
Introduction
Since the initial discovery of the predictive and prognostic significance of chromosome 1p loss [1], evidence has accumulated suggesting that the total loss of chromosomes 1p and 19q (resulting from an unbalanced centromeric translocation t (1;19) (q10; p10)), is associated with increased chemosensitivity and longer survival in oligodendroglial gliomas [2-9]. Although total 1p19q loss is a genetic hallmark of oligodendroglial tumors, it is also occasionally observed in astrocytic gliomas [10-16]. Most previous studies that have demonstrated a predictive and prognostic relevance of 1p19q loss have involved anaplastic oligodendroglial tumors. In contrast, the publication of similar studies involving 1p19q codeleted astrocytic gliomas is limited, and the conclusions reached have been inconclusive [16-18]. Furthermore, the morphological distinctions between non-classic oligodendroglioma and astrocytoma suffers from significant interobserver variation and is thus associated with limited reproducibility [4, 19]. Therefore, whether the clinical and biological significance of 1p19q loss can be generally applied to diffuse gliomas beyond histological classification remains to be validated.
In this study, we investigated a consecutive series of diffuse gliomas with an “astrocytic” appearance and that exhibited 1p19q loss, in order to assess their molecular–genetic and clinical–biological characteristics. The deletion status of 1p and 19q was analyzed by comparative genomic hybridization (CGH), which (unlike fluorescence in situ hybridization (FISH)) can accurately distinguish between complete and partial chromosome loss [16, 20]. The molecular–genetic and clinical–biological characteristics of “astrocytic” gliomas with total 1p19q loss were compared with those of oligodendroglial gliomas on the basis of the original institutional diagnosis as well as diagnosis by a single pathologist. This was done to exclude bias due to inter-observer variability or due to changes in diagnostic criteria over time [19, 21]. Moreover, because the study population was collected from consecutive cases treated at a single institution over two decades, we were able to determine the Japanese incidence of diffuse gliomas with 1p19q loss.
Results
Histopathological distribution of gliomas with total 1p19q loss
In total, 218 supratentorial, diffuse gliomas of WHO grade II or III from adult patients without prior chemotherapy or radiotherapy were found in the hospital pathology records. The status of 1p and 19q was examined for clinical purpose in 152 of 218 gliomas. Of these, 53 were oligodendroglial (oligodendroglioma, oligoastrocytoma, anaplastic oligodendroglioma, or anaplastic oligoastrocytoma), 93 were astrocytic (astrocytoma or anaplastic astrocytoma), and 6 were unclassified because of small sample size, by the original institutional diagnosis. Fifty-eight of 152 gliomas (38.2%) showed 1p19q loss. Of these, 38 were oligodendroglial (71.7% of all oligodendroglial tumors), 16 were astrocytic (17.2% of all astrocytic tumors), and 4 were unclassified. Written informed consent for translational research (institutional approval number 20050002) was obtained for 57 of the 58 codeleted gliomas, and subsequent analysis was performed on these 57 tumors. The 57 patients comprised 35 males and 22 females, with ages ranging from 22 to 63 years. Demographic factors such as age and sex were comparable between the oligodendroglial and astrocytic groups (Table S1).
The 37 codeleted oligodendroglial gliomas comprised 15 oligodendrogliomas, 9 oligoastrocytomas, 9 anaplastic oligodendogliomas, and 4 anaplastic oligoastrocytomas (Table S1). Nine of the 37 cases were diagnosed by biopsy.
The 16 codeleted “astrocytomas” comprised 7 diffuse astrocytomas and 9 anaplastic astrocytomas. Five of the 16 cases were diagnosed by biopsy (Table S1).
Histopathological features of the 57 codeleted gliomas were re-assessed by a single pathologist according to current criteria [22]. Following re-assessment, 40 were classified as oligodendroglial and 15 as “astrocytic,” while 2 were unclassified because of small sample size.
Molecular–genetic results
Of the 57 cases, CGH profiles were obtained for all, IDH mutation status was obtained for 53, and MGMT promoter methylation status was obtained for 51. Evaluation of the status of ATRX and p53 were possible in 15 of the 16 codeleted “astrocytic” gliomas. Mutational screening results along with original institutional diagnoses for each case are found in Table S1.
Among the 37 oligodendroglial gliomas, 26 revealed CNAs besides the 1p19q codeletion. The most frequent was loss on chromosome arm 4q (9 cases, 24.3%), followed by loss on 14q (7 cases, 18.9%), loss of 18q (7 cases, 18.9%), gain on 11q/11 (gain of 11q or total gain of 11, 6 cases, 16.2%), loss on 13q (5 cases, 13.5%), gain on 17q (4 cases, 10.8%), and gain on 19p (4 cases, 10.8%) (Figure 1, Table 1). IDH1 or IDH2 mutations were detected in all 34 cases (100%) for which both genes were successfully examined. All changes at IDH1 codon 132 comprised a CGT to CAT (R132H) mutation, and all IDH2 codon 172 mutations were AGG to ATG (R172M). Twenty-one of 32 analyzed cases (65.6%) displayed MGMT promoter methylation (Table 1).
Table 1: Summary of chromosomal copy number aberrations (CNAs) and status of IDH and MGMT genes in gliomas with total 1p19q loss by institutional diagnosis.
Oligo (n=37) |
Astro (n=16) |
Unclassified low-grade glioma (n=4) |
|
CNAs (detected in ≥10%) |
-1p19q (100%) -4q (24.3%) -14q (18.9%) -18q (18.9%) +11q or +11 (16.2%) -13q (13.5%) +17q (10.8%) +19p (10.8%) |
-1p19q (100%) +7q or +7 (31.2%) -4q (25%) +11q or +11 (18.8%) -18q (18.8%) +19p (18.8%) -9p (12.5%) +9q (12.5%) -10p (12.5%) -14q (12.5%) -15q (12.5%) +17q (12.5%) |
-1p19q(100%) -4(25%) -19p(25%) |
| IDH 1 mutation | 31/36(86.1%) |
12/13(92.3%) | 4/4(100%) |
| IDH 2 mutation | 3/3(100%) (Either IDH1 or IDH2 mutations in 34/34, 100%) |
0/1(0%) (Either IDH1 or IDH2 mutations in 12 of 13, 92.3%) |
Not performed |
MGMT methylation |
21/32(65.6%) | 15/15(100%) | 2/4(50%) |
| ATRX loss | Not performed | 3/15(80%) | Not performed |
| P53 accumulation | Not performed | 0/15%(0%) | Not performed |
Among the 16 “astrocytic” gliomas, 12 revealed CNAs besides the 1p19q codeletion. The most frequent were gain on chromosome 7q/7 (gain on 7q or total gain of 7, 5 cases, 31.2%), loss on 4q (4 cases, 25%), gain on 11q/11(3 cases, 18.8%), loss on 18q (3 cases, 18.8%), gain of 19p (3 cases, 18.8%), loss on 9p (2 cases, 12.5%), gain on 9q (2 cases, 12.5%), loss of 10p (2 cases, 12.5%), loss on 14q (2 cases, 12.5%), loss of 15q (2 cases, 12.5%), and gain on 17q (2 cases, 12.5%) (Figure 2, Table 1). Mutations in IDH1 or IDH2 were detected in 12 of 13 cases (92.3%) for which both genes were successfully examined. MGMT promoter methylation was found in all 15 cases for which MSP was successfully performed. Expression of p53 was negative in all of the 15 successfully evaluated cases, whereas loss of ATRX expression was detected in three of the 15 (Table 1).
Among the four gliomas that were unclassified because of small sample size, two cases had CNAs besides the 1p19q codeletion. These comprised loss on 4 (25%) and loss of 19p (25%).
Gain on 7q/7 was significantly more frequent in “astrocytic” gliomas (p = 0.0015), and loss of 10p was numerically more frequent in “astrocytic” gliomas (p = 0.0871).

Figure 1: Chromosomal copy number aberrations (CNAs) of oligodendroglial gliomas with total 1p19q loss, as determined by institutional diagnosis. Lines to the left of each idiogram represent regions of reduced relative DNA copy number, and lines to the right represent regions of increased relative DNA copy number. Each line represents a CNA found in one tumor.
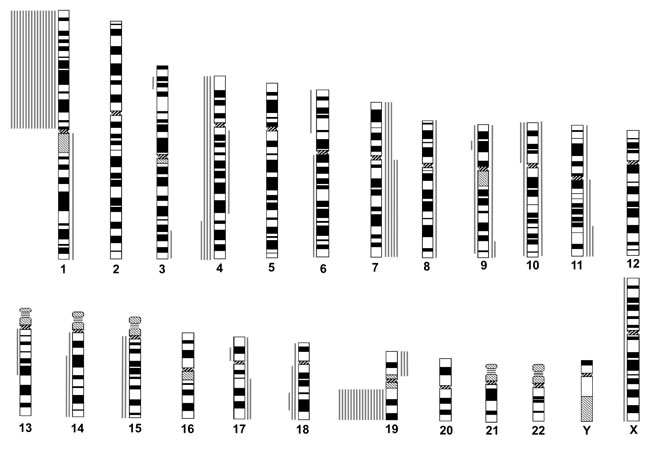
Figure 2: Chromosomal copy number aberrations (CNAs) of astrocytic gliomas with total 1p19q loss, as determined by institutional diagnosis. Idiogram features are as described for Figure. 1.
Survival analysis
To determine whether any distinct clinical or biological characteristics occurred in 1p19q codeleted gliomas with astrocytic features, the survival outcome was compared between the two groups. Although initial treatments varied (resection alone: 11 cases in oligodendroglial versus 1 case in astrocytic; radiotherapy alone: 6 in oligodendroglial versus 5 in astrocytic; chemotherapy alone: 11 in oligodendroglial versus 1 in astrocytic; chemoradiotherapy: 9 in oligodendroglial and 9 in astrocytic), treatment intensities during the overall clinical courses were similar between the two histological groups, i.e., 22 of 37 patients with oligodendroglial gliomas and 12 of 16 patients with “astrocytic” gliomas underwent both chemotherapy and radiotherapy in their clinical courses. Kaplan–Meier survival curves overlapped between the groups, and with a median follow-up of 87 months, the median OS in the oligodendroglial and “astrocytic” glioma groups was 187 and 184 months, respectively (Figure 3a, p = 0.828). Eleven patients with “astrocytic” gliomas by resection showed comparable survival times (median OS: 176 months, p = 0.5905). Moreover, histological grouping by a single pathologist using current criteria demonstrated a consistent result, again showing no significant OS difference between the two glioma categories (Figure 3b; p = 0.84). Furthermore, those results were confirmed by a comparison between oligodendroglial (33) and “astrocytic” (10) gliomas by consensus of institutional and single-pathologist diagnoses (p = 0.67).
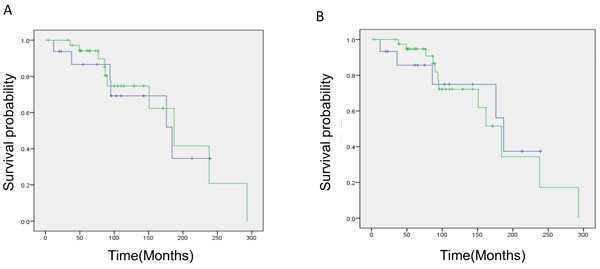
Figure 3: Kaplan–Meier survival analysis of oligodendroglial and astrocytic gliomas with total 1p19q loss. a. by institutional diagnosis: The green line represents oligodendroglial tumors and the blue line represents astrocytic tumors. Median OS: oligodendroglial 187 months, astrocytic 184 months (p = 0.828). b. by single pathologist diagnosis: The green line represents oligodendroglial tumors and the blue line represents astrocytic tumors. Median OS: oligodendroglial 184 months, astrocytic 187 months (p = 0.84). Note that there is no difference in survival estimation between the two histological groups.
Illustrative cases of “astrocytic” gliomas with total 1p19q loss
Case 1 (Table S1, case 39): A 37-year-old (yo) female had subtotal removal of a right frontal tumor. The institutional and single-pathologist diagnoses were both diffuse astrocytoma (Figure 4a, 4b). The nuclei of the tumor cells were round to oval in shape and irregular in size. Many tumor cells exhibited cellular processes and no mitosis was observed. The patient underwent radiation therapy of 58Gy after tumor removal. CGH revealed isolated 1p19q codeletion. An IDH1 mutation was detected and methylation of the MGMT promoter was observed. Expression of ATRX was retained (Figure 4c), and expression of p53 was negative. The patient remains alive 75 months from the initial surgery.
Case 2 (Table S1, case 44): A 28 yo female underwent total removal of a left frontal tumor. The institutional and single-pathologist diagnoses were both diffuse astrocytoma (Figure 4d, 4e). The nuclei of the tumor cells were round to oval in shape and irregular in size. Cytoplasmic cellular processes were present in most tumor cells. There was no mitosis observed. CNAs revealed by CGH were −1p19q, −3p24-22, and +7q. The MGMT promoter was methylated. Expression of ATRX was retained (Figure 4f), and expression of p53 was negative. The patient underwent radiation therapy of 50 Gy after tumor removal. Tumor recurrence occurred 196 months after initial surgery and the pathological finding at recurrence was anaplastic astrocytoma. The patient remains alive 239 months from the initial surgery.
Case 3 (Table S1, case 52): A 27 yo female underwent partial removal of a right frontal tumor. Institutional histopathological diagnosis was anaplastic astrocytoma, while diagnosis by a single pathologist was diffuse astrocytoma (Figure 4g, 4h). The nuclei of the tumor cells were mostly oval shaped and irregular in size. The cells displayed remarkable cellular processes and no mitosis was detected. The patient underwent chemoradiotherapy (50 Gy, ranimustine = MCNU) after initial surgery. CGH revealed isolated 1p19q codeletion. Mutations in IDH 1 and IDH2 were not detected but the MGMT promoter was methylated. Expression of ATRX was retained (Figure 4i), and expression of p53 was negative. The patient was lost during follow-up because of a change in residential address. No recurrence had occurred 23 months from the initial surgery.
Case 4 (Table S1, case 53): A 28 yo male underwent subtotal removal of a left frontal tumor. Institutional histopathological diagnosis was anaplastic astrocytoma, while the diagnosis by a single pathologist was diffuse astrocytoma (Figure 4j, 4k). The nuclei of the tumor cells were oval shaped and irregular in size. Most tumor cells displayed cytoplasmic cellular processes, and mitoses were very rare. A small area with higher cellularity was detected (not shown). The patient underwent chemoradiotherapy (60Gy, ranimustine = MCNU and vincristine = VCR) after the initial surgery. CGH revealed −1p19q, +10, and −18. Mutation of IDH1 and MGMT promoter methylation were detected. Expression of ATRX was lost (Figure 4l), whereas p53 accumulation was not detected. Tumor recurrence occurred 52 months after the initial surgery. At present, the patient’s OS is greater than 110 months.

Figure 4: Hematoxylin and eosin staining (a, b, d, e, g, h, j, k) and ATRX staining (c, f, i, l) of examples of “astrocytic” gliomas with total 1p19q loss. a, b, c. Table S1, case 39. Institutional diagnosis: diffuse astrocytoma. Single-pathologist diagnosis: diffuse astrocytoma. d, e, f. Table S1, case 44. Institutional diagnosis: diffuse astrocytoma. Single-pathologist diagnosis: diffuse astrocytoma. g, h, i. Table S1, case 52. Institutional diagnosis: anaplastic astrocytoma. Single-pathologist diagnosis: diffuse astrocytoma. j, k, l. Table S1, case 53. Institutional diagnosis: anaplastic astrocytoma. Single-pathologist diagnosis: diffuse astrocytoma. a, d, g, j: original magnification ×100. b, c, e, f, h, i, k, l: original magnification ×200.
Discussion
Some studies that have included all glioma subtypes have suggested a positive association between the presence of 1p19q codeletion and a favorable clinical outcome not only in oligodendroglial but also among diffuse gliomas [16, 23, 24]. Indeed, a recent comprehensive analysis of 293 lower grade gliomas (LGG) by The Cancer Genome Atlas (TCGA) study group suggested that LGGs could be classified into three clinically relevant groups based on IDH and 1p19q status [25]. However, probably due to their infrequency, reports that have directly investigated the association between 1p19q codeletion and patient prognosis in astrocytic gliomas are limited, and the currently available results are inconclusive [16-18]. For example, a previous study concluded that losses on 1p and 19q marked a genetic subset of tumors diagnosed as pure grade II astrocytomas. However, whether these chromosome losses were prognostic for patients with astrocytomas was a matter of further investigation [12]. Although the number of 1p19q codeleted “astrocytic” gliomas analyzed in our study is still small, to our knowledge it nonetheless represents the largest study of such tumors. An additional advantage of our study is that 1p19q codeletion was judged by CGH. CGH is one of the most common techniques to evaluate 1p19q status [26] [27], and is superior to FISH because it can accurately distinguish between true total loss of 1p and 19q and partial loss of 1p. This is of high importance because partial loss of 1p is associated with the opposite clinical outcome [16, 20]. Although copy-neutral loss of heterozygosity can not be detected by CGH, such a situation is not the case with a total loss of 1p and 19q.
The number of oligodendroglial gliomas (as assessed by our institutional diagnosis) exhibiting total 1p19q loss (71.7%) was similar to that observed in previous studies [1, 17] and even higher than that found in phase III anaplastic oligodendroglial tumor studies [2, 9]. The number of “astrocytic” gliomas, as assessed by our institutional diagnosis (17.2%), was within the range previously reported in the literature (7.2% - 44%, average 20.8%) [10-16]. However, it might be slightly higher than expected because of a recent trend that favors oligodendroglial diagnosis [21]. Moreover, diagnoses of “astrocytic” in biopsy specimens could reflect focal heterogeneity within a single tumor [19, 28]. Therefore, the “astrocytic” gliomas, as assessed by our institutional diagnosis, may comprise a mixed population of purely astrocytic gliomas and gliomas previously designated “non-classic oligodendrogliomas” [4, 29].
Prognosis
Our study demonstrated that the survival curves of 1p19q codeleted “astrocytic” gliomas and oligodendrogliomas overlapped and that there was no statistical difference in the median survival time between the two groups. The observed median OS of the “astrocytic” gliomas with 1p19q loss (15.5 years) is much longer than that reported for low-grade astrocytomas (5–7 years) [30, 31] . The survival outcomes of the patients with “astrocytic” gliomas by resection were similar (14.7 years). Moreover, the fact that over half of the codeleted “astrocytic” gliomas in the present study were grade III tumors suggests a favorable prognosis compared with astrocytic gliomas in general (Table S1). The frequency of IDH mutations and MGMT promoter methylation was similar in both the “astrocytic” and oligodendroglioma groups. Because these genetic/epigenetic alterations are strong prognostic factors in diffuse gliomas, their similar frequency supports the hypothesis that both tumor types are likely to exhibit similar biological characteristics.
A previous study suggested the prognostic relevance of classic oligodendroglial morphology, regardless of the 1p19q status [4]. In contrast, in our consecutive cohort, we found that the survival curves of “astrocytic” and oligodendroglial gliomas with total 1p19q loss were nearly identical. Furthermore, our results were confirmed by comparison between oligodendroglial and “astrocytic” gliomas by consensus of institutional and single-pathologist diagnosis. The reasons for this discrepancy are unclear, although we can suggest some possible reasons. First, the present study included grade II and III tumors, in contrast to the previous study that included only grade III tumors, which frequently possess more complex genomic abnormalities. Second, the possible false inclusion of tumors with partial 1p and/or 19q loss (due to inaccurate FISH analysis) in non-classic tumors could have biased the previous results toward a poorer survival outcome.
Molecular-genetic characteristics of “astrocytic” gliomas with total 1p19q loss
Along with 1p19q codeletion, loss of chromosome 4q, loss on 14q, and loss on 18q are commonly observed chromosomal alterations in oligodendroglial tumors [32, 33]. In this study, these alterations occurred at a similar frequency in gliomas with 1p19q loss, regardless of their histological phenotype.
On the other hand, the finding that gain on chromosome 7q/7 was associated with an “astrocytic” phenotype even within codeleted gliomas was intriguing. Gain on chromosome 7q/7 is the most frequent chromosomal alteration in astrocytic tumors, as determined by CGH [10, 12, 34, 35]. Therefore, an increased frequency of 7q/7 gains could suggest that several codeleted “astrocytic” gliomas in our cohort indeed had molecular marker of astrocytic differentiation [34]. Although gain of chromosome 7 is known to be associated with the progression of gliomas [36], the survival outcomes of the patients with these tumors were comparable to those without these tumors (unpublished data).
Mutations in the TP53 and ATRX genes represent genetic hallmarks of astrocytomas [37, 38]. Loss of ATRX expression in three of the 15 codeleted “astrocytic” gliomas could suggest the existence of their astrocytic nature. However, the absence or a low frequency of TP53 and ATRX mutations compared with those of astrocytomas in general [37, 38] suggests that codeleted “astrocytic” gliomas in the present study were genetically similar to the codeleted oligodendrogliomas.
Limitations of our study
The histopathological assessments conducted in this study were not performed by a panel of neuropathologists. Moreover, five of the 16 “astrocytic” gliomas were diagnosed by biopsy specimens. Therefore, it is possible that the “astrocytic” gliomas analyzed here could contain a population of “non-classic oligodendrogliomas” with some astrocytic features [4].
The present study was retrospective and treatments were not uniform. Nonetheless, the cohort analyzed here was collected from consecutive cases treated at a single institution, and the survival analyses were based on long-term follow-ups. Moreover, the comparable survival outcomes between the two groups were confirmed in “astrocytic” gliomas by resection, and by comparison of the two groups with consensus diagnoses.
In conclusion, gliomas with total 1p19q loss with some or more “astrocytic” features are likely to have comparable biological and prognostic characteristics to those with oligodendroglial features. Although some of the codeleted “astrocytic” gliomas showed molecular markers associated with astrocytic differentiation, such as 7q/7 gain and ATRX loss, the molecular and genetic characteristics of those gliomas were, overall, very similar to codeleted oligodendrogliomas. Thus, total loss of 1p and 19q is a robust molecular and prognostic marker for gliomas, regardless of their histological features. These findings further substantiate the importance of molecular classification of gliomas.
Materials and Methods
Study population and tissue samples
Pathology records of brain tumors treated at the Department of Neurosurgery, Keio University Hospital between 1990 and 2010 were reviewed, and patients who fulfilled the following criteria were included: a) age > 18 years; b) supra-tentorial tumor location; c) institutional histopathological diagnosis of diffuse glioma of WHO classification grade II or III; d) no prior chemotherapy or radiotherapy treatment. The institutional histopathological diagnosis was conducted according to WHO criteria [22, 39-41].
Informed consent
Written informed consent was obtained from all 57 patients finally included in the study for translational research approved by the Institutional Review Board at Keio University (Approval Number 20050002).
Clinical data
Clinical data were obtained from the patients’ records and included age at diagnosis, sex, extent of surgery, composition of initial postoperative therapy, time to progression, treatment at recurrence, and date of death or last contact. The survival outcome of patients lost during follow-up was updated for the present study.
Reassessment of original histopathological diagnoses of gliomas with total 1p19q loss
The histological criteria for defining oligodendroglial tumors have changed over time, partly because of the relevance of the diagnosis to therapeutic decision-making and the estimation of prognosis [4, 21]. Although institutional diagnoses in our hospital over the past two decades have always been based on WHO criteria [22, 39-41], we were concerned that they may have been influenced by changes in diagnostic trends over time and/or by interobserver bias. Therefore, all cases with 1p19q codeletion were re-assessed by a single pathologist (TK), who was blinded to the 1p19q status, using hematoxylin and eosin (HE) staining based on the most recent WHO criteria [22]. Attention was paid to classical oligodendroglioma features such as uniform and rounded nuclei (often with small nucleoli) surrounded by perinuclear halos, and an even tissue distribution. Attention was also paid to features commonly found in astrocytic tumors, such as the oval to elongate, mildly pleomorphic, and more hyperchromatic nuclei, often with tapering eosinophilic cell processes, and irregular distribution [4, 29].
Molecular-genetic analyses
Tumor DNA was extracted from microdissected pieces of formalin-fixed paraffin-embedded (FFPE) tissue [42]. For tissue microdissection, care was taken to exclude intermixed non-neoplastic glial/vascular cells as well as hemorrhagic/necrotic regions. This was based on HE staining and MIB-1 immunohistochemistry (Dako, Glostrup, Denmark) on consecutive sections [43].
Chromosomal number aberrations (CNAs) were assessed by metaphase CGH as described previously [36, 44]. In brief, crude tumor DNA from FFPE tissue was amplified by degenerate oligonucleotide primed-polymerase chain reaction (DOP-PCR) and labeled with another DOP-PCR using digoxigenin (DIG)-11-dUTP (Roche, Mannheim, Germany). The reference DNA was amplified from 50 ng of normal male or female DNA and labeled with biotin-dUTP (Roche). The probe mixture was denatured and hybridized to normal metaphase spreads (Vysis, Downers Grove, IL). Unhybridized probes were washed out, and the metaphase spread was incubated with a fluorescein isothiocyanate (FITC)-conjugated anti-DIG antibody (Roche) and rhodamine-conjugated avidin (Roche). Preparations were washed and counterstained with 4,6-diamino-2-phenylinodole (DAPI) in antifade solution. Red, green, and blue images were acquired, and ratios of fluorescence intensity along chromosomes were quantitated using the CytoVision® Analysis System (Applied Imaging, San Jose, CA).
MGMT promoter methylation was assessed by methylation-specific PCR (MSP) using the EZ DNA Methylation-DirectTM Kit (Zymo Research Corp., Orange, CA) as described previously [45].
Mutation of the IDH1/2 genes was assessed in three steps. First, formalin-fixed paraffin (FFP) sections were examined for IDH1R132H by immunohistochemistry with an anti-mutant IDH1 antibody (Dianova, Hamburg, Germany) [46]. For negative cases, exon 4 of the IDH1 gene was amplified with previously described primers [47]. After purifying the PCR products, DNA sequencing was performed using the 3130xL Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). Finally, in cases negative for an IDH1 exon 4 mutation, exon 4 of IDH2 was similarly amplified and sequenced [47].
Accumulation of p53 and loss of alpha-thalassemia/mental retardation syndrome X-linked (ATRX) expression were assessed by immunohistochemistry in “astrocytic” gliomas by original institutional diagnosis as molecular surrogates of astrocytic nature. FFP sections were examined for ATRX immunohistochemistry with anti-human ATRX (1:400, Sigma, HPA001906) and for p53 immunohistochemistry with anti-human p53 (1:50, Novocastra, DO-1). For the evaluation of p53, FFP sections from anaplastic astrocytoma resected in 1994 were used as positive control; staining of >5% nuclei for p53 was judged as positive. For the evaluation of ATRX, FFP sections from anaplastic oligoastrocytoma resected in 2012 were used as positive control, and staining of endothelial cells was considered as an internal positive control. Staining of >10% nuclei was judged as positive for ATRX [38].
Statistical analysis
Patients were classified into two histopathological groups (oligodendroglial and astrocytic glioma). Demographic factors and the treatment and distribution of categorical factors such as CNAs were compared between groups by Fisher’s exact test. The overall survival (OS) was calculated from the beginning of treatment, i.e., from the date of initial surgery for resection cases and from the first day of adjuvant therapy for biopsy cases. Survival curves were estimated and compared between groups using the Kaplan–Meier method and the log rank test. Significance levels for all tests were 2-sided and 0.05. All data were analyzed with IBM SPSS statistics 22.
Acknowledgments
The authors would like to thank Ms. Yuko Aikawa, Ms. Naoko Tsuzaki, and Ms. Kiyomi Koide for their technical assistance.
Conflicts of Interest
None declared.
Grant Support
Supported by a Grant-in-Aid for Scientific Research (KAKENHI) by The Ministry of Education, Culture, Sports, Science and Technology and The Japan Society for the Promotion of Science (Grant Numbers 20591721, 23592141, 25462278).
Editorial note
This paper has been accepted based in part on peer-review conducted by another journal and the authors’ response and revisions as well as expedited peer-review in Oncotarget
References
1. Cairncross JG, Ueki K, Zlatescu MC, Lisle DK, Finkelstein DM, Hammond RR, Silver JS, Stark PC, Macdonald DR, Ino Y, Ramsay DA, Louis DN. Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst. 1998; 90:1473-1479.
2. Cairncross G, Berkey B, Shaw E, Jenkins R, Scheithauer B, Brachman D, Buckner J, Fink K, Souhami L, Laperierre N, Mehta M and Curran W. Phase III Trial of Chemotherapy Plus Radiotherapy Compared With Radiotherapy Alone for Pure and Mixed Anaplastic Oligodendroglioma: Intergroup Radiation Therapy Oncology Group Trial 9402. Journal of Clinical Oncology. 2006; 24:2707-2714.
3. Cairncross G, Wang M, Shaw E, Jenkins R, Brachman D, Buckner J, Fink K, Souhami L, Laperriere N, Curran W and Mehta M. Phase III trial of chemoradiotherapy for anaplastic oligodendroglioma: long-term results of RTOG 9402. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2013; 31:337-343.
4. Giannini C, Burger PC, Berkey BA, Cairncross JG, Jenkins RB, Mehta M, Curran WJ and Aldape K. Anaplastic oligodendroglial tumors: refining the correlation among histopathology, 1p 19q deletion and clinical outcome in Intergroup Radiation Therapy Oncology Group Trial 9402. Brain pathology. 2008; 18:360-369.
5. Griffin CA, Burger P, Morsberger L, Yonescu R, Swierczynski S, Weingart JD, and Murphy KM. Identification of der(1;19)(q10;p10) in Five Oligodendrogliomas Suggests Mechanism of Concurrent 1p and 19q Loss. Journal of Neuropathology & Experimental Neurology. 2006; 65:988-994 .
6. Ino Y, Betensky RA, Zlatescu MC, Sasaki H, Macdonald DR, Stemmer-Rachamimov AO, Ramsay DA, Cairncross JG and Louis DN. Molecular Subtypes of Anaplastic Oligodendroglioma: Implications for Patient Management at Diagnosis. Clinical Cancer Research. 2001; 7:839-845.
7. Jenkins RB, Blair H, Ballman KV, Giannini C, Arusell RM, Law M, Flynn H, Passe S, Felten S, Brown PD, Shaw EG and Buckner JC. A t(1;19)(q10;p10) Mediates the Combined Deletions of 1p and 19q and Predicts a Better Prognosis of Patients with Oligodendroglioma. Cancer Research. 2006; 66:9852-9861.
8. van den Bent MJ, Brandes AA, Taphoorn MJ, Kros JM, Kouwenhoven MC, Delattre JY, Bernsen HJ, Frenay M, Tijssen CC, Grisold W, Sipos L, Enting RH, French PJ, Dinjens WN, Vecht CJ, Allgeier A, et al. Adjuvant procarbazine, lomustine, and vincristine chemotherapy in newly diagnosed anaplastic oligodendroglioma: long-term follow-up of EORTC brain tumor group study 26951. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2013; 31:344-350.
9. van den Bent MJ, Carpentier AF, Brandes AA, Sanson M, Taphoorn MJ, Bernsen HJ, Frenay M, Tijssen CC, Grisold W, Sipos L, Haaxma-Reiche H, Kros JM, van Kouwenhoven MC, Vecht CJ, Allgeier A, Lacombe D, et al. Adjuvant procarbazine, lomustine, and vincristine improves progression-free survival but not overall survival in newly diagnosed anaplastic oligodendrogliomas and oligoastrocytomas: a randomized European Organisation for Research and Treatment of Cancer phase III trial. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2006; 24:2715-2722.
10. Arslantas A AS, Oner U. Genomic alterations in low-grade, anaplastic astrocytomas and glioblastomas. Pathol Oncol Res. 2007; 13:39-46.
11. Bello MJ, Leone PE, Nebreda P, de Campos J, Cusak ME, Vaquero J, Sarasa J, García-Miguel P, Queizan A, Hernández-Moneo J, Pestaña A and Rey JA. Allelic status of chromosome 1 in neoplasms of the nervous system. Cancer Genetics and Cytogenetics. 1995; 83:160-164.
12. Hirose Y, Aldape KD, Chang S, Lamborn K, Berger MS and Feuerstein BG. Grade II astrocytomas are subgrouped by chromosome aberrations. Cancer Genetics and Cytogenetics. 2003; 142:1-7.
13. Ino Y, Zlatescu MC, Sasaki H, Macdonald DR, Stemmer-Rachamimov AO, Jhung S, Ramsay DA, von Deimling A, Louis DN and Cairncross JG. Long survival and therapeutic responses in patient histologically disparate high-grade gliomas demonstrating chromosome 1p loss. Journal of Neurosurgery. 2000; 92:983-990.
14. Maintz D, Fiedler K, Koopmann J, Rollbrocker B, Nechev S, Lenartz D, Stangl AP, Louis DN, Schramm J, Wiestler OD and von Deimling A. Molecular genetic evidence for subtypes of oligoastrocytomas. Journal of Neuropathology & Experimental Neurology. 1997; 56:1098-1104
15. Maruno M, Yoshimine T, Muhammad AKMG, Ninomiya H, Kato A and Hayakawa T. Chromosomal aberrations detected by comparative genomic hybridization (CGH) in human astrocytic tumors. Cancer Letters. 1998; 135:61-66.
16. Vogazianou AP, Chan R, Backlund LM, Pearson DM, Liu L, Langford CF, Gregory SG, Collins VP and Ichimura K. Distinct patterns of 1p and 19q alterations identify subtypes of human gliomas that have different prognoses. Neuro Oncol. 2010; 12:664-678.
17. Smith JS PA, Borell TJ, Lee HK, O’Fallon J, Hosek SM, Kimmel D, Yates A, Burger PC, Scheithauer BW, Jenkins RB. . Alterations of chromosome arms 1p and 19q as predictors of survival in oligodendrogliomas, astrocytomas, and mixed oligoastrocytomas. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2000; 18:636-645.
18. Zhao J, Ma W and Zhao H. Loss of heterozygosity 1p/19q and survival in glioma: a meta-analysis. Neuro-Oncology. 2014; 16:103-112.
19. Kros JM, Gorlia T, Kouwenhoven MC, Zheng P-P, Collins VP, Figarella-Branger D, Giangaspero F, Giannini C, Mokhtari K, Mørk SJ, Paetau A, Reifenberger G and van den Bent MJ. Panel Review of Anaplastic Oligodendroglioma From European Organization for Research and Treatment of Cancer Trial 26951: Assessment of Consensus in Diagnosis, Influence of 1p/19q Loss, and Correlations With Outcome. Journal of Neuropathology & Experimental Neurology. 2007; 66:545-551.
20. Idbaih A, Marie Y, Pierron G, Brennetot C, Hoang-Xuan K, Kujas M, Mokhtari K, Sanson M, Lejeune J, Aurias A, Delattre O and Delattre JY. Two types of chromosome 1p losses with opposite significance in gliomas. Annals of neurology. 2005; 58:483-487.
21. Burger PC. What is an Oligodendroglioma? Brain pathology. 2002; 12:257-259.
22. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK. WHO Classification of Tumours of the Central Nervous System, 4th edn.International Agency for Research on Cancer: Lyon. 2007.
23. Kaloshi G, Benouaich-Amiel A, Diakite F, Taillibert S, Lejeune J, Laigle-Donadey F, Renard M-A, Iraqi W, Idbaih A, Paris S, Capelle L, Duffau H, Cornu P, Simon J-M, Mokhtari K, Polivka M, et al. Temozolomide for low-grade gliomas: Predictive impact of 1p/19q loss on response and outcome. Neurology. 2007; 68:1831-1836.
24. Wick W, Hartmann C, Engel C, Stoffels M, Felsberg J, Stockhammer F, Sabel MC, Koeppen S, Ketter R, Meyermann R, Rapp M, Meisner C, Kortmann RD, Pietsch T, Wiestler OD, Ernemann U, et al. NOA-04 randomized phase III trial of sequential radiochemotherapy of anaplastic glioma with procarbazine, lomustine, and vincristine or temozolomide. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2009; 27:5874-5880.
25. Yung WKA, Verhaak R, Cooper L, Salama S, Aldape K and Brat D. GE-41Comprehensive and integrative genomic characterization of diffuse lower grade gliomas. Neuro-Oncology. 2014; 16:v105.
26. Talagas M, Marcorelles P, Uguen A, Redon S, Quintin-Roué I, Costa S, Férec C, Morel F, Hieu P and De Braekeleer M. Identification of a novel population in high-grade oligodendroglial tumors not deleted on 1p/19q using array CGH. Journal of neuro-oncology. 2012; 109:405-413.
27. Branle F, Lefranc F, Camby I, Jeuken J, Geurts-Moespot A, Sprenger S, Sweep F, Kiss R and Salmon I. Evaluation of the efficiency of chemotherapy in in vivo orthotopic models of human glioma cells with and without 1p19q deletions and in C6 rat orthotopic allografts serving for the evaluation of surgery combined with chemotherapy. Cancer. 2002; 95:641-655.
28. Catherine Daumas-Duport PV, 1 Marie-Louise Tucker,1 Frederic Beuvon,1 Pascale Cervera1 and and Chodkiewicz2 J-P. Oligodendrogliomas. Part I: Patterns of growth, histological diagnosis,clinical and imaging correlations: A study of 153 cases. Journal of neuro-oncology. 1997; 34:37-59.
29. Sasaki H, Zlatescu MC, Betensky RA, Johnk LB, Cutone AN, Cairncross JG and Louis DN. Histopathological-Molecular Genetic Correlations in Referral Pathologist-Diagnosed Low-Grade “Oligodendroglioma”. Journal of Neuropathology & Experimental Neurology. 2002; 61:58-63.
30. Aldape K, Simmons ML, Davis RL, Miike R, Wiencke J, Barger G, Lee M, Chen P, Wrensch M. Discrepancies in diagnoses of neuroepithelial neoplasms: the San Francisco Bay Area Adult Glioma Study. Cancer. 2000; 88:2342-2349.
31. cBioPortal for Cancer Genomics, Brain Low Grade Glioma (http://www.cbioportal.org/study.do?cancer_study_id=lgg_tcga).
32. Bigner SH, Matthews MR, Rasheed BKA, Wiltshire RN, Friedman HS, Friedman AH, Stenzel TT, Dawes DM, McLendon RE and Bigner DD. Molecular Genetic Aspects of Oligodendrogliomas Including Analysis by Comparative Genomic Hybridization. The American Journal of Pathology. 1999; 155:375-386.
33. Kitange G, Misra A, Law M, Passe S, Kollmeyer TM, Maurer M, Ballman K, Feuerstein BG and Jenkins RB. Chromosomal imbalances detected by array comparative genomic hybridization in human oligodendrogliomas and mixed oligoastrocytomas. Genes, chromosomes & cancer. 2005; 42:68-77.
34. Camelo-Piragua S, Jansen M, Ganguly A, Kim JC, Cosper AK, Dias-Santagata D, Nutt CL, Iafrate AJ and Louis DN. A Sensitive and Specific Diagnostic Panel to Distinguish Diffuse Astrocytoma from Astrocytosis: Chromosome 7 Gain with Mutant Isocitrate Dehydrogenase 1 and p53. Journal of neuropathology and experimental neurology. 2011; 70:110-115.
35. Dahlback HS, Brandal P, Gorunova L, Widing E, Meling TR, Krossnes BK and Heim S. Genomic aberrations in pediatric gliomas and embryonal tumors. Genes, chromosomes & cancer. 2011; 50:788-799.
36. Hirose Y, Aldape K, Takahashi M, Berger MS and Feuerstein BG. Tissue Microdissection and Degenerate Oligonucleotide Primed-Polymerase Chain Reaction (DOP-PCR) Is an Effective Method to Analyze Genetic Aberrations in Invasive Tumors. The Journal of Molecular Diagnostics. 2001; 3:62-67.
37. Jiao Y, Killela PJ, Reitman ZJ, Rasheed BA, Heaphy CM, de Wilde RF, Rodriguez FJ, Rosemberg S, Oba-Shinjo SM, Marie SKN, Bettegowda C, Agrawal N, Lipp E, Pirozzi CJ, Lopez GY, He Y, et al. Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget. 2012; 3:709-722.
38. Sahm F, Reuss D, Koelsche C, Capper D, Schittenhelm J, Heim S, Jones DW, Pfister S, Herold-Mende C, Wick W, Mueller W, Hartmann C, Paulus W and von Deimling A. Farewell to oligoastrocytoma: in situ molecular genetics favor classification as either oligodendroglioma or astrocytoma. Acta Neuropathol. 2014; 128:551-559.
39. Zulch KJ. (1979) Histological Typing of Tumours of the Central Nervous System. World Health Organization: Geneva. 1979.
40. Kleihues P, Burger PC, Scheithauer BW. (1993) Histological Typing of Tumours of the Central Nevous System, World Health Organization: 2nd edn. Springer-Verlag: Berlin. 1993.
41. Kleihues P, Cavenee WK. (2000) World Health Organization Classification of Tumours-Pathology and Genetics. Tumours of the Nervous System. IARC Press: Lyon. 2000.
42. Sasaki H, Zlatescu MC, Betensky RA, Ino Y, Cairncross JG and Louis DN. PTEN Is a Target of Chromosome 10q Loss in Anaplastic Oligodendrogliomas and PTEN Alterations Are Associated with Poor Prognosis. The American Journal of Pathology. 2001; 159:359-367.
43. Kitamura Y, Sasaki H, Kimura T, Miwa T, Takahashi S, Kawase T and Yoshida K. Molecular and Clinical Risk Factors for Recurrence of Skull Base Chordomas: Gain on Chromosome 2p, Expression of Brachyury, and Lack of Irradiation Negatively Correlate With Patient Prognosis. Journal of Neuropathology & Experimental Neurology. 2013; 72:816-823.
44. Miwa T, Hirose Y, Sasaki H, Ezaki T, Yoshida K and Kawase T. Single-Copy Gain of Chromosome 1q Is a Negative Prognostic Marker in Pediatric Nonependymal, Nonpilocytic Gliomas. Neurosurgery. 2011; 68:206-212.
45. Ezaki T, Sasaki H, Hirose Y, Miwa T, Yoshida K and Kawase T. Molecular characteristics of pediatric non-ependymal, nonpilocytic gliomas associated with resistance to temozolomide. Molecular medicine reports. 2011; 4:1101-1105.
46. Capper D, Zentgraf H, Balss J, Hartmann C and von Deimling A. Monoclonal antibody specific for IDH1 R132H mutation. Acta Neuropathol. 2009; 118(5):599-601.
47. Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, Friedman H, Friedman A, Reardon D, Herndon J, Kinzler KW, Velculescu VE, et al. IDH1 and IDH2 Mutations in Gliomas. New England Journal of Medicine. 2009; 360:765-773.