
INTRODUCTION
Colorectal cancer is the third most common malignancy and a leading cause of cancer-related deaths worldwide [1-4]. Early diagnosis and treatment have improved survival of colorectal cancer, but the mortality rate is still the fourth highest in males and third highest in females [4, 5]. This high mortality rate is due to recurrence and metastasis, which occur in approximately 50% of patients during the course of disease [6, 7]. Surgical resection combined with systemic chemotherapy has improved survival rates in colorectal cancer, but treatment outcomes in patients whose disease has progressed remain unsatisfactory. Newer targeted agents such as cetuximab and panitumumab are widely used to treat metastatic colorectal cancer [8, 9]. However, some patients do not respond to these targeted therapies [10], indicating the need to develop personalized treatments for these patients.
Numerous molecular investigations have been carried out to develop personalized treatments [11], requiring models that accurately represent the biologic characteristics of the individual patient. Such preclinical studies have used cancer cell lines or patient-derived xenograft (PDX) models [10, 12]. Although cell lines are practical and easy to manipulate, they generally show poorly differentiated histology and lack similarity to the original tumor [13, 14], whereas PDX models better reflect characteristics of the original tumor, including tumor heterogeneity [12, 13]. As a result, PDX models have been widely used to develop treatment strategies for patients with refractory cancer. However, not all tumors specimens from cancer patients engraft successfully in animal models, and this difference may be associated with the progressiveness of the original tumor. Despite the increased use of PDX models, few studies have reported the relationship between engraftment of tumor specimens and clinical features of patients with colorectal cancer. This relationship may be useful in the interpretation of results in preclinical studies using PDX models. Therefore, in this study we evaluated the relationship between tumor engraftment in PDX models and clinical outcomes in patients with colorectal cancer.
RESULTS
Patient characteristics and tumorigenicity
Of the 241 patients with colorectal cancer, 135 were male and 106 were female. Median age was 59.9 years (range, 24–89). Patients were classified according to cancer stage: stage I (n = 15), stage II (n = 72), stage III (n = 84), and stage IV (n = 70). In patients with stage IV cancer, sites of metastasis included the liver (n = 61), distant lymph nodes (n = 4), lung (n = 3), ovary (n = 1), and peritoneum (n = 1).
Of the 241 tumor specimens, 150 (62.2%) successfully engrafted, reaching a size of 1,000 mm3 in 90 ± 20 days. The remaining 91 tumor specimens (37.8%) failed to engraft. Tumorigenicity according to patient characteristics is shown in Table 1. Tumor take rates were significantly higher for more advanced stage primary tumors (p < 0.001), with xenografts established from four of 15 (26.7%) stage I tumors, 41 of 72 (56.9%) stage II tumors, 50 of 84 (59.5%) stage III tumors, and 55 of 70 (78.6%) stage IV tumors. Tumor take rates were significantly higher for moderately differentiated (66.5%) and poorly differentiated (66.7%) tumors compared with well-differentiated tumors (46.7%, p = 0.029).
Among the 70 patients with stage IV tumors, 50 PDX models were established using paired xenografts from primary and metastatic liver tumors. Tumorigenicity appeared to be higher for metastatic lesions than for primary tumors (84.0% vs. 78.6%), but this difference was not significant (p = 0.456).
Table 1: Patient characteristics and tumorigenicity of primary tumors in PDX models.

Clinical outcomes and tumorigenicity
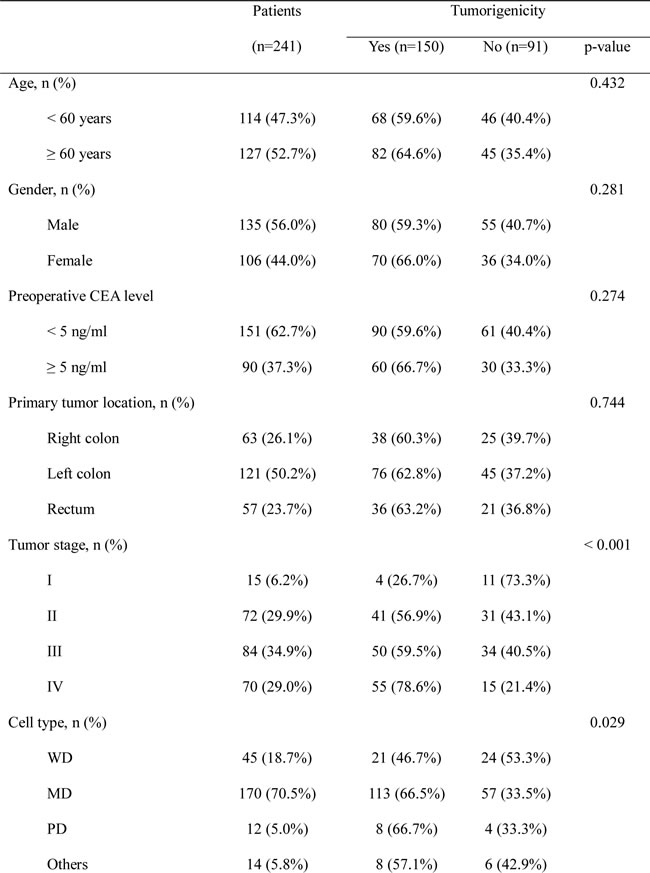
To better understand the relationship between PDX tumorigenicity and clinical outcomes, we analyzed the DFS of patients according to tumorigenicity. Median follow-up was 22.9 months (range, 0.2–51.3), and there were 58 recurrences and three deaths. The 3-year DFS rate of patients whose tumors successfully engrafted in the PDX model was significantly lower than that of patients whose tumors failed to engraft (56.1% vs. 81.5%, p = 0.011) (Figure 1A).
Further analysis of 3-year DFS rates according to cancer stage showed no significant difference between patients with stage I–II cancer whose tumors failed to engraft and those whose tumors successfully engrafted (89.5% vs. 91.5%, respectively, p = 0.861) (Figure 1B). However, 3-year DFS was significantly lower for patients with stage III cancer whose tumors engrafted in the PDX model compared with those whose tumors failed to engraft (56.5% vs. 91.1%, p = 0.012) (Figure 1C). Results of multivariate analysis revealed that tumorigenicity in the PDX model (HR, 4.966; 95% CI, 1.126–21.905; p = 0.034) and old age (HR, 0.027; 95% CI, 1.178–14.600; p = 0.027) were independent predictors of DFS in patients with stage III cancer (Table 2). In patients with stage IV cancer, 3-year DFS appeared to be lower for patients whose tumors engrafted compared with those whose tumors failed to engraft (30.4% vs. 42.4%); however, this difference was not significant (p = 0.842) (Figure 2A). Similar results were obtained for the corresponding 50 liver metastatic lesions (32.3% vs. 56.3%, p = 0.911) (Figure 2B).
Table 2: Multivariate analysis of 3-year disease-free survival in patients with stage III colorectal cancer.
Variable |
p-value |
HR (95% CI) |
| Tumorigenicity (+) | 0.034 | 4.966 (1.126–21.905) |
| Age (≥ 60 years) | 0.027 | 4.148 (1.178–14.600) |
HR, hazard ratio; CI, confidence interval.
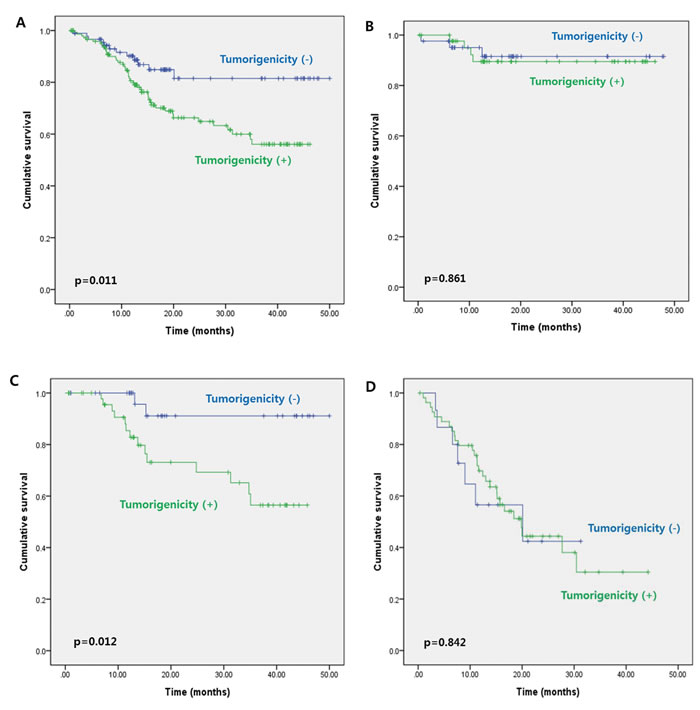
Figure 1: Three-year disease-free survival according to tumorigenicity of the primary colorectal tumor for A. all patients (stage I–IV cancer), B. patients with stage I–II cancer, C. patients with stage III cancer, and D. patients with stage IV cancer.
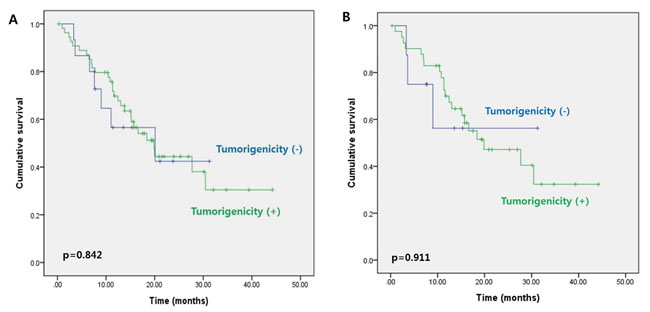
Figure 2: Three-year disease-free survival in patients with stage IV colorectal cancer according to tumorigenicity of the A. primary tumors and B. liver metastatic lesions.
Somatic DNA mutations of primary tumors with tumorigenicity
To investigate the mutational status of patients with tumorigenicity, we performed genomic profiling for successfully engrafted samples of primary tumor. We selected five tumor samples from patients with stage III disease, and analyzed the somatic DNA mutations of eight genes selected for their importance in colorectal cancer (Table 3). Nonsense and missense mutations were shown, where three of the five samples (60%) had transcription stop-gaining APC point mutations and four samples had TP53 point mutations (80%, one transcription stop-gaining mutation and four missense mutations). The mutation frequency of each gene was compared to that from 272 stage I-IV colon adenocarcinoma samples in The Cancer Genome Atlas (TCGA). The five samples from our study did not demonstrate KRAS mutation (0%), while 36% of TCGA samples had KRAS mutations. Four samples exhibited TP53 mutations (80%), while only 50% of TCGA samples had TP53 mutations. Even though the differences in mutation frequencies were not statistically significant due to the small sample size, further genomic profiling in future studies could elucidate these differences.
Table 3: Somatic DNA mutations of primary tumors from stage III patients.
Drug sensitivity to EGFR-targeted agents for PDX tumors
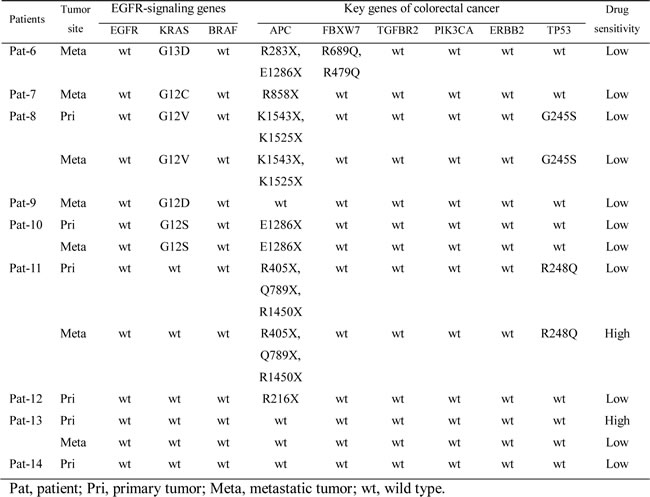
To investigate responsiveness to epidermal growth factor receptor (EGFR)-targeted agents according to mutation of EGFR-signaling genes, we performed genomic profiling for PDX tumors and drug sensitivity assay. Thirteen PDX models from nine patients with stage IV disease were selected for drug sensitivity testing. Somatic DNA mutations of selected genes, which either had known importance in colorectal cancer or belonged to the EGFR signaling pathway, were identified. Sensitivities to EGFR-targeted agents are presented for each model (Table 4). Nonsense and missense mutations are also presented, where two of 13 models showed high sensitivity to the EGFR-targeted agents, and both models belonged to the group without somatic KRAS mutations. None of the models with somatic KRAS mutations showed high sensitivity to the EGFR-targeted agents. Clearer correlation between the genomic profile of EGFR-signaling genes and responsiveness to EGFR targeted treatments can be shown with further enriched genomic profiles in the future.
Table 4: Somatic DNA mutations and drug sensitivity to EGFR targeted agents for PDX tumors.
DISCUSSION
In this study, we evaluated the relationship between successful tumor engraftment in PDX models and the clinical features of patients with colorectal cancer. Similar to the results of previous studies [10, 15], xenografts were successfully established with 62.2% of the primary tumors. Successful engraftment of the primary tumors was associated with advanced cancer stage and moderately/poorly differentiation. Primary tumor engraftment also appeared to be associated with higher preoperative carcinoembryonic antigen levels, vascular or lymphatic invasion, and microsatellite instability. For patients with stage IV cancer, the rate of engraftment was higher for metastatic lesions than for primary lesions. These results suggest that colorectal cancers with more aggressive features are better able to engraft than less aggressive cancers. In our study, 3-year DFS rates were lower for patients whose tumors successfully engrafted in the PDX model compared with patients whose tumors failed to engraft. However, when these patients were classified by cancer stage, stage III cancer showed a significant association between tumor engraftment and decreased DFS. In addition, tumorigenicity was found to be an independent predictor of DFS in stage III cancer. These findings suggest that patients with stage III cancer whose tumors successfully engraft in PDX models have an increased risk of relapse.
Few studies have evaluated the relationship between successful tumor engraftment in a PDX model and clinical outcomes in colorectal cancer [10, 12]. In a previous study to evaluate drug-response and tumor progression using 32 PDX models, tumor take rates were higher for poorly differentiated tumors and tumors from patients with lymph node invasion; however, survival did not differ among patient groups [12]. In another study, the authors compared tumor take rates of primary (n = 58) and metastatic (n = 27) lesions, reporting higher tumor take rates for metastatic lesions; however, this difference was not significant [10]. In our study, which analyzed a larger sample size than previous studies, we evaluated the relationships between tumor engraftment in the PDX model and patient characteristics and DFS. Similar studies have been carried out for non-small cell lung cancer, breast cancer, and uveal melanoma [11, 14, 16, 17]. John et al. reported that tumorigenicity correlated with the presence of KRAS mutations, poor differentiation, and larger tumor size in non-small cell lung cancer. In addition, tumorigenicity was an independent predictor of shorter DFS [14]. In contrast, Anderson et al. reported that tumorigenicity did not correlate with clinical outcomes in non-small cell lung cancer [16].
Cancer cell lines and PDX models have been widely used in the development of personalized cancer treatments. Xenografts derived from cell lines are reproducible, easy to manipulate, and well characterized; however, they do not exhibit tumor heterogeneity or the histopathologic and genetic characteristics of the tumor [8, 13, 14]. Numerous genomic mutations have been detected in patients with cancer, and high levels of oncogene mutation can accelerate the growth of tumors [18, 19]. In our study, we performed genomic profiling for primary tumor cells from patients whose tumors were successfully engrafted in the PDX models. We detected several mutations of colorectal cancer-related genes, and mutation of TP53 was the most frequently detected. According to several reports, PDX models better reflect the genetic diversity of the original tumor, and better predict clinical tumor response to new therapeutics [8, 12, 13, 20, 21]. Drug sensitivity assay with genomic profiling using PDX models can provide more information about primary tumors and new therapeutic strategies for patients, especially those with disease refractory to conventional treatments. In this study, we investigated somatic DNA mutations and accompanying sensitivity to EGFR-targeted agents in PDX models derived from patients with stage IV disease. Initial genomic profiling of PDX models and evaluation of EGFR-targeted treatments showed selective responsiveness depending on KRAS mutation. This is consistent with previous studies indicating that the mutation of EGFR-downstream KRAS can increase resistance to EGFR-targeted treatments [22-24]. However, responsiveness to EGFR-targeted agents can depend on several other factors not addressed in this study, which include DNA copy number changes, methylation status, and gene/protein expression levels. Available genomic profiles along with correlated drug response information significantly increases the applicability of PDX models, and further research is needed to complete the genomic and drug-response profiles of our PDX models.
PDX models are important for the development of novel treatments, especially for patients with refractory cancer, indicating the need for well-characterized xenograft models. In this study, our models stably established and we found that successful engraftment of patient-derived colorectal cancer cells correlates with more aggressive characteristics and worse outcomes. Therefore PDX models may provide an effective preclinical tool to evaluate cancer progression and treatment strategies. In addition, these models can be used for further genomic and pharmacologic studies to personalized treatments. Our results provide evidence that PDX models are applicable to colorectal cancer patients with a progressive disease course and high risk of relapse. We anticipate that personalized treatments using PDX models will improve survival rates for these patients.
In conclusion, our findings show that the successful engraftment of colorectal cancer tumors in PDX models is associated with more aggressive disease and worse clinical outcomes. Our PDX models maybe useful to predict disease progression in preclinical studies for personalized medicine and to improve clinical outcomes of patients. Further studies for genomic and pharmacologic information will provide novel treatments for patients with colorectal cancer.
MATERIALS AND METHODS
Patients
This study was approved by the Samsung Medical Center Institutional Review Board (no. 2010-04004). A total of 241 patients with colorectal cancer who underwent surgery from March 2010 to April 2013 at the Samsung Medical Center were included. All patients had histologically confirmed primary adenocarcinoma and underwent radical surgery for the primary tumor and synchronous metastatic lesions. Patients who underwent palliative operations and those with recurrent disease or synchronous malignancies were excluded.
Tumor samples
Specimens from primary and metastatic tumors were obtained from patients who had provided written informed consent. Tumor tissues not required for clinical diagnosis were placed in Roswell Park Memorial Institute (RPMI) medium supplemented with 250 U/ml penicillin and 250 µg/ml streptomycin. Each tumor sample was cut into 5- to 10-mm3 pieces, some of which were snap frozen in liquid nitrogen and stored in a freezer at –80°C or in liquid nitrogen for molecular analysis. Two pieces were fixed in formalin solution and paraffin-embedded for histopathologic analysis, and two pieces were coated in high concentration Matrigel (BD Biosciences, Erembodegem, Belgium) and implanted in 6- to 8-week-old female Balb/c nude mice (Orient Bio, Seongnam, Korea). A similar process of sample preservation was carried out for tumor tissues collected from mice.
Establishment of PDXs
Establishment of PDXs was performed as previously described [8]. Briefly, Matrigel-embedded tumor fragments (1–2 mm3) were implanted into subcutaneous pockets made in each side of the lower back. Tumors that reached a volume of 1,000 mm3 were considered tumorigenic. All animal experiments were carried out according to protocols approved by the appropriate institutional review boards of the Samsung Medical Center (K-B2-036) and conducted in accordance with the Institute for Laboratory Animal Research Guide for the Care and Use of Laboratory Animals.
Genomic profiling of patients and PDX tumors
Liquid nitrogen-preserved samples from patients and PDXs were mechanically dissociated and genomic DNA was extracted using the QIAamp DNA mini kit (Qiagen, Valencia, CA). Agilent Sureselect Human All Exome Kit v4 was used to capture the exon region from DNA. To obtain DNA for patients’ normal control blood cells, data from raw sequencing reads was produced by Illumina HiSeqTM2500. To obtain the DNA of tumor tissues from patients and PDXs, raw sequencing reads were produced by Illumina HiSeqTM2000. This resulted in mean coverage rates in the exome region of around 120x for the tumor tissues and around 80x for normal blood cells. Somatic mutations in tumor DNA were identified using the next generation sequencing (NGS) pipeline at Samsung Genome Institute, Seoul, Korea.
Drug sensitivity assay of PDX tumors
Dissociated tumor cells from PDXs were grown in modified neurobasal A medium (Invitrogen, Carlsbad, CA) containing N2 supplement (Invitrogen), bFGF (20 ng/ml, Invitrogen), and EGF (50 ng/ml, Invitrogen). Tumor cells were seeded in 96-well plates at 1,000 cells per well, and treated with EGFR-targeted agents (Selleckchem, Houston, TX, USA) - AEE788, Afatinib, BMS-599626, Canertinib, CO-1686, Dacomitinib, Erlotinib, Gefitinib, Lapatinib, Neratinib - under seven-point serial dilution concentrations up to 20 µM (n = 3 for each condition). After three days of incubation at 37°C in a 5% CO2 humidified incubator, cell viability was analyzed using the metabolic conversion of a water-soluble tetrazolium salt, WST-1 (Roche, Indianapolis, IN). Plates were analyzed with a spectrophotometer at 450 nm, with a reference wavelength of 630 nm. For each drug-sample pair, the drug response curve was approximated using GraphPad Prism 6.0 (GraphPad Software, San Diego, CA, USA) based on the measured cell viabilities from varying drug concentrations.
Clinicopathologic analysis
The tumor take rates of primary tumor specimens were analyzed according to patient characteristics. In addition, patients were grouped according to cancer stage, and survival rates of each group were analyzed according to tumorigenicity (i.e., tumor engraftment in the PDX model). In stage IV cases, survival rates were evaluated according to tumorigenicity of the primary tumors and corresponding liver metastatic lesions. The primary endpoint of this study was clinical outcome according to tumorigenicity. The secondary endpoint was response of PDX tumors to EGFR-targeted agents according to mutational status.
Statistical analysis
Statistical analysis was performed using SPSS for Windows version 20.0 (IBM SPSS Statistics, IBM Corporation, Armonk, NY). Patient characteristics were compared using the chi-squared test or linear-by-linear association. Survival rates were analyzed using the Kaplan–Meier method and log-rank test. Multivariate analysis was performed using logistic regression to identify predictors of survival. Factors that were significant or near significant (p < 0.1) in univariate analysis were included in the multivariate model; p < 0.05 was considered significant.
ACKNOWLEDGEMENTS
This research was supported by Samsung Medical Center grant and by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea. (HI14C3418).
ROLE OF THE FUNDING SOURCE
The funding source did not have any role in the design of the study, in the analysis of the data, or in the writing of the paper.
CONFLICTs OF INTEREST STATEMENT
None declared.
REFERENCES
1. Huang MY, Wu CH, Huang CM, Chung FY, Huang CW, Tsai HL, Chen CF, Lin SR and Wang JY. DPYD, TYMS, TYMP, TK1, and TK2 genetic expressions as response markers in locally advanced rectal cancer patients treated with fluoropyrimidine-based chemoradiotherapy. Biomed Res Int. 2013; 2013: 931028.
2. Zhang ZJ, Zheng ZJ, Kan H, Song Y, Cui W, Zhao G and Kip KE. Reduced risk of colorectal cancer with metformin therapy in patients with type 2 diabetes: a meta-analysis. Diabetes Care. 2011; 34: 2323-2328.
3. Sui X, Xu Y, Yang J, Fang Y, Lou H, Han W, Zhang M, Chen W, Wang K, Li D, Jin W, Lou F, Zheng Y, et al. Use of metformin alone is not associated with survival outcomes of colorectal cancer cell but AMPK activator AICAR sensitizes anticancer effect of 5-fluorouracil through AMPK activation. PLoS One. 2014; 9: e97781.
4. Jemal A, Bray F, Center MM, Ferlay J, Ward E and Forman D. Global cancer statistics. CA Cancer J Clin. 2011; 61: 69-90.
5. Oh BY, Kim KH, Chung SS, Hong KS and Lee RA. Role of beta1-Integrin in Colorectal Cancer: Case-Control Study. Ann Coloproctol. 2014; 30: 61-70.
6. Kamiyama H, Noda H, Konishi F and Rikiyama T. Molecular biomarkers for the detection of metastatic colorectal cancer cells. World J Gastroenterol. 2014; 20: 8928-8938.
7. Taieb J, Tabernero J, Mini E, Subtil F, Folprecht G, Van Laethem JL, Thaler J, Bridgewater J, Petersen LN, Blons H, Collette L, Van Cutsem E, Rougier P, et al. Oxaliplatin, fluorouracil, and leucovorin with or without cetuximab in patients with resected stage III colon cancer (PETACC-8): an open-label, randomised phase 3 trial. Lancet Oncol. 2014; 15: 862-873.
8. Cho YB, Hong HK, Choi YL, Oh E, Joo KM, Jin J, Nam DH, Ko YH and Lee WY. Colorectal cancer patient-derived xenografted tumors maintain characteristic features of the original tumors. J Surg Res. 2014; 187: 502-509.
9. Bokemeyer C, Bondarenko I, Hartmann JT, de Braud F, Schuch G, Zubel A, Celik I, Schlichting M and Koralewski P. Efficacy according to biomarker status of cetuximab plus FOLFOX-4 as first-line treatment for metastatic colorectal cancer: the OPUS study. Ann Oncol. 2011; 22: 1535-1546.
10. Julien S, Merino-Trigo A, Lacroix L, Pocard M, Goere D, Mariani P, Landron S, Bigot L, Nemati F, Dartigues P, Weiswald LB, Lantuas D, Morgand L, et al. Characterization of a large panel of patient-derived tumor xenografts representing the clinical heterogeneity of human colorectal cancer. Clin Cancer Res. 2012; 18: 5314-5328.
11. Nemati F, Sastre-Garau X, Laurent C, Couturier J, Mariani P, Desjardins L, Piperno-Neumann S, Lantz O, Asselain B, Plancher C, Robert D, Peguillet I, Donnadieu MH, et al. Establishment and characterization of a panel of human uveal melanoma xenografts derived from primary and/or metastatic tumors. Clin Cancer Res. 2010; 16: 2352-2362.
12. Puig I, Chicote I, Tenbaum SP, Arques O, Herance JR, Gispert JD, Jimenez J, Landolfi S, Caci K, Allende H, Mendizabal L, Moreno D, Charco R, et al. A personalized preclinical model to evaluate the metastatic potential of patient-derived colon cancer initiating cells. Clin Cancer Res. 2013; 19: 6787-6801.
13. Marangoni E, Vincent-Salomon A, Auger N, Degeorges A, Assayag F, de Cremoux P, de Plater L, Guyader C, De Pinieux G, Judde JG, Rebucci M, Tran-Perennou C, Sastre-Garau X, et al. A new model of patient tumor-derived breast cancer xenografts for preclinical assays. Clin Cancer Res. 2007; 13: 3989-3998.
14. John T, Kohler D, Pintilie M, Yanagawa N, Pham NA, Li M, Panchal D, Hui F, Meng F, Shepherd FA and Tsao MS. The ability to form primary tumor xenografts is predictive of increased risk of disease recurrence in early-stage non-small cell lung cancer. Clin Cancer Res. 2011; 17: 134-141.
15. Tokunaga T, Nakamura M, Oshika Y, Ohnishi Y and Ueyama Y. Is xenotransplantability of human colon cancers in SCID mice affected by angiogenic factors? J Natl Cancer Inst. 1998; 90: 400-401.
16. Anderson TM, Hess SD, Egilmez NK, Nwogu CE, Lenox JM and Bankert RB. Comparison of human lung cancer/SCID mouse tumor xenografts and cell culture growth with patient clinical outcomes. J Cancer Res Clin Oncol. 2003; 129: 565-568.
17. DeRose YS, Wang G, Lin YC, Bernard PS, Buys SS, Ebbert MT, Factor R, Matsen C, Milash BA, Nelson E, Neumayer L, Randall RL, Stijleman IJ, et al. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat Med. 2011; 17: 1514-1520.
18. Lee WS, Kim HY, Seok JY, Jang HH, Park YH, Kim SY, Shin DB and Hong S. Genomic profiling of patient-derived colon cancer xenograft models. Medicine (Baltimore). 2014; 93: e298.
19. Burgenske DM, Monsma DJ, Dylewski D, Scott SB, Sayfie AD, Kim DG, Luchtefeld M, Martin KR, Stephenson P, Hostetter G, Dujovny N and MacKeigan JP. Establishment of genetically diverse patient-derived xenografts of colorectal cancer. Am J Cancer Res. 2014; 4: 824-837.
20. Kopetz S, Lemos R and Powis G. The promise of patient-derived xenografts: the best laid plans of mice and men. Clin Cancer Res. 2012; 18: 5160-5162.
21. Siolas D and Hannon GJ. Patient-derived tumor xenografts: transforming clinical samples into mouse models. Cancer Res. 2013; 73: 5315-5319.
22. De Roock W, Piessevaux H, De Schutter J, Janssens M, De Hertogh G, Personeni N, Biesmans B, Van Laethem JL, Peeters M, Humblet Y, Van Cutsem E and Tejpar S. KRAS wild-type state predicts survival and is associated to early radiological response in metastatic colorectal cancer treated with cetuximab. Ann Oncol. 2008; 19: 508-515.
23. Di Fiore F, Blanchard F, Charbonnier F, Le Pessot F, Lamy A, Galais MP, Bastit L, Killian A, Sesboue R, Tuech JJ, Queuniet AM, Paillot B, Sabourin JC, et al. Clinical relevance of KRAS mutation detection in metastatic colorectal cancer treated by Cetuximab plus chemotherapy. Br J Cancer. 2007; 96: 1166-1169.
24. Lievre A, Bachet JB, Boige V, Cayre A, Le Corre D, Buc E, Ychou M, Bouche O, Landi B, Louvet C, Andre T, Bibeau F, Diebold MD, et al. KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J Clin Oncol. 2008; 26: 374-379.