
INTRODUCTION
Lung cancer has high incidence rates worldwide, and its 5-year survival is dismal as most cases are diagnosed at late stages. Chemotherapy, although with limited efficacy, used to be the main treatment option for patients with advanced lung cancer [1]. In 2004, somatic mutations were reported to exist in the tyrosine kinase domain of epidermal growth factor receptor (EGFR) in tumors of a subset of patients with non-small cell lung cancer (NSCLC) who responded dramatically to EGFR tyrosine kinase inhibitors (TKIs) [2, 3]. This discovery has opened a new era of targeted therapy for NSCLC. Nowadays, EGFR-TKIs are used as the standard first-line therapy for patients with advanced lung adenocarcinoma harboring activating EGFR mutations [4, 5], and they remarkably improve the survival and quality of life in patients with these driver mutations [6].
Drug resistance is a major obstacle in targeted cancer therapy, and understanding the mechanisms of resistance is pivotal for developing more effective treatment strategies. Around 70% of patients with lung adenocarcinoma that has activating EGFR mutations (mostly a small in-frame deletion in exon 19 and a substitution mutation L858R) display objective clinical response to EGFR-TKI treatment [7-11]. However, despite the initial disease control, tumor relapse is inevitably observed after a median of 9-14 months, indicating the development of acquired resistance to EGFR-TKIs in these patients [7-9, 11]. Studies have identified different mechanisms of acquired EGFR-TKI resistance, including a second-site EGFR T790M mutation [12, 13], MET amplification [14, 15], PIK3CA mutations [16], FGFR1 activation [17] , epithelial-to-mesenchymal transitions and conversion to small cell carcinoma [16, 18-19]. On the other hand, ~30% patients with TKI-sensitive EGFR mutations fail to demonstrate objective tumor regression on initial EGFR-TKI therapy and are defined as having primary or intrinsic resistance [7,9, 11]. Some co-existing genetic variations have been implicated in the mechanism of TKI insensitivity in EGFR-mutant patients, including de novo presence of EGFR T790M or MET amplification [20, 21], KRAS mutations [22], loss of PTEN [23], and a germline deletion polymorphism of BIM [24]. However, the majority of resistant cases cannot be explained by these variations and the mechanistic basis for intrinsic EGFR-TKI resistance in patients supposed to be responsive is still largely unknown.
In this study, we hypothesized that specific genetic alterations may underlie the primary resistance to EGFR-TKIs in lung adenocarcinoma harboring activating EGFR mutations. Towards uncovering such genetic determinants of treatment resistance, we performed next-generation sequencing (NGS)-based mutation profiling of lung adenocarcinomas with the EGFR L858R mutation from patients who received EGFR-TKI therapy, and searched for genetic variants/mutations that could differentiate patients displaying primary resistance to EGFR-TKIs from those having a durable response.
RESULTS
Forty-six-gene mutation profiles of EGFR L858R-positive lung adenocarcinomas
NGS was used to interrogate mutations within hotspot regions of 46 cancer-related genes in lung adenocarcinoma samples from 13 and 16 EGFR-TKI-treated patients who had short (< 3 months) and long (> 1 year) PFS, respectively. Differential mutation patterns were revealed in these two groups (Fig. 1 and more details in the Supplementary Appendix). All 29 tumors were confirmed to harbor the activating EGFR L858R mutation without the simultaneous presence of the T790M allele that predicts EGFR-TKI resistance. Among the 46 genes, KDR (which encodes for vascular endothelial growth factor receptor 2) was the most commonly mutated gene coexisting with EGFR L858R, regardless of the patient’s treatment response. Mutation rates of ABL1, APC, and PDGFRA were disproportionately high in the patient with long PFS. In contrast, mutations in FGFR2 (K368E), KRAS (G12D), MLH1 (V384D), and TP53 occurred more often in patients with short PFS. Derepression of FGFR2 expression has been implicated in the mechanism for rapidly acquired EGFR-TKI resistance in NSCLC cells [12]. KRAS G12C is linked to poor outcomes of EGFR-TKI therapy in NSCLC patients [13]. A study shows higher p53 mutation rates in advanced-stage than in early-stage lung cancers and suggests that the concurrent occurrence of p53 mutations with EGFR mutations may foster the development of therapeutic resistance [25]. However, the DNA mismatch repair gene MLH1 has not been associated with EGFR-TKI resistance yet.
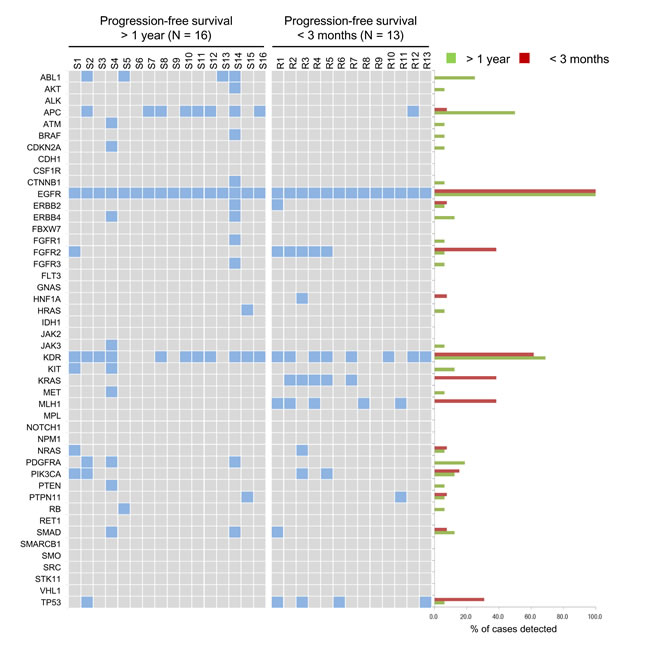
Figure 1: Genetic variations of EGFR L858R lung adenocarcinomas in 46 cancer-related genes. The mutation profiles in hotspot regions of 46 cancer-related genes in individual EGFR L858R tumors are shown on the left in 2 groups, according to the progression-free survival of patients. A positive result for mutation is indicated by a filled box. The percentage of mutation identified in each group for each gene is shown on the right.
MLH1 V384D in patients with primary lung adenocarcinoma
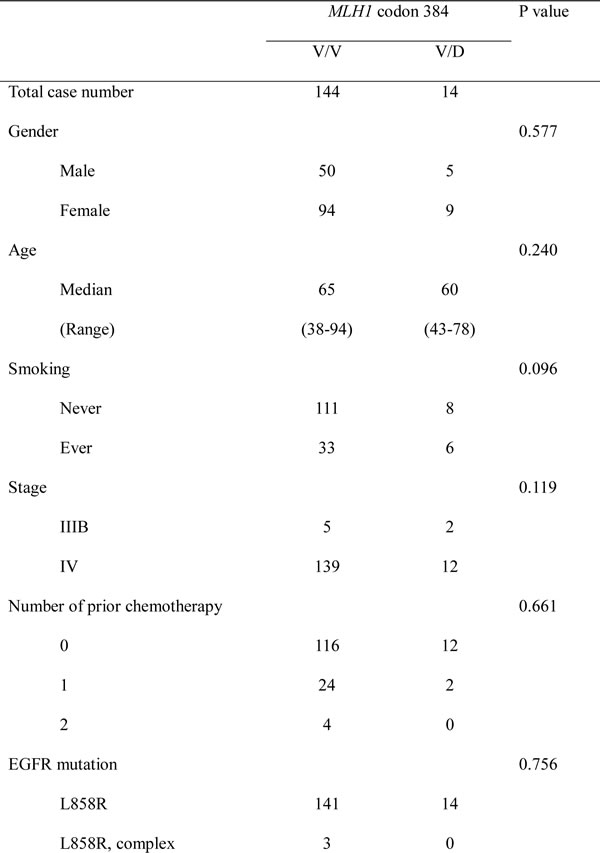
We examined the mutation status of MLH1 in a larger set of EGFR L858R-positive lung adenocarcinomas. A total of 158 tumors were subjected to MLH1 mutation analysis by direct sequencing of PCR products. Fourteen of the 158 tumors (8.9%) had a heterozygous T→A change at nucleotide 1151 (Fig. 2A) which results in the same V384D substitution in MLH1 as discovered in NGS screening. We were able to analyze genomic DNA from blood specimens of 4 patients and non-tumor tissue specimens from 1 patient, and all of these samples were tested positive for MLH1 V384D (as in Fig. 2A). Clinical characteristics of patients with or without MLH1 V384D were analyzed (Table 1), and no statistically significant demographic differences between the two groups were noted. We also performed sequencing analysis of MLH1 exon 12 in 51 EGFR-wildtype lung adenocarcinomas and found a comparable incidence (4/51, 7.8%) of the MLH1 V384D allele.
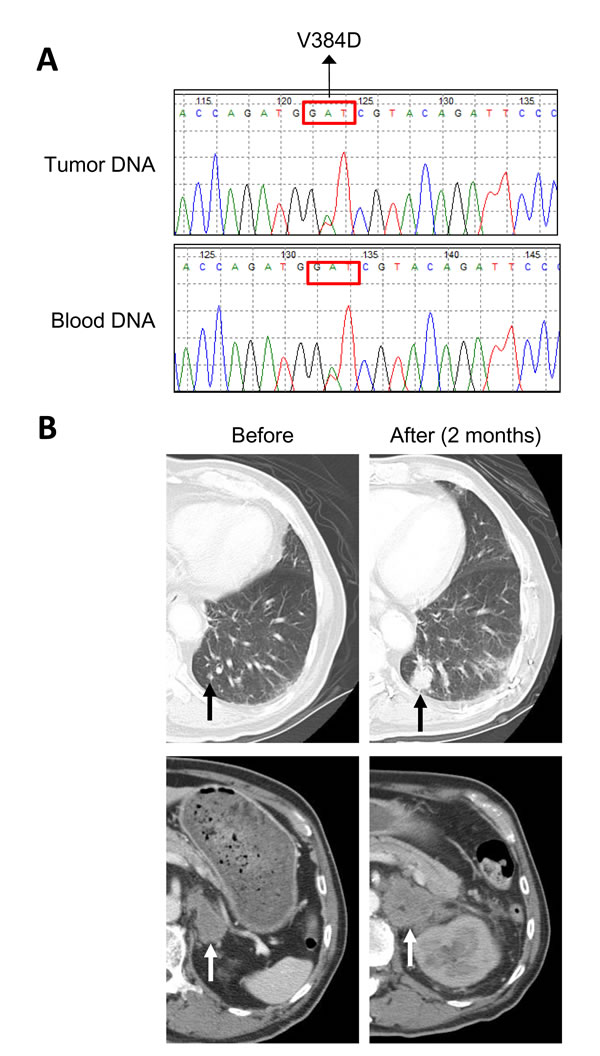
Figure 2: MLH1 V384D polymorphism in a lung adenocarcinoma patient with EGFR-TKI resistance. Panel A shows the results of Sanger sequencing of the MLH1 gene in both tumor and blood DNA specimens from a representative patient with a heterozygous T→A substitution at nucleotide 1151, which results in a Val384Asp substitution in MLH1. Panel B shows the chest CT scans of a representative patient with the MLH1 V384D polymorphism, demonstrating the persistent growth of metastatic lesions in lung (black arrows) and adrenal gland (white arrows) during erlotinib treatment.
Table 1: Patient characteristics (n= 158)

Tumor response to EGFR-TKI
A representative tumor with concurrent EGFR L858R and MLH1 V384D mutations displayed primary resistance to EGFR-TKI treatment (Fig. 2B). We evaluated individual tumor responses to EGFR-TKIs in patients whose tumors were of measurable sizes. Twenty-four of the NGS-screened 29 patients were monitored, and the tumor responses and PFS clustered correspondingly (Fig. 3A); 5 of 10 (50%) patients with short PFS had progressive disease whilst on EGFR-TKI treatment and 13 of 14 (92.9%) patients with long PFS had a partial response to EGFR-TKIs. Among the 4 patients with the MLH1 V384D allele, 2 had progressive disease and 2 had stable disease; none of the patients with partial response carried the MLH1 V384D allele.
In the 158 patients examined for MLH1 mutations by PCR and Sanger sequencing, 155 had measurable tumors and their responses to EGFR-TKIs were monitored (Fig. 3B). The overall response rate was 69.7%; 108, 39 and 8 patients achieved partial response, stable disease and progressive disease, respectively. The response rates for tumors with and without MLH1 V384D mutation were 50% and 71.6%, respectively (P= 0.088). MLH1 V384D-positive tumors had a smaller size reduction in response to EGFR-TKI treatment than that in tumors without the allele (median size change -28.2% vs. -40.5%, P= 0.015, Mann-Whitney U test). The MLH1 V384D allele was over-represented in patients with EGFR-TKI resistance. Only 11 of 155 (7.1%) EGFR L858R-positive tumors showed disease progression under EGFR-TKI treatment, and 4 of these 11 (36.4%) had MLH1 V384D. Among the 144 tumors either showing a partial response or being stable on treatment, only 10 (6.9%) were MLH1 V384D-positive.
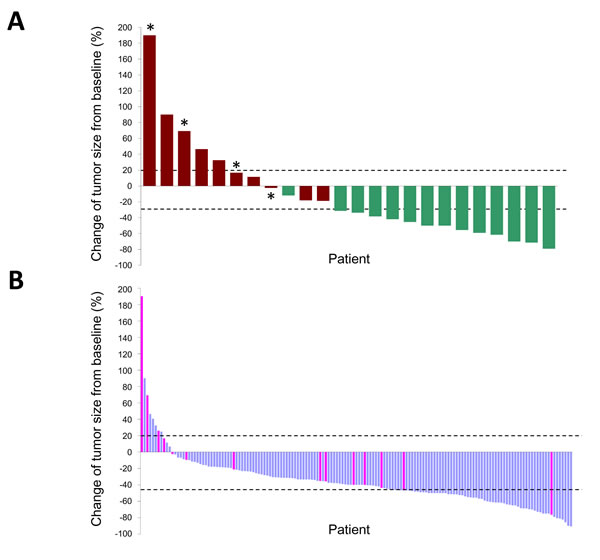
Figure 3: Waterfall plots of the maximum percentage change in tumor size of individual EGFR L858R lung adenocarcinomas treated by EGFR-TKIs. Tumors are listed in order of increasing extent of response to EGFR-TKIs; only those with measurable sizes before and after EGFR-TKI treatment are shown. The upper (20%) and lower (-30%) dashed lines indicate the thresholds used to define a progressive disease and a partial response, respectively, by the RECIST criteria. Panel A shows individual tumor responses in 24 patients analyzed by NGS. Red bars, PFS < 3 months; green bars, PFS < 1 year; asterisks, positive for MHL1 V384D. Panel B shows 155 EGFR L858R tumors analyzed for MLH1 status by direct sequencing of PCR products. Pink bars, positive for MLH1 V384D.
Survival analysis
At the time of analysis, with a median follow-up of 47.4 months, 51 patients remained in use of an EGFR-TKI and 107 patients (67.7%) had experienced PFS. The overall median PFS was 10.5 months (95% CI, 8.1 to 12.8 months). Patients with the MLH1 V384D mutation had a significant shorter PFS (median, 5.1 months; 95% CI, 1.5 to 8.7 months) than that of those without (median, 10.6 months; 95% CI, 8.8 to 12.5 months) (P= 0.001) (Fig. 4). Gender (male vs. female, P= 0.031) and the number of prior chemotherapy (0 vs. ≥ 1, P= 0.002) were also predictor variables for PFS. In the multivariate analysis using the Cox regression model, only the number of prior treatment (HR= 2.3, 95% CI, 1.4 to 3.8; in favor of none; P= 0.001) and the MLH1 mutation status (HR= 3.5, 95% CI, 1.7 to 7.2; in favor of no V384D mutation; P= 0.001) were independent predictors for PFS.
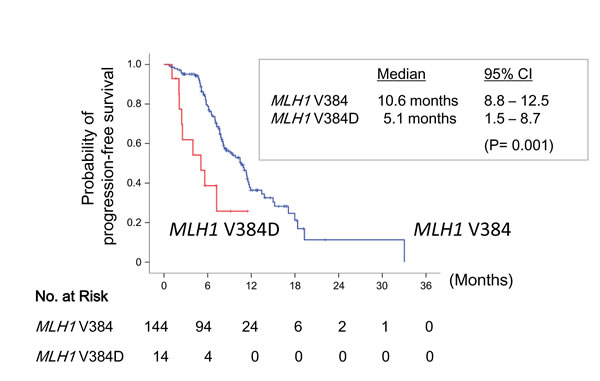
Figure 4: Progression-free survival stratified by MLH1 V384 status. Kaplan-Meier curves of progression-free survival in patients with EGFR L858R lung adenocarcinoma who were treated by EGFR-TKIs, according to the presence (red line) or absence (blue line) of the MLH1 V384D polymorphism.
DISCUSSION
Our NGS-based multi-gene mutation profiling of lung adenocarcinomas carrying the activating EGFR mutation L858R has uncovered an MLH1 V384D allele that is over-represented in a subset of tumors showing primary resistance to EGFR-TKIs. We have validated the disproportionately high occurrence of the MLH1 V384D allele in patients showing poor response to EGFR-TKI treatment, and demonstrated that MLH1 V384D is associated with a shorter PFS.
MLH1 is a component of the cellular DNA mismatch repair (MMR) machinery [26]. The MMR system is responsible for the recognition and repair of single-base mismatches and short insertions/deletions that may arise during DNA replication and recombination. Inherited mutations in the MMR system can cause genomic instability and human diseases such as Lynch syndrome; individuals with this syndrome are at high risk of colon cancer and other malignancies. Recognition of DNA damage by the MMR system is instrumental for activation of the apoptosis cascade; therefore, cells with MMR dysfunction may not properly induce apoptosis in response to DNA damage. Indeed, dysfunctional MMR has been implicated in the mechanism of resistance to DNA-damaging chemotherapeutics such as cisplatin [27]. However, to our knowledge, MLH1 and other MMR genes have never been linked to EGFR-TKI resistance.
Although many MLH1 variants are known [28, 29], only the V384 variant was identified in this study. MLH1 V384D was first identified in Chinese colorectal cancer patients and shown to be a common (2.5-2.67% in normal individuals) germline polymorphism in the East-Asian population [29, 30]. The detection of MLH1 V384D in blood or non-tumor tissue samples of patients with EGFR L858R-mutant lung adenocarcinoma also suggests that it is not a somatic mutation but a germline mutation/polymorphism. Similar to the reported 7.7% allele frequency in Chinese colorectal cancer patients [30], we found an incidence of MLH1 V384D around 7-8% in tumors with wild-type or L858R mutant EGFR, suggesting that MLH1 V384D may not be a mutation secondary to the EGFR L858R mutation. Whether the presence of MLH1 V384D increases the rates of activating EGFR mutations requires further investigation to determine.
The exact mechanism of how the MLH1 V384D variant influences EGFR-TKI treatment response on EGFR L858R-mutant tumors is not clear at present. A few possible scenarios are hereby presented: i) The MLH1 V384D variant is, at least partially, impaired in its protein function, displaying decreased efficiency in the interaction with its partner protein PMS2 and showing reduced MMR activity in vitro [28, 29]. As MMR contributes to genome stability, cancer cells with MLH1 V384D may hence be prone to accumulating mutations during cancer progression. This mutator phenotype could facilitate the acquisition of additional driver mutations in alternative proliferation or survival pathways, and enable cancer cells to become resistant to EGFR-targeting therapies. ii) Activated EGFR can translocate from cell membrane into the nucleus to execute several important functions, including DNA repair [31]. EGFR-TKI therapy inhibits the kinase activity and interferes with nuclear translocation of EGFR, which may possibly increase DNA damage. Because MLH1 is responsible for recognizing DNA damage and activating the apoptotic pathway, cells expressing the functionally impaired MLH1 V384D variant may not enter apoptosis efficiently and thus still display unchecked proliferation under EGFR-TKI treatment. iii) Besides mechanisms attributable to MMR dysfunction, we cannot exclude the possibility that MLH1 may have MMR-independent functions involved in EGFR signaling or other proliferation or survival pathways. Further investigation is warranted to elucidate the principal mechanism underlying the MLH1 V384D-associated EGFR-TKI resistance.
This study has important clinical implications. Instead of discovering additional “driver” mutations, we have identified a germline polymorphism associating with primary resistance to EGFR-TKIs. The first implication of this study is that: not only pathway-activating mutations in tumors but also germline genetic variations in individuals should be considered for molecular testing in the era of “personalized therapy”. In line with this notion, both tumor and blood samples should be archived to facilitate future translational studies. Secondly, our findings suggest that the combination of EGFR-TKIs with other anti-cancer therapeutics may be a rational treatment strategy for patients with lung adenocarcinomas concurrently harboring a somatic EGFR L858R mutation in the tumor and a germline MLH1 V384D polymorphism. In fact, combination therapy has been proved to be a successful strategy to overcome other types of EGFR-TKI resistance mechanism [32, 33].
There are some limitations of this study. Firstly, although all imaging studies were interpreted by two independent reviewers who were not aware of patients’ molecular profiles, all clinical data were collected retrospectively. Secondly, only a small number of tissue samples were in adequate amounts for molecular analysis during the study period; therefore, unintentional selection bias might exist as it could not have been prevented. Thirdly, although the NGS-based cancer panel is a powerful tool for detecting hundreds of mutations, this screening platform cannot identify certain genetic alterations, such as gene amplification and rearrangement. There exist examples of these types of genomic variations, e.g., MET amplification and ALK rearrangement, which are associated with EGFR-TKI resistance [14, 34, 35]. Finally, we only included EGFR L858R-mutant tumors in this study. Patients with L858R and those with another prevalent EGFR-activating mutation, a small deletion in exon 19, have different EGFR-TKI treatment outcomes [36, 37]. It is conceivable that these two different activating EGFR mutations may not associate with the same mechanism of primary resistance to TKIs.
In conclusion, this study has identified a novel candidate for genetic predictor of primary EGFR-TKI resistance in EGFR L858R-positive lung adenocarcinomas. Patients with EGFR L858R-mutant lung adenocarcinoma have inferior EGFR-TKI treatment outcomes if they have a co-existing MLH1 V384D variant. Future prospective clinical studies are warranted to confirm the prognostic importance of MLH1 V384D, as well as to define appropriate combinations of anti-cancer therapeutics in treating tumors with concomitant existence of EGFR L858R and MLH1 V384D alleles.
METHODS
Patients and study design
Patients were included if they had primary lung adenocarcinoma harboring the L858R mutation without a co-existing T790M mutation in EGFR and received their first-time EGFR-TKI treatment at Taipei Veterans General Hospital during the period from January 2009 to January 2013. Patients who had prior EGFR-TKI therapy or received EGFR-TKI in combination with other anti-cancer treatment were excluded. Patients who had adequate tumor specimens for further molecular testing were enrolled. This study was approved by the Institution Review Board of Taipei Veterans General Hospital.
If tumors progressed within 3 months of the initiation of EGFR-TKI therapy, we considered that the treatment was clinically ineffective and that these patients presented primary (or intrinsic) resistance. To discover candidate genetic variations that may associate with primary EGF-TKI resistance in EGFR mutant tumors, we performed genomic profiling of EGFR L858R tumors from 16 patients with long (> 1 year) progression-free survival (PFS) and 13 patients with short (< 3 months) PFS. NGS was performed to screen through a cancer-related gene mutation panel (Ion AmpliSeq Cancer Panel, Ion Torrent, Life Technologies); 739 mutation hotspot regions within 46 key cancer-related genes from the COSMIC database were examined. Distributions of genomic variants in the two groups of patients were compared. Genes with differential mutation status between two groups were further investigated in a total of 158 EGFR L858R tumors by PCR amplification and direct Sanger sequencing, and the association of candidate variants with differential tumor response to EGFR-TKIs was explored.
Histopathology review and sample preparation
Consecutive tissue sections were prepared from each archived formalin-fixed paraffin-embedded (FFPE) pathology specimen and reviewed by pathologists; tumor areas were marked on deparaffinized unstained sections and manually dissected. Proteinase K-digested tissue extracts were subjected to genomic profiling tests. Genomic DNA was also prepared from available blood samples using the illustra blood genomicPrep Mini Spin Kit (GE Healthcare Life Sciences) according to the manufacturer’s protocol.
Next-generation sequencing
Genomic DNA from FFPE tumor tissues was quantified using the Qubit® dsDNA HS Assay Kit and the Qubit® fluorometer (Life Technologies); 10 nanograms were amplified by multiplex PCR using the Ion AmpliSeq Cancer Panel Primers Pool (Life Technologies). PCR amplicons were ligated with barcode adaptors using the Ion Xpress Barcode Adapters 1-16 Kit (Life Technologies), and subjected to emulsion PCR. Template was prepared by the automated Ion OneTouch System using the Ion OneTouch 200 Template Kit v2 DL, and DNA was sequenced on a 316 chip using the Ion PGM Sequencing Kit v2 and the Ion Torrent Personal Genome Machine (PGM, Ion Torrent, Life Technologies). Data were analyzed using the Torrent Suite software v3.0 and the Ion Torrent Variant Caller software v3.0. Variants were called when a minimum coverage of 500 reads was achieved and at least 5% of variant reads were identified.
PCR and sanger sequencing
Exon 12 of the MLH1 gene was amplified from genomic DNA by PCR using a forward primer (5’-CAGACTTTGCTACCAGGACTTGC-3’) and a reverse primer (5’-CTGCCTAGCCCTGCCACTAG-3’). PCR products were sequenced using the Sanger method. DNA sequences were analyzed by the Mutation Surveyor software (SoftGenetics, State College, PA).
Statistical analysis
The objective tumor response was evaluated according to the revised RECIST criteria [34]. PFS was calculated from the date of starting EGFR-TKI therapy to the date of disease progression or death. The association between patient characteristics and MLH1 mutation status was analyzed by chi-square and Fisher’s exact tests. Kaplan-Meier survival curves were constructed and compared using the log-rank test. Cox regression models were built using a backward stepwise procedure for multivariate survival analysis. Analyses were carried out using PASW Statistics 18.0 (SPSS Inc., Chicago, IL).
ACKNOWLEDGEMENTS
This study was sponsored by grants from the Taipei Veterans General Hospital (V102C-195, V102C-135 and R1200102), the National Science Council (NSC99-2320-B-010-023-MY3 and NSC101-2314-B-075-062-MY2), the Department of Health (MOHW104-TDU-B-211-124-001) “Center of Excellence for Cancer Research at Taipei Veterans General Hospital - Health and welfare surcharge of tobacco products”, and the Ministry of Education (“Aim for the Top University Plan”), Taiwan. The authors thank Yi-Ching Sun, Yi-Ting Wan, Yu-Wei Liu, Yi-Chun Chang Chien, and Zih-Ying Wu for providing technical assistance.
Authors’ disclosures of potential conflicts of interest
C.-H. Chiu, T.-Y. Chou and C.-M. Tsai have received honoraria from AstraZeneca, Boehringer Ingelheim, Eli Lilly and Company, Pfizer and Roche. All remaining authors have declared no conflicts of interest.
REFERENCES
1. Carney DN. Lung cancer--time to move on from chemotherapy. N Engl J Med. 2002; 346: 126-128.
2. Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004; 350: 2129-2139.
3. Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004;304:1497-1500.
4. Felip E, Gridelli C, Baas P, Rosell R, Stahel R; Panel Members. Metastatic non-small-cell lung cancer: consensus on pathology and molecular tests, first-line, second-line, and third-line therapy: 1st ESMO Consensus Conference in Lung Cancer; Lugano 2010. Ann Oncol. 2011; 22: 1507-1519.
5. Keedy VL, Temin S, Somerfield MR, Beasley MB, Johnson DH, McShane LM, Milton DT, Strawn JR, Wakelee HA, Giaccone G. American Society of Clinical Oncology provisional clinical opinion: epidermal growth factor receptor (EGFR) Mutation testing for patients with advanced non-small-cell lung cancer considering first-line EGFR tyrosine kinase inhibitor therapy. J Clin Oncol. 2011; 29: 2121-2127.
6. Ohashi K, Maruvka YE, Michor F, Pao W. Epidermal growth factor receptor tyrosine kinase inhibitor-resistant disease. J Clin Oncol. 2013; 31: 1070-1080.
7. Zhou C, Wu YL, Chen G, Feng J, Liu XQ, Wang C, Zhang S, Wang J, Zhou S, Ren S, Lu S, Zhang L, Hu C, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011; 12: 735-742.
8. Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, Palmero R, Garcia-Gomez R, Pallares C, Sanchez JM, Porta R, Cobo M, Garrido P, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012; 13: 239-246.
9. Mitsudomi T, Morita S, Yatabe Y, Negoro S, Okamoto I, Tsurutani J, Seto T, Satouchi M, Tada H, Hirashima T, Asami K, Katakami N, Takada M, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol. 2010; 11: 121-128.
10. Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y, Nishiwaki Y, Ohe Y, Yang JJ, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009; 361: 947-957.
11. Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, Gemma A, Harada M, Yoshizawa H, Kinoshita I, Fujita Y, Okinaga S, Hirano H, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010; 362: 2380-2388.
12. Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, Halmos B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005; 352: 786-792.
13. Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H.. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005; 2: e73.
14. Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, Kosaka T, Holmes AJ, Rogers AM, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007; 316: 1039-43.
15. Bean J, Brennan C, Shih JY, Riely G, Viale A, Wang L, Chitale D, Motoi N, Szoke J, Broderick S, Balak M, Chang WC, Yu CJ, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A. 2007; 104: 20932-20937.
16. Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, Bergethon K, Shaw AT, Gettinger S, Cosper AK, Akhavanfard S, Heist RS, Temel J, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011; 3: 75ra26.
17. Azuma K, Kawahara A, Sonoda K, Nakashima K, Tashiro K, Watari K, Izumi H, Kage M, Kuwano M, Ono M, Hoshino T. FGFR1 activation is an escape mechanism in human lung cancer cells resistant to afatinib, a pan-EGFR family kinase inhibitor. Oncotarget. 2014; 5: 5908-5919.
18. Suda K, Tomizawa K, Fujii M, Murakami H, Osada H, Maehara Y, Yatabe Y, Sekido Y, Mitsudomi T. Epithelial to mesenchymal transition in an epidermal growth factor receptor-mutant lung cancer cell line with acquired resistance to erlotinib. J Thorac Oncol. 2011; 6: 1152-1161.
19. Watanabe S, Sone T, Matsui T, Yamamura K, Tani M, Okazaki A, Kurokawa K, Tambo Y, Takato H, Ohkura N, Waseda Y, Katayama N, Kasahara K. Transformation to small-cell lung cancer following treatment with EGFR tyrosine kinase inhibitors in a patient with lung adenocarcinoma. Lung Cancer. 2013; 82: 370-372.
20. Cappuzzo F, Janne PA, Skokan M, Finocchiaro G, Rossi E, Ligorio C, Zucali PA, Terracciano L, Toschi L, Roncalli M, Destro A, Incarbone M, Alloisio M, et al. MET increased gene copy number and primary resistance to gefitinib therapy in non-small-cell lung cancer patients. Ann Oncol. 2009; 20: 298-304.
21. Su KY, Chen HY, Li KC, Kuo ML, Yang JC, Chan WK, Ho BC, Chang GC, Shih JY, Yu SL, Yang PC. Pretreatment epidermal growth factor receptor (EGFR) T790M mutation predicts shorter EGFR tyrosine kinase inhibitor response duration in patients with non-small-cell lung cancer. J Clin Oncol. 2012; 30: 433-440.
22. Takeda M, Okamoto I, Fujita Y, Arao T, Ito H, Fukuoka M, Nishio K, Nakagawa K. De novo resistance to epidermal growth factor receptor-tyrosine kinase inhibitors in EGFR mutation-positive patients with non-small cell lung cancer. J Thorac Oncol. 2010; 5: 399-400.
23. Sos ML, Koker M, Weir BA, Heynck S, Rabinovsky R, Zander T, Seeger JM, Weiss J, Fischer F, Frommolt P, Michel K, Peifer M, Mermel C, et al. PTEN loss contributes to erlotinib resistance in EGFR-mutant lung cancer by activation of Akt and EGFR. Cancer Res. 2009; 69: 3256-3261.
24. Ng KP, Hillmer AM, Chuah CT, Juan WC, Ko TK, Teo AS, Ariyaratne PN, Takahashi N, Sawada K, Fei Y, Soh S, Lee WH, Huang JW, et al. A common BIM deletion polymorphism mediates intrinsic resistance and inferior responses to tyrosine kinase inhibitors in cancer. Nat Med. 2012; 18: 521-528.
25. Soh S, Ong ST. A novel BIM deletion polymorphism: implications and lessons for cancer targeted therapies. Rinsho Ketsueki. 2013; 54: 1714-1719.
26. Harfe BD, Jinks-Robertson S. DNA mismatch repair and genetic instability. Annu Rev Genet. 2000; 34: 359-399.
27. Fink D, Aebi S, Howell SB. The role of DNA mismatch repair in drug resistance. Clin Cancer Res. 1998; 4: 1-6.
28. Takahashi M, Shimodaira H, Andreutti-Zaugg C, Iggo R, Kolodner RD, Ishioka C. Functional analysis of human MLH1 variants using yeast and in vitro mismatch repair assays. Cancer Res. 2007; 67: 4595-604.
29. Fan Y, Wang W, Zhu M, Zhou J, Peng J, Xu L, Hua Z, Gao X, Wang Y. Analysis of hMLH1 missense mutations in East Asian patients with suspected hereditary nonpolyposis colorectal cancer. Clin Cancer Res. 2007; 13: 7515-7521.
30. Wang Y, Friedl W, Lamberti C, Nothen MM, Kruse R, Propping P. A novel missense mutation in the DNA mismatch repair gene hMLH1 present among East Asians but not among Europeans. Hum Hered. 1998; 48: 87-91.
31. Chen DJ, Nirodi CS. The epidermal growth factor receptor: a role in repair of radiation-induced DNA damage. Clin Cancer Res. 2007; 13: 6555-6560.
32. Noto A, De Vitis C, Roscilli G, Fattore L, Malpicci D, Marra E, Luberto L, D’Andrilli A, Coluccia P, Giovagnoli MR, Normanno N, Ruco L, Aurisicchio L, et al. Combination therapy with anti-ErbB3 monoclonal antibodies and EGFR TKIs potently inhibits non-small cell lung cancer. Oncotarget. 2013; 4: 1253-1265.
33. Wheler J, Falchook G, Tsimberidou AM, Hong D, Naing A, Piha-Paul S, Chen SS, Heymach J, Fu S, Stephen B, Fok JY, Janku F, Kurzrock R. Revisiting clinical trials using EGFR inhibitor-based regimens in patients with advanced non-small cell lung cancer: a retrospective analysis of an MD Anderson Cancer Center phase I population. Oncotarget. 2013; 4: 772-784.
34. Tiseo M, Gelsomino F, Boggiani D, Bortesi B, Bartolotti M, Bozzetti C, Sammarelli G, Thai E, Ardizzoni A. EGFR and EML4-ALK gene mutations in NSCLC: a case report of erlotinib-resistant patient with both concomitant mutations. Lung Cancer. 2011; 71: 241-243.
35. Tanimoto A, Yamada T, Nanjo S, Takeuchi S, Ebi H, Kita K, Matsumoto K, Yano S. Receptor ligand-triggered resistance to alectinib and its circumvention by Hsp90 inhibition in EML4-ALK lung cancer cells. Oncotarget. 2014; 5: 4920-4928.
36. Riely GJ, Pao W, Pham D, Li AR, Rizvi N, Venkatraman ES, Zakowski MF, Kris MG, Ladanyi M, Miller VA. Clinical course of patients with non-small cell lung cancer and epidermal growth factor receptor exon 19 and exon 21 mutations treated with gefitinib or erlotinib. Clin Cancer Res. 2006; 12: 839-844.
37. Jackman DM, Yeap BY, Sequist LV, Lindeman N, Holmes AJ, Joshi VA, Bell DW, Huberman MS, Halmos B, Rabin MS, Haber DA, Lynch TJ, Meyerson M, et al. Exon 19 deletion mutations of epidermal growth factor receptor are associated with prolonged survival in non-small cell lung cancer patients treated with gefitinib or erlotinib. Clin Cancer Res. 2006; 12: 3908-3914.
38. Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009; 45: 228-247.