
INTRODUCTION
Although the molecular mechanisms that are involved in the regulation and execution of cell death have been studied extensively during the last decades, the definition of cell death itself is still a matter of debate [1, 2]. It has been proposed that the demise of proliferating cells corresponds to the loss of their clonogenic potential [3]. However, this definition cannot be extended to post-mitotic cells (e.g., neurons and cardiomyocytes), which definitively exited the cell cycle along with terminal differentiation [4], and is rather inaccurate, as proliferating cells can enter a temporary or irreversible cell cycle arrest (and hence fail to replicate), while maintaining biological functions (for instance the secretion of soluble mediators), for very long periods [5, 6]. Other, more accurate definitions of cell death should therefore be employed. Recently, a large committee of experts in the field agreed on pragmatically identifying the transition between a reversible perturbation of cellular homeostasis and cell death (which is irreversible, by definition) with the permanent loss of plasma membrane integrity [7]. This process can be conveniently monitored by the uptake of so-called exclusion dyes, i.e., colored or fluorescent molecules that are excluded by healthy cells owing to the integrity of their plasma membrane [2, 8]. A commonly employed exclusion dye is 4′,6-diamidino-2-phenylindole (DAPI) [8].
However, plasma membrane permeabilization is a late process in the cascade of biochemical events that bridges the delivery of a lethal stimulus to the execution of cell death [7, 9]. This implies that, at least in some instances, other processes well upstream of plasma membrane permeabilization may irreversibly commit cells to die, including the complete loss of mitochondrial functions [10–12], and the massive activation of cysteine proteases of the caspase family [13, 14]. Thus, although plasma membrane permeabilization constitutes the gold standard indicator of cell death, at least in vitro, there are many experimental settings in which it is important to quantify the proportion of cells that still possess an intact plasma membrane yet are doomed to die.
Mitochondrial functions are usually monitored by means of cationic lipophilic fluorochromes that accumulate within the mitochondrial matrix driven by the mitochondrial transmembrane potential (Δψm), such as 3,3′-dihexyloxacarbocyanine iodide (DiOC6(3)) or tetramethylrhodamine ethyl ester (TMRE) [15] At odds with their functional counterparts, dysfunctional mitochondria exhibit indeed a reduced Δψm, resulting in an impaired accumulation of these probes [16–18]. One economic way of monitoring caspase activation relies on YO-PRO®-3, a fluorochome that accumulates in the nucleus upon crossing the plasma membrane through pannexin 1 (PANX1) channels [19–21]. PANX1 channels are activated by caspase-3 before the onset of plasma membrane permeabilization, a setting in which cells do not take up vital dyes yet, but secrete small metabolites (such as ATP) and accumulate YO-PRO®-3 [22–24]. Of note, cells with permeabilized plasma membrane may exhibit profound nuclear alterations, including chromatin condensation and shrinkage (which are associated with apoptosis) or nuclear dilation (which is associated with necrosis). These changes can be monitored by epifluorescence microscopy using a plasma-membrane chromatinophilic dye such as Hoechst 33342 [2, 8].
Based on these premises, we decided to combine an exclusion dye (DAPI) or a dye for monitoring nuclear integrity (Hoechst 33342) (both of which emit a blue fluorescence) with DiOC6(3) (which emits a green fluorescence) and YO-PRO®-3 (which emits a red/orange fluorescence) to monitor the evolution of cells exposed to lethal stimuli. Here, we demonstrate that such a co-staining approach allows for monitoring the absolute number and percentage of truly viable cells (which exhibit an intact plasma membrane with closed PANX1 channels and high Δψm). This procedure is compatible with a fully robotized workflow in which cellular samples are processed, stained and analyzed by epifluorescence microscopy or flow cytometry.
RESULTS AND DISCUSSION
Epifluorescence microscopy-based fine analysis of cellular viability
To develop an automatable method for the simultaneous assessment of plasma membrane integrity, mitochondrial functions and caspase activation, we maintained human non-small cell lung carcinoma (NSCLC) A549 cells in control conditions or we exposed them to a lethal dose of the multikinase inhibitor staurosporine (a well-known inducer of apoptosis) [25] or the platinum derivative oxaliplatin [26, 27] for 24 hrs. Thereafter, we simultaneously stained cells with Hoechst 33342, DiOC6(3) and YO-PRO®-3 and analyzed them by epifluorescence microscopy. Upon automated image segmentation (Supplementary Figure 1), we identified five possible cellular phenotypes: (1) cells that stained weakly for Hoechst 33342 (Hoechst 33342dim cells, manifesting normal nuclear morphology) and negatively for YO-PRO®-3 (and hence retained normal plasma membrane impermeability) while exhibiting a high DiOC6(3) signal (preserving a high Δψm); (2) cells that stained intensely for both Hoechst 33342 (Hoechst 33342bright cells, manifesting nuclear condensation) and YO-PRO®-3 (manifesting a complete loss of plasma membrane barrier functions) while exhibiting a low DiOC6(3) signal (a sign of complete impairment of mitochondrial functions); and (3–5) Hoechst 33342dim cells bearing either YO-PRO®-3 positivity or low DiOC6(3) signal, or both (three intermediate situations) (Figure 1A). In control conditions, most cells exhibited a uniform Hoechst 33342dimDiOC6(3)highYO-PRO®-3− phenotype (Figure 1B and Supplementary Figure 2), a staining profile that clearly changed in response to cell death induction. Exposing A549 cells to 500 μM oxaliplatin or 4 μM staurosporine for 24 hrs, indeed, provoked an increase in the frequency of Hoechst 33342bright cells as well as of Hoechst 33342dimDiOC6(3)lowYO-PRO®-3+, Hoechst 33342dimDiOC6(3)highYO-PRO®-3+ or Hoechst 33342dimDiOC6(3)lowYO-PRO®-3− cells as it reduced the frequency and absolute number of healthy Hoechst 33342dimDiOC6(3)highYO-PRO®-3− cells (Figure 1C, 1D and Supplementary Figure 2).
Implementation of the triple straining procedure on flow cytometry
Next, we implemented the triple staining protocol described above on a cytofluorometric platform amenable to automation. To this aim, human NSCLC A549 cells maintained in control conditions or exposed to lethal stimuli for 24 hrs were collected and then co-stained with DAPI, DiOC6(3) and YO-PRO®-3. The vast majority of untreated A549 cells exhibited a DAPI−DiOC6(3)high YO-PRO®-3− phenotype, indicating intact plasma membranes, normal mitochondrial functions and no caspase activation (Figure 2A and Supplementary Figure 3). In contrast, exposing A549 cells to 500 μM oxaliplatin or 4 μM staurosporine for 24 hrs caused the accumulation of DAPI+ cells, as well as of cells exhibiting a DAPI−DiOC6(3)lowYO-PRO®-3+, DAPI−DiOC6(3)highYO-PRO®-3+ or DAPI−DiOC6(3)low YO-PRO®-3− staining pattern (Figure 2B, 2C and Supplementary Figure 3). Hence, both oxaliplatin and staurosporine cause a drop in the absolute amount of DAPI−DiOC6(3)high YO-PRO®-3− (viable) A549 cells in a dose-dependent fashion (Figure 3A). The pan-caspase inhibitor N-benzyloxycarbonyl-Val-Ala-Asp(O-Me) fluoromethylketone (Z-VAD-fmk) was unable to prevent the loss of viability among cells cultured in the presence of oxaliplatin or staurosporine. However, caspase inhibition with Z-VAD-fmk substantially reduced the surge in DAPI−DiOC6(3)lowYO-PRO®-3+ and DAPI−DiOC6(3)highYO-PRO®-3+ cells, without eliminating the DAPI−DiOC6(3)lowYO-PRO®-3− subset. This was particularly evident when all events in control, oxaliplatin or staurosporine-treated conditions were analyzed upon aggregation, as if they derived from a single sample, either in the absence (Figure 3B, left panels) or in the presence of Z-VAD-fmk (Figure 3B, right panels). These results confirm the stringent requirement for caspase activation for the opening of YO-PRO®-3-permeable PANX1 channels [19, 20], and validate the feasibility and sensitivity of our viability test.
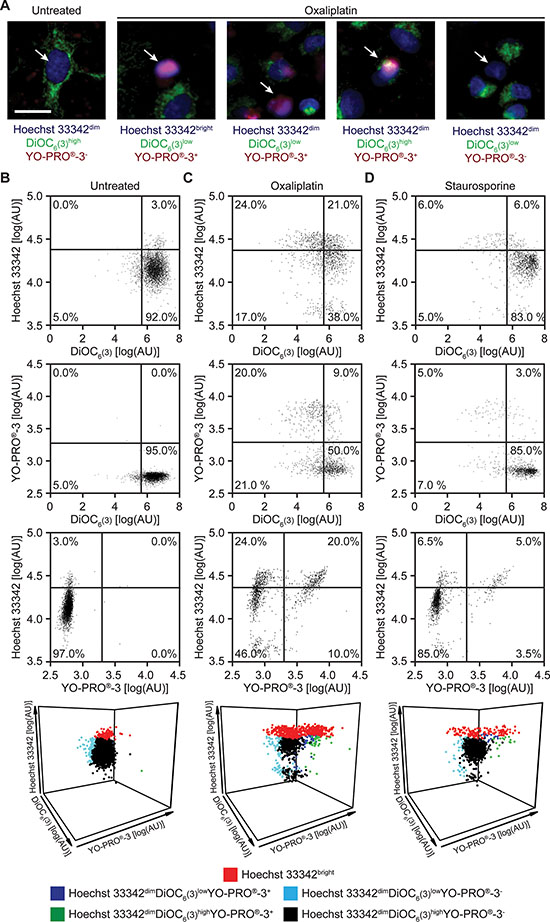
Figure 1: Epifluorescence microscopy-based assessment of cellular viability in response to standard inducers of apoptosis. (A–D) Human non-small cell lung carcinoma A549 cells were maintained in control conditions or exposed to 500 μM oxaliplatin or 4 μM staurosporine for 24 hrs, then imaged by epifluorescence microscopy upon co-staining with Hoechst 33342, DiOC6(3) and YO-PRO®-3. Representative images and dot plots obtained upon automated image segmentation and analysis are reported. Scale bar = 10 μm. In panels B, C and D, numbers indicate the percentage of events within each gate. See also Supplementary Figure 1.
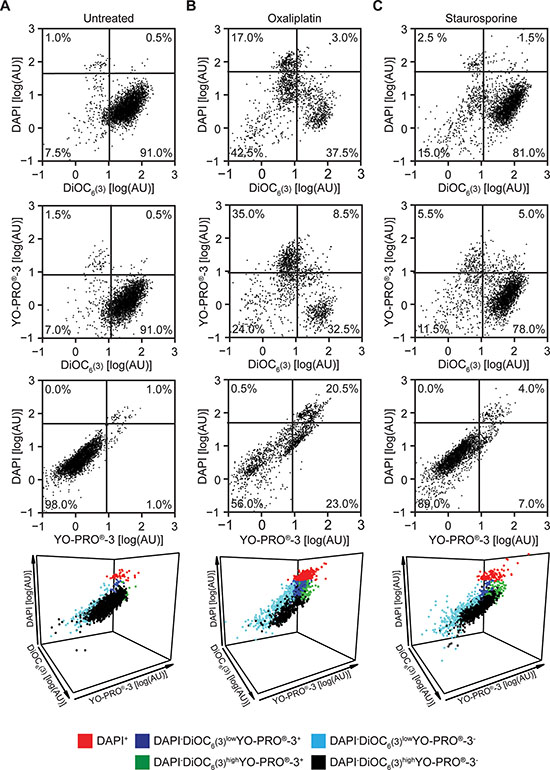
Figure 2: Flow cytometry-based assessment of cellular viability to standard inducers of apoptosis. (A–C) Human non-small cell lung carcinoma A549 cells were cultured in control conditions or treated with 500 μM oxaliplatin or 4 μM staurosporine for 24 hrs, then co-stained with DAPI, DiOC6(3) and YO-PRO®-3 and analyzed by flow cytometry. Representative dot plots are reported. In panels A, B and C, numbers indicate the percentage of events within each gate. See also Supplementary Figure 2.
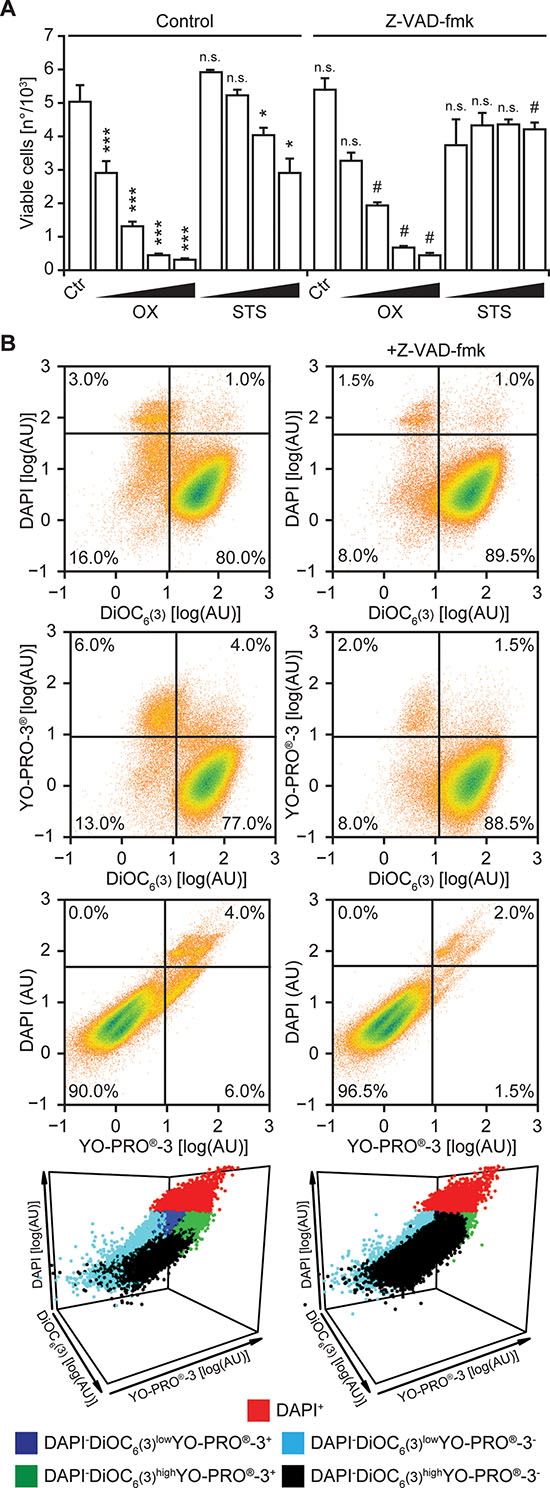
Figure 3: Flow cytometry-based assessment of cell death modulation. (A–B) Human non-small cell lung carcinoma A549 cells were maintained in control conditions (Ctr) or incubated with increasing concentrations of staurosporine (STS; 0.5 –1.0 – 2.0 – 4.0 μM), oxaliplatin (OX; 0.125–0.250–0.500–1 mM) and 50 μM Z-VAD-fmk, alone or in combination for 24 hrs. Thereafter, cells co-stained with DAPI, DiOC6(3) and YO-PRO®-3 and analyzed by flow cytometry. In panel (A) quantitative data on the absolute number of DAPI−DiOC6(3)highYO-PRO®-3− (viable) cells are reported (means ± SD, n = 3 replicate assessments from one representative experiment; *p < 0.05, ***p < 0.001, as compared to untreated cells; #p < 0.05, as compared to cells maintained in the same conditions in the absence of Z-VAD-fmk; n.s., non-significant, as compared to untreated cells or cells maintained in the same conditions in the absence of Z-VAD-fmk). In panel (B) dot plots depict the aggregate analysis of cells maintained in control conditions and exposed to 4 μM STS or 500 μM OX, alone (left panels, upon pooling data from 3 distinct samples) or in the presence of Z-VAD.fmk (right panels, upon pooling data from 3 distinct samples). Numbers indicate the percentage of events within each gate.
Detection of synergistic interactions by automatic viability measurements
To further investigate the utility of our fully automated viability test, we measured its performance on the pharmacological interaction between another platinum derivative, i.e., cis-diamminedichloroplatinum(II) (CDDP, best known as cisplatin) and the poly(ADP-ribose) polymerase 1 (PARP1) inhibitor PJ-34 hydrochloride hydrate (hereafter referred to as PJ-34) [28, 29]. CDDP reduced the number of DAPI−DiOC6(3)highYO-PRO®-3− (viable) A549 cells at much lower concentrations than did PJ-34 (Figure 4A). Moreover, at certain concentrations, CDDP and PJ-34 exhibited a synergistic interaction, meaning that their co-administration reduced the number of DAPI−DiOC6(3)highYO-PRO®-3− A549 cells in a hyperadditive fashion, and hence was much more effective than the administration of either the two agents alone (Figure 4B). Importantly, these results were obtained by means of a completely automated workflow that did not require any manual intervention.
Concluding remarks
Here, we describe a straightforward workflow that allows for the accurate and automatable quantification of living cells based on several parameters, namely, plasma membrane integrity, PANX1 channel impermeability and normal Δψm. The protocol that has been developed to this aim is robust (reagents are not particularly prone to degradation or sensitive to unpredictable environmental conditions), simple (it does not involve washing steps), rapid (staining time = 30 min), can be readily scaled up (as shown for 96-well plates) and is adaptable to different readouts (as shown for epifluorescence microscopy and flow cytometry). This implies that our protocol can be implemented on virtually any robotized platform that allows for automatic pipetting (for trypsinization and staining) and incubation, and is coupled to any reader (be it an epifluorescence microscope or a flow cytometer) that can automatically handle multi-well plates.
One advantage of modern microscopic and cytofluorometric systems is that they are compatible with the quantification of the absolute number of cells per area or volume. Thus, the staining protocol described here allows for the precise discrimination of live, dead and dying cells in a variety of experimental samples, including transformed cells growing on plates or in suspension, as well as primary cells of animal or human derivation. This informs on cytostatic and/or cytotoxic effects induced by anticancer agents, promising ameliorations for future drug screening campaigns and mechanistic explorations.
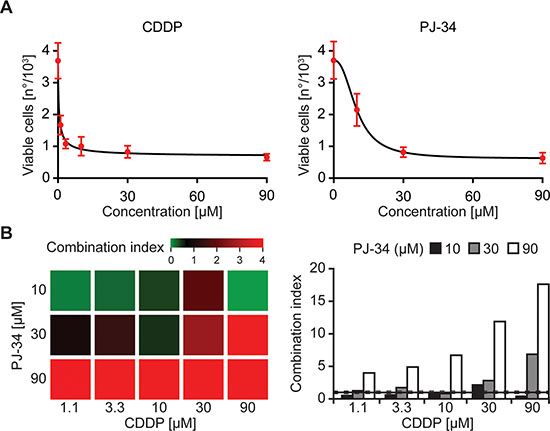
Figure 4: Flow cytometry-based assessment of pharmacological interactions. (A, B) Human non-small cell lung carcinoma A549 cells were left untreated or treated with the indicated concentrations of cisplatin (CDDP) or PJ-34 for 48 hrs, then stained with DAPI, DiOC6(3) and YO-PRO®-3 and analyzed by flow cytometry. Panel A reports quantitative data on DAPI−DiOC6(3)highYO-PRO®-3− (viable) cells (means ± SD, n = 3 replicate assessments from one representative experiment), while panel B illustrates combination indexes (CIs) calculated according the Harbron’s method. Please note that CIs < 0.8 (green on the heatmap) and > 1.2 (red on the heatmap) represent synergistic and antagonistic interactions, respectively.
MATERIALS AND METHODS
Chemicals and cell cultures
Except otherwise indicated, cell culture media and supplements were obtained from Gibco-Life Technologies (Carlsbad, CA, USA), chemicals from Sigma-Aldrich (St. Louis, MO, USA), and plasticware from Greiner Bio-One (Monroe, CA, USA). Human non-small cell lung carcinoma (NSCLC) A549 cells were cultured in Glutamax®-containing DMEM/F12 medium supplemented with 10% fetal calf serum (FCS), and 10 mM HEPES buffer. Cells were grown at 37°C in a humidified incubator generating a 5% CO2 atmosphere.
Automated experimental workflow
All experimental steps were conducted on the PACRI HTS cell biology platform that integrates a Biomek FXP automated liquid handling workstation (Beckman–Coulter, Fullerton, CA, USA), a multidrop combi automated dispenser (ThermoScientific, Whaltam, MA, USA), a Cytomat6000 automated cell culture incubator (ThermoScientific), 3 ImageXpress Micro XL automated microscopes (Molecular Devices, Sunnyvale, CA, USA), a CyAn ADP cytofluorometer (Beckman–Coulter), a Hypercyt loader (Intellicyt, Albuquerque, NM, USA) and a Motoman HP3JC industrial robot (Motoman, West Carrollton, OH, USA). A fully automated workflow entailing cell culture, drug treatment, sample preparation and cytofluorometric or microscopic acquisition was generated by means of the SAMI automation control software (Beckman–Coulter). Multiplex assays were prepared in parallel for microscopic and cytofluorometric acquisition.
Cell death assays
Five × 103 human NSCLC A549 cells were seeded in 96-well clear cell culture plates or black imaging plates for flow cytometry- or epifluorescence microscopy-based assays, respectively. Cells were allowed to adapt for 24 hrs and then maintained in control conditions or exposed to lethal stimuli for 24 or 48 hrs. For flow cytometry, culture supernatants were discarded and cells were detached with 30 μL TrypLE™ Express per well, then resuspended in 30 μL of medium supplemented with 40 nM DiOC6(3), 1 μM YO-PRO®-3 iodide and 2 μM DAPI (all from Molecular Probes®−Life Technologies, Carlsbad, CA, USA). Subsequently, cells were transferred to a 96-well V-shape plate and incubated for 30 min at 37°C before acquisition. Cytofluorometric acquisitions were performed on a CyanADP or a MACSQuant cytometer (Miltenyi Biotec, Bergisch Gladbach, Germany). For epifluorescence microscopy, supernatants were removed and cells were incubated in 30 μL of medium supplemented with 40 nM DiOC6(3), 1 μM YO-PRO®-3 iodide and 2 μM Hoechst 33342 for 30 min at 37°C before acquisition. Images were acquired on ImageXpress Micro XL automated microscopes (4 view fields per well).
Data processing and statistical analyses
Unless otherwise specified, experiments were performed in quadruplicate parallel instances, and data were analyzed with the R software (http://www.r-project.org/). The first-line flow cytometry data analysis was performed using the flowcore package for R (http://www.bioconductor.org) upon gating on events with normal forward and side scatter parameters. Microscopy images were analyzed by means of the MetaXpress (Molecular Devices) software. In particular, images were segmented using the built-in custom module editor to identify nuclei (based on Hoechst 33342 fluorescence), and cytoplasmic regions (based on DiOC6(3) fluorescence). Thereafter, the average nuclear signal of Hoechst 33342 and YO-PRO®-3 as well as the average cytoplasmic signal of DiOC6(3) were measured. The pharmacological interaction between PJ-34 and CDDP was evaluated computing combination indexes (CI) following Harbron’s method [30, 31]. Unless otherwise specified, data are presented as means ± SD. Statistical significance was assessed with two-tailed, unpaired Student’s t-tests.
ACKNOWLEDGMENTS
GK is supported by the Ligue contre le Cancer (équipe labelisée); Agence National de la Recherche (ANR); Association pour la recherche sur le cancer (ARC); Cancéropôle Ile-de-France; Institut National du Cancer (INCa); Fondation Bettencourt–Schueller; Fondation de France; Fondation pour la Recherche Médicale (FRM); the European Commission (ArtForce); the European Research Council (ERC); the LabEx Immuno-Oncology; the SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); the SIRIC Cancer Research and Personalized Medicine (CARPEM); and the Paris Alliance of Cancer Research Institutes (PACRI).
REFERENCES
1. Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, Blagosklonny MV, El-Deiry WS, Golstein P, Green DR, Hengartner M, Knight RA, Kumar S, et al. Classification of cell death: recommendations of the nomenclature committee on cell Death. Cell Death Differ. 2009; 16:3–11.
2. Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El-Deiry WS, Fulda S, Gottlieb E, Green DR, Hengartner MO, et al. Molecular definitions of cell death subroutines: recommendations of the nomenclature committee on cell death. Cell Death Differ. 2012; 19:107–120.
3. Galluzzi L, Maiuri MC, Vitale I, Zischka H, Castedo M, Zitvogel L, Kroemer G. Cell death modalities: classification and pathophysiological implications. Cell Death Differ. 2007; 14:1237–1243.
4. Musacchio A, Helin K. Cell cycle, differentiation and disease. Curr Opin Cell Biol. 2013; 25:673–675.
5. Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013; 153:1194–1217.
6. Gitenay D, Lallet-Daher H, Bernard D. Caspase-2 regulates oncogene-induced senescence. Oncotarget. 2014; 5:5845–5847.
7. Galluzzi L, Bravo-San Pedro JM, Vitale I, Aaronson SA, Abrams JM, Adam D, Alnemri ES, Altucci L, Andrews D, Annicchiarico-Petruzzelli M, Baehrecke EH, Bazan NG, Bertrand MJ, et al. Essential versus accessory aspects of cell death: recommendations of the NCCD. Cell Death Differ. 2015; 22:58–73.
8. Kepp O, Galluzzi L, Lipinski M, Yuan J, Kroemer G. Cell death assays for drug discovery. Nat Rev Drug Discov. 2011; 10:221–237.
9. Galluzzi L, Bravo-San Pedro JM, Kroemer G. Organelle-specific initiation of cell death. Nat Cell Biol. 2014; 16:728–736.
10. Zamzami N, Marchetti P, Castedo M, Zanin C, Vayssiere JL, Petit PX, Kroemer G. Reduction in mitochondrial potential constitutes an early irreversible step of programmed lymphocyte death in vivo. J Exp Med. 1995; 181:1661–1672.
11. Castedo M, Macho A, Zamzami N, Hirsch T, Marchetti P, Uriel J, Kroemer G. Mitochondrial perturbations define lymphocytes undergoing apoptotic depletion in vivo. Eur J Immunol. 1995; 25:3277–3284.
12. Metivier D, Dallaporta B, Zamzami N, Larochette N, Susin SA, Marzo I, Kroemer G. Cytofluorometric detection of mitochondrial alterations in early CD95/Fas/APO-1-triggered apoptosis of Jurkat T lymphoma cells. Comparison of seven mitochondrion-specific fluorochromes. Immunol Lett. 1998; 61:157–163.
13. Fernandes-Alnemri T, Litwack G, Alnemri ES. CPP32, a novel human apoptotic protein with homology to Caenorhabditis elegans cell death protein Ced-3 and mammalian interleukin-1 beta-converting enzyme. J Biol Chem. 1994; 269:30761–30764.
14. Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle M, Lazebnik YA, et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995; 376:37–43.
15. Galluzzi L, Aaronson SA, Abrams J, Alnemri ES, Andrews DW, Baehrecke EH, Bazan NG, Blagosklonny MV, Blomgren K, Borner C, Bredesen DE, Brenner C, Castedo M, et al. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ. 2009; 16:1093–1107.
16. Macho A, Castedo M, Marchetti P, Aguilar JJ, Decaudin D, Zamzami N, Girard PM, Uriel J, Kroemer G. Mitochondrial dysfunctions in circulating T lymphocytes from human immunodeficiency virus-1 carriers. Blood. 1995; 86:2481–2487.
17. Zamzami N, Maisse C, Metivier D, Kroemer G. Measurement of membrane permeability and the permeability transition of mitochondria. Methods Cell Biol. 2007; 80:327–340.
18. Qian W, Wang J, Roginskaya V, McDermott LA, Edwards RP, Stolz DB, Llambi F, Green DR, Van Houten B. Novel combination of mitochondrial division inhibitor 1 (mdivi-1) and platinum agents produces synergistic pro-apoptotic effect in drug resistant tumor cells. Oncotarget. 2014; 5:4180–4194.
19. Chekeni FB, Elliott MR, Sandilos JK, Walk SF, Kinchen JM, Lazarowski ER, Armstrong AJ, Penuela S, Laird DW, Salvesen GS, Isakson BE, Bayliss DA, Ravichandran KS. Pannexin 1 channels mediate ‘find-me’ signal release and membrane permeability during apoptosis. Nature. 2010; 467:863–867.
20. Qu Y, Misaghi S, Newton K, Gilmour LL, Louie S, Cupp JE, Dubyak GR, Hackos D, Dixit VM. Pannexin-1 is required for ATP release during apoptosis but not for inflammasome activation. J Immunol. 2011; 186:6553–6561.
21. Bink K, Walch A, Feuchtinger A, Eisenmann H, Hutzler P, Hofler H, Werner M. TO-PRO-3 is an optimal fluorescent dye for nuclear counterstaining in dual-colour FISH on paraffin sections. Histochem Cell Biol. 2001; 115:293–299.
22. Sandilos JK, Chiu YH, Chekeni FB, Armstrong AJ, Walk SF, Ravichandran KS, Bayliss DA. Pannexin 1, an ATP release channel, is activated by caspase cleavage of its pore-associated C-terminal autoinhibitory region. J Biol Chem. 2012; 287:11303–11311.
23. Martins I, Wang Y, Michaud M, Ma Y, Sukkurwala AQ, Shen S, Kepp O, Metivier D, Galluzzi L, Perfettini JL, Zitvogel L, Kroemer G. Molecular mechanisms of ATP secretion during immunogenic cell death. Cell Death Differ. 2014; 21:79–91.
24. Joo YN, Jin H, Eun SY, Park SW, Chang KC, Kim HJ. P2Y2R activation by nucleotides released from the highly metastatic breast cancer cell MDA-MB-231 contributes to pre-metastatic niche formation by mediating lysyl oxidase secretion, collagen crosslinking, and monocyte recruitment. Oncotarget. 2014; 5:9322–9334.
25. Green DR, Galluzzi L, Kroemer G. Cell biology. Metabolic control of cell death. Science. 2014; 345:1250256.
26. Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, Castedo M, Kroemer G. Molecular mechanisms of cisplatin resistance. Oncogene. 2012; 31:1869–1883.
27. Liang S, Peng X, Li X, Yang P, Xie L, Li Y, Du C, Zhang G. Silencing of CXCR4 sensitizes triple-negative breast cancer cells to cisplatin. Oncotarget. 2014; 6:1020–1030.
28. Michels J, Vitale I, Galluzzi L, Adam J, Olaussen KA, Kepp O, Senovilla L, Talhaoui I, Guegan J, Enot DP, Talbot M, Robin A, Girard P, et al. Cisplatin resistance associated with PARP hyperactivation. Cancer Res. 2013; 73:2271–2280.
29. Michels J, Obrist F, Castedo M, Vitale I, Kroemer G. PARP and other prospective targets for poisoning cancer cell metabolism. Biochem Pharmacol. 2014; 92:164–171.
30. Lainey E, Wolfromm A, Marie N, Enot D, Scoazec M, Bouteloup C, Leroy C, Micol JB, De Botton S, Galluzzi L, Fenaux P, Kroemer G. Azacytidine and erlotinib exert synergistic effects against acute myeloid leukemia. Oncogene. 2013; 32:4331–4342.
31. Montraveta A, Xargay-Torrent S, Lopez-Guerra M, Rosich L, Perez-Galan P, Salaverria I, Bea S, Kalko SG, de Frias M, Campas C, Roue G, Colomer D. Synergistic anti-tumor activity of acadesine (AICAR) in combination with the anti-CD20 monoclonal antibody rituximab in in vivo and in vitro models of mantle cell lymphoma. Oncotarget. 2014; 5:726–739.