 67, 68], have all been reported in meningiomas. Moreover, COX-2 expression has been correlated with a greater degree of invasiveness to the brain or the adjacent soft tissues [67], tumor recurrence [26], a higher MIB-1 labeling index [68] and VEGF levels [67, 68], suggesting it may play an important role in the development and growth of meningiomas.
The mTOR signaling pathway
Recent studies have found the mammalian target of rapamycin (mTOR) to be also involved in the signaling pathways associated with meningioma tumorigenesis [69, 70]. mTOR is a protein kinase that may be expressed in two distinct complexes (mTORC1 and mTORC2). mTORC1 regulates cell growth by promoting increased translation and protein synthesis through phosphorylation of the p70S6K and 4EBP1 (eukaryotic translation initiation factor 4E-binding protein 1) effector proteins; in turn, mTORC2 directly phosphorylates Akt, a step required for its full activation [1, 3]. Recently, merlin has been identified as a negative regulator of mTORC1 and, James et al. [69] have demonstrated that mTORC1 levels are elevated in tumors derived from patients with NF2 disease and in fibroblasts from an NF2-deficient mouse model. Thus, activation of mTORC1 has been associated with meningioma growth [69], and mTORC1 inhibitors have been shown to suppress meningioma growth in mouse models [70]. However, the exact mechanism through which merlin inhibits mTORC1 still remains unclear. In contrast to its effects on mTORC1, merlin positively regulates the kinase activity of mTORC2, downstream phosphorylation of mTORC2 substrates, including Akt, being reduced upon acute merlin deficiency in cells. Nevertheless, the attenuated mTORC2 signaling profiles reported in response to the loss of merlin, could not be detected in NF2-deficient meningiomas [69].
The WNT/β-catenin pathway
The wingless (wnt)/β-catenin pathway has also been implicated in meningioma progression, through an altered expression of several of its genes. Thus, early studies based on microarray gene expression profiling identified increased expression of genes such as CTNNB1, CDK5R1, ENC1 and CCND1 [59]. Subsequently, Pecina-Slaus et al. reported LOH of the E-cadherin (CDH1) [71] and the adenomatous polyposis coli (APC) tumor suppressor genes in about one-third and half of the cases, respectively. Downregulation of E-cadherin expression in clinically aggressive and invasive meningiomas had already been described [72] in association with upregulation and nuclear/perinuclear localization of β-catenin [71], suggesting an important role for the WNT/β catenin pathway in meningioma tumorigenesis. Interestingly, Zhou et al. [73] suggested a model in which active merlin would inhibit Wnt/β-catenin signaling and maintain β-catenin and N-cadherin complexed at the plasma membrane; loss of merlin would then lead to loss of contact inhibition and activation of Wnt/β-catenin signaling, translocation of β-catenin to the nucleus and expression of intracellular growth-associated proteins such as c-myc and cyclin D1.
Interestingly, Pérez-Magán et al. [74] recently reported a gene expression profiling (GEP) signature of advanced and recurrent meningiomas, which included aberrant expression of genes of the Wnt pathway; thus, these authors found downregulation of SFRP1, a gene from the SFRPs (secreted frizzled-related proteins) protein family which are able to downregulate Wnt signaling, in recurrent and atypical meningiomas. Similarly, the BCR gene, which typically shows lower expression in meningiomas with LOH at chromosome 22q [16], has also been shown to act as a negative regulator of the Wnt pathway [75].
The notch signaling pathway
The notch signaling pathway is involved in extracellular-to-intracellular signaling via the notch1-4 transmembrane proteins. Ligand proteins bind to the extracellular portion of the Notch proteins, resulting in proteolytic cleavage and release of the intracellular portion, which is translocated to the nucleus and initiates expression of the hairy/enhancer of split (HES) family of transcriptional regulators [3]. Cuevas et al. [76] comparatively analyzed the GEP of normal/reactive meninges and meningiomas of all histopathological grades, and demonstrated the potential involvement of the notch signaling pathway in meningiomas. Thus, HES1 expression was increased in all meningioma grades and it correlated with increased expression of notch1, notch2, and the jagged ligand [76]; in contrast, transducin-like enhancer of split (TLE) 2 and TLE3, two co-repressors that modulate HES1 activity, were specifically upregulated in malignant meningiomas [76]. Furthermore, deregulation of notch in meningiomas results in tetraploidy and chromosomal instability [77], further studies being required to elucidate the precise mechanism by which abnormal notch activation induces such genetic changes in meningiomas.
The hedgehog (Hh) signaling pathway
When Hh binds its patched (PTCH) receptor, the smoothened (SMO) transmembrane protein is activated and initiates a signaling cascade that results in the activation of the GLI transcription factors (e.g. GLI1 and GLI2) and subsequent transcription of genes involved in cell growth, proliferation, angiogenesis, matrix remodeling, and stem cell homeostasis [3]. Recently, Laurendeau et al. [78] have analyzed the mRNA expression patterns of 32 Hh pathway-related genes in 36 meningiomas and found increased levels of 16 genes involved in the activation of the Hh pathway (e.g. SMO, GLI1, GLI2, GLIS2, FOXM1, IGF2 and SPP1) and cell growth, together with decreased expression of 7 genes involved in the inhibition of the Hh pathway (e.g. the PTCH1 tumor suppressor); some of these genes further showed different expression profiles among tumors of different histopathological grades, suggesting distinctly altered profiles early during tumorigenesis vs. progression to more aggressive tumor lesions. Interestingly, recent reports have identified SMO mutations in meningiomas lacking NF2 mutations [14, 23], which further supports the potentially relevant role of this pathway in the development of at least some meningiomas.
Cytogenetic subgroups of meningiomas and their association with tumor progression
Based on all what has been described above, at present it is well-established that meningiomas are cytogenetically heterogenous tumors. Consequently, for decades now, attempts have been made to classify meningiomas on cytogenetic grounds (Figure 2). The first cytogenetic classifications and cytogenetic models of progression of meningiomas were proposed in the 1990s [25]. Weber et al. [25] proposed a model of genomic alterations associated with meningioma progression based on comparative genomic hybridization (CGH) analysis of tumors of different grades (Figure 2). Later on, Ketter et al. [19] and Zang et al. [20], subdivided meningiomas into four subgroups based on their cytogenetic findings: Group 0, included meningiomas with a normal diploid chromosomal set; Group 1, consisted of tumors with monosomy 22 as the sole cytogenetic alteration; Group 2, was composed of tumors showing marked hypodiploidy with loss of additional autosomes, and finally; Group 3 included meningiomas with deletions of the short arm of chromosome 1, in association with other chromosomal aberrations such as loss of chromosome 22. Later on, these same authors applied oncogenetic tree mixtures to identify recurrent oncogenetic routes based on specific sequences of accumulation of somatic chromosomal changes in tumor cells (Figure 2); based on the routes identified, a genetic progression score (GPS) was built and used to categorize meningiomas into three groups of increasingly higher genetic complexity [79]. Subsequently, intratumoral cytogenetic patterns of clonal evolution have also been evaluated to establish recurrent pathways of cytogenetic progression at the single level, within individual tumors (Figure 2). Based on these later studies, complete or partial loss of either chromosome 22, a sex chromosome (i.e. chromosome Y in males and chromosome X in females), del(1p) or less frequently, monosomy 14/14q- alone or in combination with other chromosomal losses (e.g., monosomy 10/10q- and 18/18q-), would frequently represent the earliest detectable chromosomal alteration in meningioma cells. Interestingly, despite the clear association observed between a more advanced tumor grade and a higher number of tumor cell clones and complex karyotypes, authors found that the pathways of intratumoral clonal evolution observed in benign meningiomas were markedly different from those most frequently found in atypical/anaplastic tumors; altogether, these results suggest that the latter tumors might not always represent a more advanced stage of histologically benign meningiomas, but they could more likely correspond to stages of distinct clonal evolution pathways (Figure 2). For example, while monosomy 22 is commonly present in the earliest tumor cell clone observed in grade I meningiomas, it was only detected in a minor proportion of all atypical/anaplastic tumors; in contrast, presence of isolated losses of a sex chromosome, del(1p) and monosomy 14 in the ancestral tumor cell clone were characteristic of atypical/anaplastic tumors (Figure 2). In fact, although cytogenetic progression from low- to high-grade meningiomas may occur [22], it remains controversial [1] and the precise pathways of clonal evolution remain to be identified. This is due, at least in part, to the fact that data used to explain stepwise progression (e.g. cumulative acquisition of genetic alterations leading to more aggressive subclones), typically derives from cytogenetic analyses of different tumors of distinct grades, and from different patients. In contrast, studies addressing this question through the follow-up of patients showing tumor recurrence mostly reported no (or only minor) cytogenetic changes at recurrence, even after progression to a higher histopathological tumor grade [80].
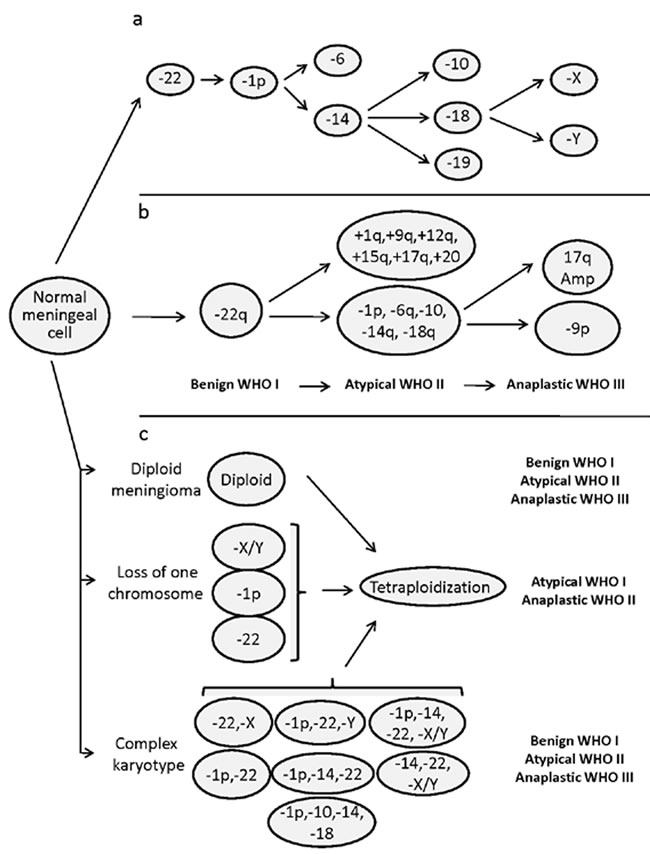
Figure 2: Patterns of cytogenetic alterations proposed to reflect cytogenetic progression of meningiomas, according to Ketter et al. [79] (panel a), Weber et al. [25] (panel b), and Sayagues et al. [86] (panel c). Panel a, oncogenetic tree mixture model for the acquisition of chromosomal alterations in the development of meningiomas; the first two critical steps in the progression model correspond to monosomy 22, followed by loss of the short arm of chromosome 1. In the Weber et al model (panel b), progression from grade I to grade III is proposed to occur in parallel to the acquisition of specific chromosomal gains and losses at frequencies of more than 30% of cases; nevertheless, chromosomal changes may already have occurred in a lower grade in a smaller percentage of tumors. Finally, hypothetical intratumoral aneuploidization pathways defined on the basis of the patterns of clonal evolution observed for 11 chromosomes analyzed at the single cell level for individual tumors by Sayagues et al. (panel c).
From a clinical point of view, both the cytogenetic and genomic profile of meningiomas has recurrently been associated with the behavior of the disease [50, 80-86] (Table 2). Thus, both tumors carrying monosomy 22 and those displaying diploid karyotypes, have been associated with a better outcome [87] than that of cases with more complex karyotypes who show shorter recurrence-free survival rates [41, 88, 89]. Most interestingly, simultaneous presence of monosomy 14 and del (1p) in the ancestral tumor cell clones has been shown to be an adverse independent prognostic marker for disease-free survival in meningiomas [41].
Table 2: Genetic markers of meningiomas which have been associated with an independent prognostic value on patient relapse-free survival (RFS).
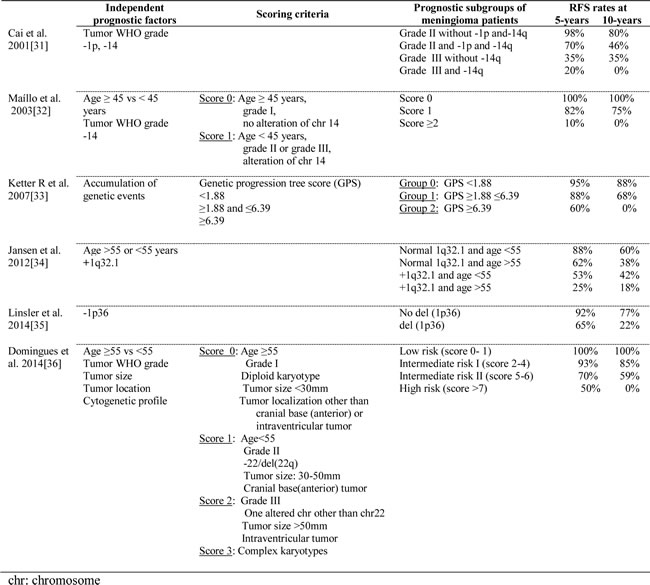
In parallel, single nucleotide polymorphism (SNP)-array studies have confirmed and extended the available information about the distinct cytogenetic subgroups of meningiomas [4, 21, 34]. Thus, Lee et al. [21] described 5 ‘classes’ of meningiomas based on gene expression analyses that showed a high correlation with their copy number alteration profile by SNP-arrays, as well as the tumor recurrence status and histopathology. Similarly, Tabernero et al. [4] confirmed by SNP-arrays that meningiomas can be classified into 3 major cytogenetic subgroups: diploid cases, meningiomas with a single chromosomal change [e.g. monosomy 22/22q-] and tumors with complex karyotypes including cases with ≥2 altered chromosomes. Of note, such cytogenetic classification of meningiomas was recently shown to be the most powerful independent predictor of outcome (e.g. 10–year recurrence-free survival rates of 91±6%, 90±4%, 67±8%, respectively) particularly when evaluated in combination with patient age and tumor size, localization and WHO grade [90].
Regarding GEP, Watson et al. [91], in a pioneering study in meningiomas, reported GEP of tumor cells to be associated with the WHO grade. Such association has been subsequently confirmed by others [59, 91, 92], and Fevre-Montange et al [92] and Serna et al [84] also reported an association of GEP with the main histopathological subtypes of grade I meningiomas. Similarly, unique GEP of meningioma cells have also been associated with tumor localization (e.g. spinal vs. intracranial tumors) [93], patient gender [94] and clinically relevant cytogenetic subgroups of meningiomas (i.e. diploid tumors vs cases with isolated monosomy 22/del(22q) vs meningiomas with complex karyotypes) [50].
Most interestingly, GEP of meningiomas have shown the existence of heterogeneous profiles which are associated with a different tumor behavior, even within the same WHO grade; thus, GEP of tumor cells from aggressive and/or invasive meningiomas emerged as being unique [44, 74, 84, 92] and typically associated with both high-proliferative gene expression signatures [95] and a high-risk of recurrence [50, 74, 84]. In this regard, Carvalho et al. [95] showed that meningiomas fall into two main molecular subgroups designated as ‘low-proliferative’ and ‘high-proliferative’ meningiomas, according to their different GEP and median MIB-1 labeling indices. Similarly, Perez-Magan et al. [74] identified a 49-gene signature of meningioma aggressiveness that characterizes histologically benign meningiomas which may recur.
Concluding remarks
Important advances have been achieved in the last decades in our understanding of the genetic/chromosomal alterations of meningiomas. Thus, current knowledge point out to the existence of several different pathways of tumorigenesis and clonal evolution which may target different genes in low vs high-grade tumors, as well as among low grade meningiomas. Thus, although NF2 mutation in association with monosomy 22 is the most frequently observed alteration of a single gene, early mutations involving genes other than NF2 (e.g. AKT1, SMO, TRAF7, KLF4) have recently emerged as alternative oncogenic pathways in NF2 non-mutated tumors. In addition, other genetic and/or chromosomal alterations are present in around one third of cases typically in the context of more complex karyotypes; such complex karyotypes appear to develop through multiple different pathways of clonal evolution which typically implicate several intracellular signaling pathways associated with cell proliferation, migration and apoptosis. Independently of the precise pathways of clonal evolution involved, accumulation of cytogenetic alterations, as reflected by more complex karyotypes with ≥2 altered chromosomes, usually leads to more aggressive disease, higher tumor grade and a poorer outcome in terms of recurrence-free survival. Consequently, assessment of tumor cytogenetics at diagnosis together with other clinical and histophatological features of the disease, becomes critical for both the establishment of optimal follow-up strategies and the definition of the most appropriate treatment for individual patients.
Acknowledgments
This work was partially supported by grants from the Fundação para a Ciência e Tecnologia (PIC/IC/83108/2007, FCT, Portugal), Fondo de Investigaciones Sanitarias (RD12/0036/0048, Instituto de Salud Carlos III (ISCIII/FEDER), Ministerio de Sanidad y Consumo, Madrid, Spain), and Consejeria Sanidad Junta de Castilla y León, Gerencia Regional de Salud: GRS689/A/11, and Proyecto Intramural-IBSAL IB14-05. Patrícia Domingues is supported by grant (SFRH/BD/64799/2009) from FCT. Maria Dolores Tabernero is supported by IECSCYL (Soria, Spain).
Conflicts of Interest
The authors declare that they have no conflict of interest.
Funding
Patrícia Domingues is partially supported by a grant (SFRH/BD/64799/2009) from FCT.
Maria Dolores Tabernero is supported by IECSCYL (Soria, Spain).
Author's contribution
PD, MGT, AO and MDT have been involved in the review design, generation of figures and data collection, writing the paper and they have given final approval of the version to be published.
AlO, DP, LR, DM, PS, JMG and MCL have been involved in drafting the manuscript and have given final approval of the version to be published.
References
1. Mawrin C and Perry A. Pathological classification and molecular genetics of meningiomas. J Neurooncol. 2010; 99:379-391.
2. Perry A, Louis DN, Scheithauer BW, Budka H and von Deimling A. (2007). Meningiomas. In: Louis DN, Ohgaki H, Wiestler OT and Cavenee WK, eds. WHO Classification of Tumors of the Central Nervous System. (Lyon, France: IARC press), pp. 164-172.
3. Choy W, Kim W, Nagasawa D, Stramotas S, Yew A, Gopen Q, Parsa AT and Yang I. The molecular genetics and tumor pathogenesis of meningiomas and the future directions of meningioma treatments. Neurosurg Focus. 2011; 30:E6.
4. Tabernero MD, Maillo A, Nieto AB, Diez-Tascon C, Lara M, Sousa P, Otero A, Castrillo A, Patino-Alonso Mdel C, Espinosa A, Mackintosh C, de Alava E and Orfao A. Delineation of commonly deleted chromosomal regions in meningiomas by high-density single nucleotide polymorphism genotyping arrays. Genes Chromosomes Cancer. 2012; 51:606-617.
5. Pecina-Slaus N. Merlin, the NF2 gene product. Pathol Oncol Res. 2013; 19:365-373.
6. Tabernero M, Jara-Acevedo M, Nieto AB, Caballero AR, Otero A, Sousa P, Goncalves J, Domingues PH and Orfao A. Association between mutation of the NF2 gene and monosomy 22 in menopausal women with sporadic meningiomas. BMC Med Genet. 2013; 14:114.
7. Lomas J, Bello MJ, Arjona D, Alonso ME, Martinez-Glez V, Lopez-Marin I, Aminoso C, de Campos JM, Isla A, Vaquero J and Rey JA. Genetic and epigenetic alteration of the NF2 gene in sporadic meningiomas. Genes Chromosomes Cancer. 2005; 42:314-319.
8. van Tilborg AA, Morolli B, Giphart-Gassler M, de Vries A, van Geenen DA, Lurkin I, Kros JM and Zwarthoff EC. Lack of genetic and epigenetic changes in meningiomas without NF2 loss. J Pathol. 2006; 208:564-573.
9. He S, Pham MH, Pease M, Zada G, Giannotta SL, Wang K and Mack WJ. A review of epigenetic and gene expression alterations associated with intracranial meningiomas. Neurosurg Focus. 2013; 35:E5.
10. Lallemand D, Manent J, Couvelard A, Watilliaux A, Siena M, Chareyre F, Lampin A, Niwa-Kawakita M, Kalamarides M and Giovannini M. Merlin regulates transmembrane receptor accumulation and signaling at the plasma membrane in primary mouse Schwann cells and in human schwannomas. Oncogene. 2009; 28:854-865.
11. Stamenkovic I and Yu Q. Merlin, a “magic” linker between extracellular cues and intracellular signaling pathways that regulate cell motility, proliferation, and survival. Curr Protein Pept Sci. 2010; 11:471-484.
12. Hartmann C, Sieberns J, Gehlhaar C, Simon M, Paulus W and von Deimling A. NF2 mutations in secretory and other rare variants of meningiomas. Brain Pathol. 2006; 16:15-19.
13. Kros J, de Greve K, van Tilborg A, Hop W, Pieterman H, Avezaat C, Lekanne Dit Deprez R and Zwarthoff E. NF2 status of meningiomas is associated with tumour localization and histology. J Pathol. 2001; 194:367-372.
14. Clark VE, Erson-Omay EZ, Serin A, Yin J, Cotney J, Ozduman K, Avsar T, Li J, Murray PB, Henegariu O, Yilmaz S, Gunel JM, Carrion-Grant G, Yilmaz B, Grady C, Tanrikulu B, et al. Genomic analysis of non-NF2 meningiomas reveals mutations in TRAF7, KLF4, AKT1, and SMO. Science. 2013; 339:1077-1080.
15. Peyrard M, Fransson I, Xie YG, Han FY, Ruttledge MH, Swahn S, Collins JE, Dunham I, Collins VP and Dumanski JP. Characterization of a new member of the human beta-adaptin gene family from chromosome 22q12, a candidate meningioma gene. Hum Mol Genet. 1994; 3:1393-1399.
16. Wozniak K, Piaskowski S, Gresner SM, Golanska E, Bieniek E, Bigoszewska K, Sikorska B, Szybka M, Kulczycka-Wojdala D, Zakrzewska M, Zawlik I, Papierz W, Stawski R, Jaskolski DJ, Och W, Sieruta M, et al. BCR expression is decreased in meningiomas showing loss of heterozygosity of 22q within a new minimal deletion region. Cancer Genet Cytogenet. 2008; 183:14-20.
17. Bello MJ, Aminoso C, Lopez-Marin I, Arjona D, Gonzalez-Gomez P, Alonso ME, Lomas J, de Campos JM, Kusak ME, Vaquero J, Isla A, Gutierrez M, Sarasa JL and Rey JA. DNA methylation of multiple promoter-associated CpG islands in meningiomas: relationship with the allelic status at 1p and 22q. Acta Neuropathol. 2004; 108:413-421.
18. Barski D, Wolter M, Reifenberger G and Riemenschneider MJ. Hypermethylation and transcriptional downregulation of the TIMP3 gene is associated with allelic loss on 22q12.3 and malignancy in meningiomas. Brain pathology (Zurich, Switzerland). 2010; 20:623-631.
19. Ketter R, Henn W, Niedermayer I, Steilen-Gimbel H, Konig J, Zang KD and Steudel WI. Predictive value of progression-associated chromosomal aberrations for the prognosis of meningiomas: a retrospective study of 198 cases. J Neurosurg. 2001; 95:601-607.
20. Zang KD. Meningioma: a cytogenetic model of a complex benign human tumor, including data on 394 karyotyped cases. Cytogenet Cell Genet. 2001; 93:207-220.
21. Lee Y, Liu J, Patel S, Cloughesy T, Lai A, Farooqi H, Seligson D, Dong J, Liau L, Becker D, Mischel P, Shams S and Nelson S. Genomic landscape of meningiomas. Brain Pathol. 2010; 20:751-762.
22. Al-Mefty O, Kadri PA, Pravdenkova S, Sawyer JR, Stangeby C and Husain M. Malignant progression in meningioma: documentation of a series and analysis of cytogenetic findings. J Neurosurg. 2004; 101:210-218.
23. Brastianos PK, Horowitz PM, Santagata S, Jones RT, McKenna A, Getz G, Ligon KL, Palescandolo E, Van Hummelen P, Ducar MD, Raza A, Sunkavalli A, Macconaill LE, Stemmer-Rachamimov AO, Louis DN, Hahn WC, et al. Genomic sequencing of meningiomas identifies oncogenic SMO and AKT1 mutations. Nat Genet. 2013; 45:285-289.
24. Reuss DE, Piro RM, Jones DT, Simon M, Ketter R, Kool M, Becker A, Sahm F, Pusch S, Meyer J, Hagenlocher C, Schweizer L, Capper D, Kickingereder P, Mucha J, Koelsche C, et al. Secretory meningiomas are defined by combined KLF4 K409Q and TRAF7 mutations. Acta neuropathologica. 2013; 125:351-358.
25. Weber RG, Bostrom J, Wolter M, Baudis M, Collins VP, Reifenberger G and Lichter P. Analysis of genomic alterations in benign, atypical, and anaplastic meningiomas: toward a genetic model of meningioma progression. Proc Natl Acad Sci U S A. 1997; 94:14719-14724.
26. Ruiz J, Martinez A, Hernandez S, Zimman H, Ferrer M, Fernandez C, Saez M, Lopez-Asenjo JA and Sanz-Ortega J. Clinicopathological variables, immunophenotype, chromosome 1p36 loss and tumour recurrence of 247 meningiomas grade I and II. Histol Histopathol. 2010; 25:341-349.
27. Maillo A, Orfao A, Espinosa AB, Sayagues JM, Merino M, Sousa P, Lara M and Tabernero MD. Early recurrences in histologically benign/grade I meningiomas are associated with large tumors and coexistence of monosomy 14 and del(1p36) in the ancestral tumor cell clone. Neuro Oncol. 2007; 9:438-446.
28. Nakane Y, Natsume A, Wakabayashi T, Oi S, Ito M, Inao S, Saito K and Yoshida J. Malignant transformation-related genes in meningiomas: allelic loss on 1p36 and methylation status of p73 and RASSF1A. J Neurosurg. 2007; 107:398-404.
29. Bouvier C, Liprandi A, Colin C, Giorgi R, Quilichini B, Metellus P and Figarella-Branger D. Lack of alkaline phosphatase activity predicts meningioma recurrence. Am J Clin Pathol. 2005; 124:252-258.
30. Jansen M, Mohapatra G, Betensky RA, Keohane C and Louis DN. Gain of chromosome arm 1q in atypical meningioma correlates with shorter progression-free survival. Neuropathol Appl Neurobiol. 2012; 38:213-219.
31. Perez-Magan E, Rodriguez de Lope A, Ribalta T, Ruano Y, Campos-Martin Y, Perez-Bautista G, Garcia JF, Garcia-Claver A, Fiano C, Hernandez-Moneo JL, Mollejo M and Melendez B. Differential expression profiling analyses identifies downregulation of 1p, 6q, and 14q genes and overexpression of 6p histone cluster 1 genes as markers of recurrence in meningiomas. Neuro Oncol. 2010; 12:1278-1290.
32. Amatya VJ, Takeshima Y and Inai K. Methylation of p14(ARF) gene in meningiomas and its correlation to the p53 expression and mutation. Mod Pathol. 2004; 17:705-710.
33. Bostrom J, Meyer-Puttlitz B, Wolter M, Blaschke B, Weber RG, Lichter P, Ichimura K, Collins VP and Reifenberger G. Alterations of the tumor suppressor genes CDKN2A (p16(INK4a)), p14(ARF), CDKN2B (p15(INK4b)), and CDKN2C (p18(INK4c)) in atypical and anaplastic meningiomas. Am J Pathol. 2001; 159:661-669.
34. Goutagny S, Yang HW, Zucman-Rossi J, Chan J, Dreyfuss JM, Park PJ, Black PM, Giovannini M, Carroll RS and Kalamarides M. Genomic profiling reveals alternative genetic pathways of meningioma malignant progression dependent on the underlying NF2 status. Clin Cancer Res. 2010; 16:4155-4164.
35. Aydemir F, Yurtcu E, Balci TB, Sahin FI, Gulsen S and Altinors N. Identification of promoter region methylation patterns of MGMT, CDKN2A, GSTP1, and THBS1 genes in intracranial meningioma patients. Genet Test Mol Biomarkers. 2012; 16:335-340.
36. Perry A, Banerjee R, Lohse CM, Kleinschmidt-DeMasters BK and Scheithauer BW. A role for chromosome 9p21 deletions in the malignant progression of meningiomas and the prognosis of anaplastic meningiomas. Brain Pathol. 2002; 12:183-190.
37. Mihaila D, Jankowski M, Gutierrez JA, Rosenblum ML, Newsham IF, Bogler O and Rempel SA. Meningiomas: loss of heterozygosity on chromosome 10 and marker-specific correlations with grade, recurrence, and survival. Clin Cancer Res. 2003; 9:4443-4451.
38. Joachim T, Ram Z, Rappaport ZH, Simon M, Schramm J, Wiestler OD and von Deimling A. Comparative analysis of the NF2, TP53, PTEN, KRAS, NRAS and HRAS genes in sporadic and radiation-induced human meningiomas. Int J Cancer. 2001; 94:218-221.
39. Dobbins SE, Broderick P, Melin B, Feychting M, Johansen C, Andersson U, Brannstrom T, Schramm J, Olver B, Lloyd A, Ma YP, Hosking FJ, Lonn S, Ahlbom A, Henriksson R, Schoemaker MJ, et al. Common variation at 10p12.31 near MLLT10 influences meningioma risk. Nat Genet. 2011; 43:825-827.
40. Tabernero MD, Espinosa AB, Maillo A, Sayagues JM, Alguero Mdel C, Lumbreras E, Diaz P, Goncalves JM, Onzain I, Merino M, Morales F and Orfao A. Characterization of chromosome 14 abnormalities by interphase in situ hybridization and comparative genomic hybridization in 124 meningiomas: correlation with clinical, histopathologic, and prognostic features. Am J Clin Pathol. 2005; 123:744-751.
41. Maillo A, Orfao A, Sayagues JM, Diaz P, Gomez-Moreta JA, Caballero M, Santamarta D, Santos-Briz A, Morales F and Tabernero MD. New classification scheme for the prognostic stratification of meningioma on the basis of chromosome 14 abnormalities, patient age, and tumor histopathology. J Clin Oncol. 2003; 21:3285-3295.
42. Zhang X, Gejman R, Mahta A, Zhong Y, Rice KA, Zhou Y, Cheunsuchon P, Louis DN and Klibanski A. Maternally expressed gene 3, an imprinted noncoding RNA gene, is associated with meningioma pathogenesis and progression. Cancer research. 2010; 70:2350-2358.
43. Balik V, Srovnal J, Sulla I, Kalita O, Foltanova T, Vaverka M, Hrabalek L and Hajduch M. MEG3: a novel long noncoding potentially tumour-suppressing RNA in meningiomas. Journal of neuro-oncology. 2013; 112:1-8.
44. Lusis EA, Watson MA, Chicoine MR, Lyman M, Roerig P, Reifenberger G, Gutmann DH and Perry A. Integrative genomic analysis identifies NDRG2 as a candidate tumor suppressor gene frequently inactivated in clinically aggressive meningioma. Cancer Res. 2005; 65:7121-7126.
45. Skiriute D, Tamasauskas S, Asmoniene V, Saferis V, Skauminas K, Deltuva V and Tamasauskas A. Tumor grade-related NDRG2 gene expression in primary and recurrent intracranial meningiomas. J Neurooncol. 2011; 102:89-94.
46. Surace EI, Lusis E, Haipek CA and Gutmann DH. Functional significance of S6K overexpression in meningioma progression. Ann Neurol. 2004; 56:295-298.
47. Buschges R, Ichimura K, Weber RG, Reifenberger G and Collins VP. Allelic gain and amplification on the long arm of chromosome 17 in anaplastic meningiomas. Brain Pathol. 2002; 12:145-153.
48. Zhang MX, Zhao X, Wang ZG, Zhao WM and Wang YS. Constitutive activation of signal transducer and activator of transcription 3 regulates expression of vascular endothelial growth factor in human meningioma differentiation. J Cancer Res Clin Oncol. 2010; 136:981-988.
49. Johnson MD, O’Connell M, Facik M, Maurer P, Jahromi B and Pilcher W. Cerebrospinal fluid stimulates leptomeningeal and meningioma cell proliferation and activation of STAT3. J Neurooncol. 2012; 107:121-131.
50. Tabernero MD, Maillo A, Gil-Bellosta CJ, Castrillo A, Sousa P, Merino M and Orfao A. Gene expression profiles of meningiomas are associated with tumor cytogenetics and patient outcome. Brain Pathol. 2009; 19:409-420.
51. Uzum N and Ataoglu GA. Histopathological parameters with Ki-67 and bcl-2 in the prognosis of meningiomas according to WHO 2000 classification. Tumori. 2008; 94:389-397.
52. Cheng G, Zhang L, Lv W, Dong C, Wang Y and Zhang J. Overexpression of cyclin D1 in meningioma is associated with malignancy grade and causes abnormalities in apoptosis, invasion and cell cycle progression. Med Oncol 2015; 32:439.
53. Gerber MA, Bahr SM and Gutmann DH. Protein 4.1B/differentially expressed in adenocarcinoma of the lung-1 functions as a growth suppressor in meningioma cells by activating Rac1-dependent c-Jun-NH(2)-kinase signaling. Cancer Res. 2006; 66:5295-5303.
54. Gutmann DH, Donahoe J, Perry A, Lemke N, Gorse K, Kittiniyom K, Rempel SA, Gutierrez JA and Newsham IF. Loss of DAL-1, a protein 4.1-related tumor suppressor, is an important early event in the pathogenesis of meningiomas. Human molecular genetics. 2000; 9:1495-1500.
55. Yi C, McCarty JH, Troutman SA, Eckman MS, Bronson RT and Kissil JL. Loss of the putative tumor suppressor band 4.1B/Dal1 gene is dispensable for normal development and does not predispose to cancer. Mol Cell Biol. 2005; 25:10052-10059.
56. Nunes F, Shen Y, Niida Y, Beauchamp R, Stemmer-Rachamimov AO, Ramesh V, Gusella J and MacCollin M. Inactivation patterns of NF2 and DAL-1/4.1B (EPB41L3) in sporadic meningioma. Cancer Genet Cytogenet. 2005; 162:135-139.
57. Martinez-Glez V, Bello MJ, Franco-Hernandez C, De Campos JM, Isla A, Vaquero J and Rey JA. Mutational analysis of the DAL-1/4.1B tumour-suppressor gene locus in meningiomas. Int J Mol Med. 2005; 16:771-774.
58. Liu Y, Pang JC, Dong S, Mao B, Poon WS and Ng HK. Aberrant CpG island hypermethylation profile is associated with atypical and anaplastic meningiomas. Hum Pathol. 2005; 36:416-425.
59. Wrobel G, Roerig P, Kokocinski F, Neben K, Hahn M, Reifenberger G and Lichter P. Microarray-based gene expression profiling of benign, atypical and anaplastic meningiomas identifies novel genes associated with meningioma progression. Int J Cancer. 2005; 114:249-256.
60. Otsuka S, Tamiya T, Ono Y, Michiue H, Kurozumi K, Daido S, Kambara H, Date I and Ohmoto T. The relationship between peritumoral brain edema and the expression of vascular endothelial growth factor and its receptors in intracranial meningiomas. J Neurooncol. 2004; 70:349-357.
61. Preusser M, Hassler M, Birner P, Rudas M, Acker T, Plate KH, Widhalm G, Knosp E, Breitschopf H, Berger J and Marosi C. Microvascularization and expression of VEGF and its receptors in recurring meningiomas: pathobiological data in favor of anti-angiogenic therapy approaches. Clin Neuropathol. 2012; 31:352-360.
62. Bajetto A, Barbieri F, Pattarozzi A, Dorcaratto A, Porcile C, Ravetti JL, Zona G, Spaziante R, Schettini G and Florio T. CXCR4 and SDF1 expression in human meningiomas: a proliferative role in tumoral meningothelial cells in vitro. Neuro Oncol. 2007; 9:3-11.
63. Johnson MD, O’Connell MJ, Pilcher W and Reeder JE. Fibroblast growth factor receptor-3 expression in meningiomas with stimulation of proliferation by the phosphoinositide 3 kinase-Akt pathway. J Neurosurg. 2010; 112:934-939.
64. Johnson M and Toms S. Mitogenic signal transduction pathways in meningiomas: novel targets for meningioma chemotherapy? J Neuropathol Exp Neurol. 2005; 64:1029-1036.
65. Mawrin C, Sasse T, Kirches E, Kropf S, Schneider T, Grimm C, Pambor C, Vorwerk CK, Firsching R, Lendeckel U and Dietzmann K. Different activation of mitogen-activated protein kinase and Akt signaling is associated with aggressive phenotype of human meningiomas. Clin Cancer Res. 2005; 11:4074-4082.
66. Jensen RL and Wurster RD. Calcium channel antagonists inhibit growth of subcutaneous xenograft meningiomas in nude mice. Surg Neurol. 2001; 55(5):275-283.
67. Lee SH, Lee YS, Hong YG and Kang CS. Significance of COX-2 and VEGF expression in histopathologic grading and invasiveness of meningiomas. APMIS. 2014; 122:16-24.
68. Kato Y, Nishihara H, Mohri H, Kanno H, Kobayashi H, Kimura T, Tanino M, Terasaka S and Tanaka S. Clinicopathological evaluation of cyclooxygenase-2 expression in meningioma: immunohistochemical analysis of 76 cases of low and high-grade meningioma. Brain Tumor Pathol. 2014; 31:23-30.
69. James MF, Han S, Polizzano C, Plotkin SR, Manning BD, Stemmer-Rachamimov AO, Gusella JF and Ramesh V. NF2/merlin is a novel negative regulator of mTOR complex 1, and activation of mTORC1 is associated with meningioma and schwannoma growth. Mol Cell Biol. 2009; 29:4250-4261.
70. Pachow D, Andrae N, Kliese N, Angenstein F, Stork O, Wilisch-Neumann A, Kirches E and Mawrin C. mTORC1 inhibitors suppress meningioma growth in mouse models. Clin Cancer Res. 2013; 19:1180-1189.
71. Pecina-Slaus N, Nikuseva Martic T, Deak AJ, Zeljko M, Hrascan R, Tomas D and Musani V. Genetic and protein changes of E-cadherin in meningiomas. J Cancer Res Clin Oncol. 2010; 136:695-702.
72. Zhou K, Wang G, Wang Y, Jin H, Yang S and Liu C. The potential involvement of E-cadherin and beta-catenins in meningioma. PloS one. 2010; 5:e11231.
73. Zhou L, Ercolano E, Ammoun S, Schmid MC, Barczyk MA and Hanemann CO. Merlin-deficient human tumors show loss of contact inhibition and activation of Wnt/beta-catenin signaling linked to the PDGFR/Src and Rac/PAK pathways. Neoplasia. 2010; 13:1101-1112.
74. Perez-Magan E, Campos-Martin Y, Mur P, Fiano C, Ribalta T, Garcia JF, Rey JA, Rodriguez de Lope A, Mollejo M and Melendez B. Genetic alterations associated with progression and recurrence in meningiomas. J Neuropathol Exp Neurol. 2012; 71:882-893.
75. Ress A and Moelling K. Bcr is a negative regulator of the Wnt signalling pathway. EMBO Rep. 2005; 6:1095-1100.
76. Cuevas IC, Slocum AL, Jun P, Costello JF, Bollen AW, Riggins GJ, McDermott MW and Lal A. Meningioma transcript profiles reveal deregulated Notch signaling pathway. Cancer Res. 2005; 65:5070-5075.
77. Baia GS, Stifani S, Kimura ET, McDermott MW, Pieper RO and Lal A. Notch activation is associated with tetraploidy and enhanced chromosomal instability in meningiomas. Neoplasia. 2008; 10:604-612.
78. Laurendeau I, Ferrer M, Garrido D, D’Haene N, Ciavarelli P, Basso A, Vidaud M, Bieche I, Salmon I and Szijan I. Gene expression profiling of the hedgehog signaling pathway in human meningiomas. Mol Med. 2010; 16:262-270.
79. Ketter R, Urbschat S, Henn W, Feiden W, Beerenwinkel N, Lengauer T, Steudel WI, Zang KD and Rahnenfuhrer J. Application of oncogenetic trees mixtures as a biostatistical model of the clonal cytogenetic evolution of meningiomas. Int J Cancer. 2007; 121:1473-1480.
80. Espinosa AB, Tabernero MD, Maillo A, Sayagues JM, Ciudad J, Merino M, Alguero MC, Lubombo AM, Sousa P, Santos-Briz A and Orfao A. The cytogenetic relationship between primary and recurrent meningiomas points to the need for new treatment strategies in cases at high risk of relapse. Clin Cancer Res. 2006; 12:772-780.
81. Barbera S, San Miguel T, Gil-Benso R, Munoz-Hidalgo L, Roldan P, Gonzalez-Darder J, Cerda-Nicolas M and Lopez-Gines C. Genetic changes with prognostic value in histologically benign meningiomas. Clinical neuropathology. 2013; 32:311-317.
82. Urbschat S, Rahnenfuhrer J, Henn W, Feiden W, Wemmert S, Linsler S, Zang KD, Oertel J and Ketter R. Clonal cytogenetic progression within intratumorally heterogeneous meningiomas predicts tumor recurrence. International journal of oncology. 2011; 39:1601-1608.
83. Ketter R, Rahnenfuhrer J, Henn W, Kim YJ, Feiden W, Steudel WI, Zang KD and Urbschat S. Correspondence of tumor localization with tumor recurrence and cytogenetic progression in meningiomas. Neurosurgery. 2008; 62:61-69; discussion 69-70.
84. Serna E, Morales JM, Mata M, Gonzalez-Darder J, San Miguel T, Gil-Benso R, Lopez-Gines C, Cerda-Nicolas M and Monleon D. Gene expression profiles of metabolic aggressiveness and tumor recurrence in benign meningioma. PloS one. 2013; 8:e67291.
85. Monleon D, Morales JM, Gonzalez-Segura A, Gonzalez-Darder JM, Gil-Benso R, Cerda-Nicolas M and Lopez-Gines C. Metabolic aggressiveness in benign meningiomas with chromosomal instabilities. Cancer research. 2010; 70:8426-8434.
86. Sayagues JM, Tabernero MD, Maillo A, Espinosa A, Rasillo A, Diaz P, Ciudad J, Lopez A, Merino M, Goncalves JM, Santos-Briz A, Morales F and Orfao A. Intratumoral patterns of clonal evolution in meningiomas as defined by multicolor interphase fluorescence in situ hybridization (FISH): is there a relationship between histopathologically benign and atypical/anaplastic lesions? J Mol Diagn. 2004; 6:316-325.
87. Domingues PH, Teodosio C, Otero A, Sousa P, Ortiz J, Macias Mdel C, Goncalves JM, Nieto AB, Lopes MC, de Oliveira C, Orfao A and Tabernero MD. Association between inflammatory infiltrates and isolated monosomy 22/del(22q) in meningiomas. PloS one. 2013; 8:e74798.
88. Ketter R, Kim YJ, Storck S, Rahnenfuhrer J, Romeike BF, Steudel WI, Zang KD and Henn W. Hyperdiploidy defines a distinct cytogenetic entity of meningiomas. Journal of neuro-oncology. 2007; 83:213-221.
89. Cerda-Nicolas M, Lopez-Gines C, Perez-Bacete M, Barcia-Salorio JL and Llombart-Bosch A. Histopathological and cytogenetic findings in benign, atypical and anaplastic human meningiomas: a study of 60 tumors. Journal of neuro-oncology. 2000; 47:99-108.
90. Domingues PH, Sousa P, Otero A, Goncalves JM, Ruiz L, Lopes MC, de Oliveira C, Orfao A and Tabernero MD. Proposal for a new risk stratification classification for meningioma based on patient age, WHO tumor grade, size, localization, and karyotype. Neuro Oncol. 2014; 16:735-747.
91. Watson MA, Gutmann DH, Peterson K, Chicoine MR, Kleinschmidt-DeMasters BK, Brown HG and Perry A. Molecular characterization of human meningiomas by gene expression profiling using high-density oligonucleotide microarrays. Am J Pathol. 2002; 161:665-672.
92. Fevre-Montange M, Champier J, Durand A, Wierinckx A, Honnorat J, Guyotat J and Jouvet A. Microarray gene expression profiling in meningiomas: differential expression according to grade or histopathological subtype. Int J Oncol. 2009; 35:1395-1407.
93. Sayagues JM, Tabernero MD, Maillo A, Trelles O, Espinosa AB, Sarasquete ME, Merino M, Rasillo A, Vera JF, Santos-Briz A, de Alava E, Garcia-Macias MC and Orfao A. Microarray-based analysis of spinal versus intracranial meningiomas: different clinical, biological, and genetic characteristics associated with distinct patterns of gene expression. J Neuropathol Exp Neurol. 2006; 65:445-454.
94. Tabernero MD, Espinosa AB, Maillo A, Rebelo O, Vera JF, Sayagues JM, Merino M, Diaz P, Sousa P and Orfao A. Patient gender is associated with distinct patterns of chromosomal abnormalities and sex chromosome linked gene-expression profiles in meningiomas. Oncologist. 2007; 12:1225-1236.
95. Carvalho LH, Smirnov I, Baia GS, Modrusan Z, Smith JS, Jun P, Costello JF, McDermott MW, Vandenberg SR and Lal A. Molecular signatures define two main classes of meningiomas. Mol Cancer. 2007; 6:64.
96. Surace EI, Lusis E, Murakami Y, Scheithauer BW, Perry A and Gutmann DH. Loss of tumor suppressor in lung cancer-1 (TSLC1) expression in meningioma correlates with increased malignancy grade and reduced patient survival. Journal of neuropathology and experimental neurology. 2004; 63:1015-1027.
97. Utsuki S, Oka H, Sato Y, Kawano N, Tsuchiya B, Kobayashi I and Fujii K. Invasive meningioma is associated with a low expression of E-cadherin and beta-catenin. Clin Neuropathol. 2005; 24:8-12.
98. Das A, Tan WL and Smith DR. Expression of extracellular matrix markers in benign meningiomas. Neuropathology. 2003; 23:275-281.
99. Cai DX, Banerjee R, Scheithauer BW, Lohse CM, Kleinschmidt-Demasters BK and Perry A. Chromosome 1p and 14q FISH analysis in clinicopathologic subsets of meningioma: diagnostic and prognostic implications. J Neuropathol Exp Neurol. 2001; 60:628-636.
100. Linsler S, Kraemer D, Driess C, Oertel J, Kammers K, Rahnenfuhrer J, Ketter R and Urbschat S. Molecular biological determinations of meningioma progression and recurrence. PloS one. 2014; 9:e94987.