
INTRODUCTION
The identification of genes that participate in oncogenesis has facilitated recent development of molecular targeted therapies against cancer (reviewed in [1-3]). In the past several years, the identification of mutations in the BRAF gene in melanomas has led to the development of molecular therapies targeting those mutations in metastatic melanomas, which is a highly lethal disease [4-6]. However, recent studies have also shown that the frequency of mutation of the BRAF gene in melanomas has clear discrepancies depending on the subtype of melanomas [7-11]. A high frequency of aberrations in the BRAF gene is observed in Non-CSD melanomas (CSD:chronic sun-induced damage) [8, 12], while the BRAF V599E mutation has a relatively low frequency in other subtypes of melanomas, including frequencies ranging from 15% to 33% in acral melanomas, which occur on the palms, soles and subungual sites [8, 10], but almost 0% in mucosal melanomas [9, 10]. These discrepancies suggest the hypothesis that other genomic aberrations may play important roles in melanomagenesis in subtypes of melanomas other than Non-CSD melanomas and that further explorations of genomic aberrations, which can be ideal and effective targets for molecular targeted therapies against melanomas, are required to develop more effective and alternative therapies targeting a wide range of melanomas. In acral melanomas, several genomic aberrations, including amplification of 11q13 and KIT mutations, have been found [13-15]. The elucidation of genomic aberrations in melanomas using detailed analyses of public array-CGH databases suggests that genomic gain at chromosome 1q32 has profound effects on acral melanomagenesis. NUAK2, a gene at this locus, has regulatory impacts on the proliferation and migration of melanoma cells [16].
The SNF1/AMP-activated protein kinase (AMPK) family functions to control the balance of cellular metabolisms, and is activated by the cellular AMP:ATP ratio that is regulated by metabolic stresses such as hypoxia and glucose deprivation [17, 18]. AMPK is composed of 3 subunits (α, β and γ), and the α-subunit is the catalytic subunit. This catalytic subunit family includes 5 members: AMPK-α1, AMPK-α2 [19-21], MELK [22], NUAK2/SNARK [23, 24] and NUAK1/ARK5 [25]. Twelve protein kinases (BRSK1, BRSK2, NUAK1, NUAK2, QIK, QSK, SIK, MARK1, MARK2, MARK3, MARK4 and MELK) have been identified as AMPK-α1 and AMPK-α2 related kinases in the human kinome [26, 27]. The function of each member of this catalytic subunit are also closely connected to tumor formation and metastasis [18, 28, 29]. However, those functions participating in tumor formation and metastasis are different depending on each subunit. As shown in previous studies, AMPK-α1 and AMPK-α2 have anti-oncogenic properties [30-32] while ARK5 has pro-oncogenic properties [33]. The exact function(s) and mechanism(s) of NUAK2 (also known as SNARK), the fourth member of catalytic subunit of AMPK, remain unknown. NUAK2 has been speculated to have contradictory functions on tumorigenesis as a tumor suppressor or as an oncogene [24, 34]. In this review, we discuss the oncogenic role of NUAK2 and its clinical significance in melanoma patients, as well as its regulation by intracellular signaling pathways including LKB1 and CaMKKβ in melanoma cells.
ACRAL MELANOMA ONCOGENES AND ITS CLINICAL ASPECTS
Recent tenacious efforts by investigators have accelerated the elucidation of genomic alterations in melanoma cells. The first step to decipher enigmas in complex genomic alterations in melanoma cells had started by taking advantage of techniques that had been utilized to elucidate chromosomal abnormalities in hematopoietic malignancies [35-39]. Those techniques, which examine structural and numerical chromosomal aberrations in hematopoietic malignancies and sarcomas, had elucidated a reciprocal translocation between the long arms of chromosomes 9 and 22 (e.g., the Philadelphia chromosome) in chronic myelogenous leukemia (CML) [40-41] and resulted in identifying the oncogenic fusion protein of BCR-Abl [42-44]. Those discoveries eventually led to the development of a BCR-Abl inhibitor (imatinib mesylate; Gleevec), which improved the management of patients who suffer from CML [45-48]. As this example has clearly indicated, the identification of oncogenes and tumor suppressor genes, which can be guided by the characterization of structural and numerical aberrations of chromosomes in cancer cells, is an important step and an efficient approach to develop effective therapeutic modalities to control lethal diseases and improve the quality of life of cancer patients. For malignant melanoma, early studies pointed out nonrandom chromosomal aberrations involving chromosomes 1, 6, 7, 10 and 19 [35-39], and those aberrations had been confirmed by a larger number of cases [49]. A series of those early studies emphasizes that chromosomal aberrations involving chromosomes 1 and 6 are important in melanomagenesis, and particularly a recurring translocation of t(1;6) was noted (Fig. 1a) [50]. However, progress in the elucidation of oncogenes and tumor suppressor genes in melanomas by cytogenetic analyses were hampered technologically due to difficulties in obtaining metaphase preparations that are suitable for karyotyping from primary tumors. Comparative genomic hybridization (CGH) had overcome those technical obstacles by using genomic DNA as source materials to analyze genomic alterations of primary tumors [51], and microarray technologies extensively increased the resolutions of CGH analyses [52]. Those novel technologies were swiftly applied to analyses of genomic alterations of melanoma cells [13, 53]. Analyses of a large number of cases with different clinical subtypes of primary melanomas using array-CGH revealed that melanomas have four different subtypes from a genomic point of view: melanomas on skin with chronic sun-induced damage (CSD melanoma), melanomas on skin without chronic sun-induced damage (Non-CSD melanoma), Acral melanomas and Mucosal melanomas [54]. Those novel findings obtained by a series of CGH studies in addition to mutation analyses including the BRAF gene enabled progress in the research fields of molecular pathogenesis of melanomas in contrast to the previous concept, which was speculated from clinical and histopathological observations, that melanomas do not have enough evidence to subcategorize them into different subsets [55].
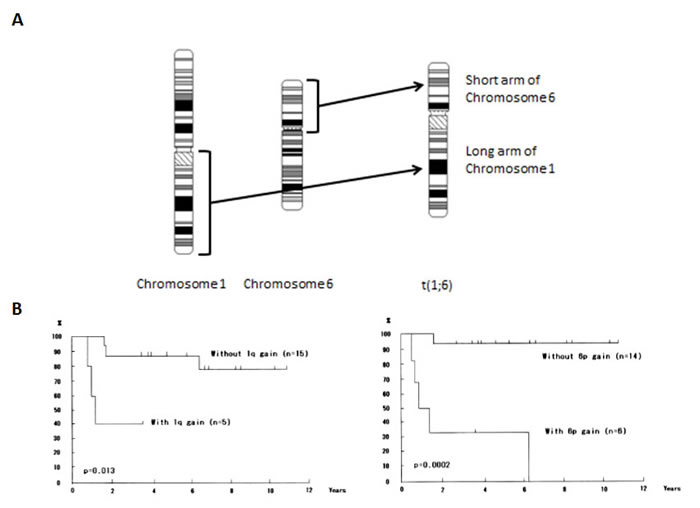
Figure 1: Chromosome rearrangements and impact on survival of melanoma patients. A) Translocations in melanoma cells. The long arm of chromosome 1 is translocated to the short arm of chromosome 6 to result in the t(1;6) chromosome. B) Metaphase CGH analyses revealed that both chromosomal gains of 1q and 6p significantly correlate with the survival of melanoma patients (ref. 62). Kaplan-Meier survival analyses for overall survival are shown. P values are indicated.
Acral melanomas occur on nailbeds and plantar regions such as the palms of the hands and soles of the feet [56]. The histopathological expression of acral melanomas is not always acral lentiginous melanoma, and other histopathological types of superficial spreading melanoma and nodular melanoma also exist [57]. Although the aggressiveness of acral melanomas had been disputed due to small sample size [58], recent data with a large cohort supports the aggressiveness of acral melanomas [59]. CGH analyses have shown unique genomic changes of acral melanomas that differ from those of other melanomas. At the chromosomal level, numbers of genomic aberrations in the whole genome were higher in acral melanomas. Particularly numbers of amplifications are significantly high, and amplifications at 11q13 and 5p15 are noted in acral melanomas [60, 61]. Those changes were also verified by array-CGH analyses [53]. High resolution mapping of an amplicon at 11q13-14 in breast cancer suggests CCND1, S6K2 and GAB2 as candidate genes in that region [62]. Fluorescence in situ hybridization (FISH) and/or immunohistochemical analyses showed that amplification and high expression of CCND1 were also observed in acral melanomas [63, 64]. Interestingly, the amplification of CCND1 is relatively rare in melanomas with BRAF-NRAS mutations and may have similar effects on melanoma cell growth as the activation of the mitogen-activated protein kinase (MAPK) signaling pathway resulting from BRAF and/or NRAS mutations [65, 66]. Another array-CGH study indicated the importance of the 4q12 region, where a narrow amplification is observed in acral, mucosal and CSD melanomas and the KIT gene resides at that locus [67]. Activation of KIT results from amplification and/or mutation, and mutations are frequently observed in exons 11, 13 and 17 [15, 68]. This KIT activation is intriguing, because it could be a direct target of therapies against acral melanomas using inhibitors targeting KIT (such as imatinib mesylate) [69, 70]. A particularly important point in the tumorigenesis of acral melanomas that is different from other subtypes of melanomas such as Non-CSD melanomas is that BRAF mutations occur at lower frequencies in acral melanomas [71]. These observations imply that other genetic aberrations may have a profound effects on the tumorigenesis of acral melanomas.
CGH combined with analyses of clinical parameters are a powerful approach to identify genomic loci that have significant impact on the clinical outcome of melanoma patients. Both the classical cytogenetic approach and the CGH approach share the same implication that the long arm of chromosome 1 and the short arm of chromosome 6 may have a profound effect on the tumorigenesis of melanomas since both chromosomal regions have a significant impact on the clinical outcome of patient survival (Fig.1b) [50, 61]. Statistical analyses comparing tumor thickness, which is a predominant factor for clinical outcome in primary melanomas, by taking advantage of public array-CGH databases have revealed that 1q32, among the 4 loci of 1q21-23, 1q32, 6p23-25 and 6p21, is significantly correlated with tumor thickness in acral melanomas. Using the candidate gene approach, NUAK2 at this locus has been revealed as a promising gene that participates in the clinical outcome of acral melanomas (Fig. 2) [16].
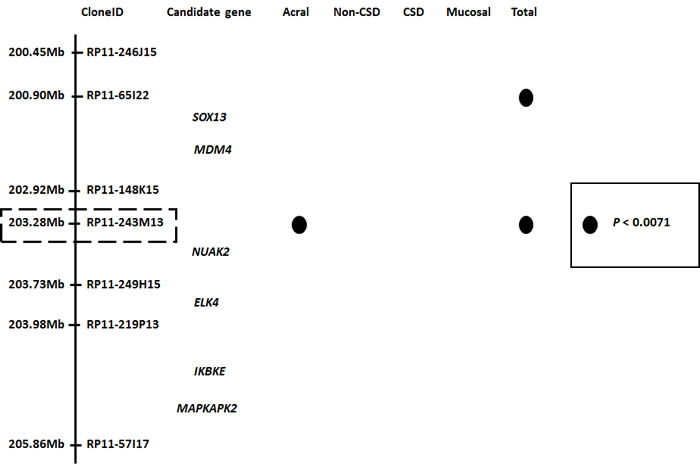
Figure 2: Candidate genes at the 1q32 locus. Analyses of a public array-CGH database indicated that a gene around clone RP11-243M13 is the most promising candidate oncogene at this locus. NUAK2 resides at the vicinity of this clone. Filled circles represent genomic clones with P values <0.0071 in each subset of melanoma. Adapted from [16].
PHYSIOLOGICAL ROLES OF NUAK2 AND THE AMPK FAMILY
NUAK2 is the fourth member of the AMPK family of kinases and shares a similar catalytic domain of the sucrose-non-fermenting protein kinase (SNF1)/AMP-activated protein kinase (AMPK) family of serine/threonine protein kinases. The NUAK2 gene resides at 1q32 and encodes 630 amino acid residues that are translated into a protein of approximately 76 kDa [23]. In general, AMPK family members are made up as heterotrimeric complexes of a catalytic α-subunit and regulatory β- and γ-subunits, and they act as an energy sensor to monitor energy homeostasis by binding AMP to the γ-subunit [17]. The binding of AMP to the γ-subunit stimulates the kinase activity of the α-subunit and promotes the phosphorylation of a Thr residue in the kinase domain, which results in the boost of kinase activity by additive (and/or synergic) effects of both stimulations [72]. Upstream regulators of the AMPK family have been identified including LKB1 and the calmodulin-dependent protein kinase kinases (CaMKKα and CaMKKβ) [73, 74]. LKB1 regulates 13 AMPK related kinases including NUAK2 as an upstream regulator [26]. Downstream effects of the AMPK family are diverse. The main effects of the AMPK family are functions relating to cellular metabolisms including the regulation of glucose intake [75]. The AMPK family has also diverse effects via regulation of the transcription of various genes related to the control of cell proliferation and cell polarity [76, 77]. Some of these effects are suspected to be tissue specific [17].
The amount of knowledge about the regulation and function(s) of NUAK2 is quite limited compared to AMPK-α1 and AMPK-α2. However, several important functions related to myosin filaments and cytoskeleton organization have been revealed. Myosin phosphatase target subunit 1 (MYPT1) was identified as a specific substrate for NUAK2. MYPT1 is phosphorylated by NUAK2 at sites other than Thr696 and The853, which are known as Rho-kinase (ROCK) phosphorylation sites [78]. Further, a study showed that unc-82, which encodes a serine/threonine kinase orthologous to human NUAK1/NUAK2 (ARK5/SNARK) in Caenorhabditis elegans, participates in maintaining the integrity of components of myosin filaments. Disruption of unc-82 by mutations causes defects in cytoskeleton reorganization during embryogenesis [79]. Another study showed that NUAK2 is able to associate with myosin phosphatase Rho-interacting protein (MRIP) and this association results in increased levels of myosin regulatory light chain (MLC) phosphorylation and facilitates the formation of stress fibers. Activities resulting from those associations of NUAK2 and MRIP are independent of NUAK2 kinase activity and those associations inhibit fiber disassembly and MYPT1-mediated MLC dephosphorylation. Important roles of NUAK2 on fiber maintenance in proliferating cells and the existence of a positive –feedback loop regulating actin stress fibers independent of the MLC kinase Rho-associated protein kinase (ROCK) have been indicated [80]. As the AMPK family of kinases in general functions as a sensor of metabolic homeostasis in cells, NUAK2 is activated by cellular stresses such as glucose deprivation, rotenone and sorbitol [23, 81]. Muscle contraction increases glucose transport by increasing NUAK2 activity in skeletal muscle cells, which suggests that NUAK2 functions to connect to the cytoskeleton modulation induced by muscle contraction with energy homeostasis in cells [82]. Those results indicate that NUAK2 plays a pivotal role in regulating the cytoskeleton, which is also important in cell proliferation and motility, and suggests that NUAK2 connects metabolic homeostasis and cell motility. The disruption of those mechanisms in normal cells may reflect cell proliferation and migration in cancer cells.
POTENTIAL ONCOGENIC ROLES OF NUAK2 AND ITS SIGNIFICANCE IN ACRAL MELANOMAS
Disruption of the normal regulation and function(s) of NUAK2 may lead to the dysregulation of proliferation and migration in cancer cells. However, the exact effects and mechanisms participating in tumorigenesis remain to be elucidated due to the lack of sufficient molecular studies of NUAK2. Several conflicting results on tumorigenesis including cell proliferation, apoptosis and migration have been reported. The knockdown of NUAK2 using siRNA or shRNA reduces cell proliferation in vitro and tumor growth in vivo in melanomas, and the extent of those reductions of cell proliferation varies depending on the different genomic aberrations of melanoma cells [16]. Another study with a carcinogen (azoxymethane) induced colorectal tumorigenesis model using NUAK2-deficient mice has shown that hemiallelic loss of NUAK2 contributes to carcinogen-induced neoplastic and preneoplastic lesions of colorectal carcinomas, which suggests there are tumor suppressive roles of NUAK2 in the early phase of tumorigenesis and suggests the minor effects of NUAK2 deficiency on cell proliferation in vivo from the profile of the proliferating cell population [83]. Over-expression of NUAK2 induced prolongation of cell survival of HepG2 cells in nutrient-deprived circumstances [24]. This function of NUAK2 is partly dependent on anti-apoptotic properties against apoptosis induced by death ligand such as the CD95 ligand, TRAIL and TNF-α which shows that NUAK2 is a kinase induced by TNF-α [78, 84]. However, NUAK2 functions during apoptosis are different depending on the melanoma cell lines [16]. NUAK2 also has effects on the migration of cancer cells as speculated from studies on myosin filaments and cytoskeleton organization in normal cells. An initial study revealed that the over-expression of NUAK2 has effects on cell-cell detachment in glucose deprived circumstances and suggested that over-expression of NUAK2 induced dysregulation of mechanisms to maintain the cytoskeleton and to coordinate its attachment to the cell membrane [24]. CD95 stimulation facilitates cell motility and invasiveness of MCF7-FB cells, which up-regulates NUAK2 expression by stimulation of the CD95 ligand [84]. That evidence suggests that the effects of NUAK2 on tumorigenesis are different depending on the tissue and the phase of tumorigenesis, and that NUAK2 participates in increased cell motility and invasiveness.
UV irradiation is one of the major causes of cutaneous melanomas, but acral and mucosal melanomas are protected from exposure to UV irradiation due to their anatomical locations. Thus, the molecular pathogenesis of acral and mucosal melanomas should be different from that of cutaneous melanomas arising from sun-exposed areas such as Non-CSD melanomas, and causes other than UV irradiation, such as reactive oxygen species (ROS), may play an important role in the melanomagenesis of acral and/or mucosal melanomas. The HGF/SF transgenic mouse model is prone to develop cutaneous melanomas following UV irradiation [85]. In that mouse model, LKB1 is one of the major downstream targets and uncoupling of the LKB1-AMPK pathway by oncogenic BRAF is one possible mechanism to promote the proliferation of melanoma cells with BRAF mutations [86]. Another study has substantiated the pivotal role of the LKB1-AMPK pathway in melanomagenesis [87]. The anatomical distribution of lentigines in Peutz-Jeghers syndrome, which is caused by mutations in the LKB1 gene, is almost identical to the distribution of both acral and mucosal melanomas. Although those observations imply that the LKB1-AMPK pathway may also play a role in the neoplastic formation of melanocytes distributed in those areas, melanomas are relatively rare with a few exceptions in those areas of patients with Peutz-Jeghers syndrome [88, 89]. Recent studies have shown that both ROS and hypoxia can activate AMPK through calcium release-activated calcium (CRAC) channels and CaMKKβ independent of LKB1 [90, 91]. Melanocytes are speculated to reside both in ROS abundant and in hypoxic conditions from observations that ROS are constantly generated as a byproduct of melanin synthesis in melanocytes [92], and that the epidermis where melanocytes reside is a relatively hypoxic environment (with oxygen levels ranging from 1.5% to 5.0%) [93]. Although the exact mechanism(s) that connects ROS and/or hypoxia to NUAK2 are still under investigation, those mechanisms should be further elucidated to explain melanomagenesis arising from acral areas. The downstream pathways by which NUAK2 regulates the cell cycle machinery have been examined with knockdown experiments of NUAK2 by siRNA and suggest that NUAK2 regulates Cyclin D1 and Cyclin D3 expression through the mTOR pathway to control cell proliferation (Fig. 3) [16]. The mTOR pathway participates in controlling progression through the cell cycle. Several studies using melanoma cells suggest that the mTOR pathway also participates in controlling the balance between senescence and quiescence under oncogenic and/or tumor suppressive stimuli. These intricate molecular mechanisms may partly explain the dual functions of NUAK2 as an oncogene and as a tumor suppressor gene [94, 95]. Clinical data also suggest that NUAK2 plays a pivotal role in melanomagenesis in acral areas and has effects on the survival of patients with acral melanomas. Univariate analysis using Kaplan-Meier curves showed that high expression of NUAK2 has an impact on relapse-free survival of acral melanoma patients (P = 0.0036), and multivariate analysis using multiple Cox regression analysis also showed the impact of NUAK2 on relapse-free survival (hazard ratio = 3.88, 95% confidence interval = 1.44-10.50, P = 0.0075). Interestingly, those impacts of NUAK2 on melanoma patient survival are significant at relapse-free survival of acral melanomas compared to weak significances at Non-CSD melanomas and overall survival. Those observations lead to the speculation that NUAK2 has more profound effects on cell migration resulting in worsening relapse-free survival of acral melanoma patients as suggested by both a migration assay and a wound healing assay [16].
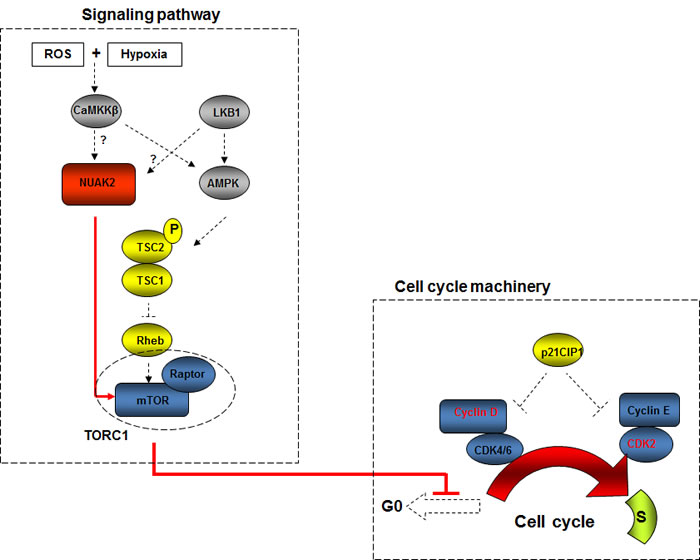
Figure 3: Hypothetical schematic of the regulation of NUAK2 in acral melanomas. Hypothetically, both reactive oxygen species (ROS) and hypoxia activate NUAK2 through CaMKKβ in acral melanoma cells, and result in dysregulation of cell cycle machinery through the mTOR pathway. Hypothetical pathways, which are activated in acral melanoma cells, are depicted as red line and/or arrows.
FUTURE PERSPECTIVES FOR THERAPEUTIC IMPLICATIONS
The recent identification of KIT mutations in acral, mucosal and CSD melanomas led to the development of therapeutic modalities using imatinib mesylate to target KIT activation. Although initial studies achieved only poor responses using imatinib mesylate against melanomas, the stratification and appropriate selection of patients based on KIT activation by mutations and amplifications have improved responses of melanoma patients to imatinib mesylate [70, 98]. As this example has clearly shown, the use of molecular targeting agents should be based on a better understanding of molecular carcinogenesis and an appropriate selection of patients [96, 97]. In acral melanomas, the frequency of KIT mutations is relatively low and therapeutic strategies targeting other mutations and amplifications are required to improve overall efficacy and response taking advantage of inhibitors targeting those genetic aberrations. A better understanding of the roles of AMPK family members including NUAK2 in acral melanomagenesis should be a necessary step to improve the management of acral melanoma patients.
ACKNOWLEDGEMENTS
This research was supported in part by the Intramural Research Program of the NIH, National Cancer Institute.
REFERENCES
1. Haber DA, Gray NS, Baselga J. The evolving war on cancer. Cell. 2011; 145: 19-24.
2. Sidransky D. Emerging molecular markers of cancer. Nat Rev Cancer. 2002; 2: 210-19.
3. Hunter T. Treatment for chronic myelogenous leukemia: the long road to imatinib. J Clin Invest. 2007; 117: 2036-43.
4. Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teaque J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002; 417: 949-54.
5. Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, O’Dwyer PJ, Lee RJ, Grippo JF, Nolop K, Chapman PB. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010; 363: 809-19.
6. Solit DB, Rosen N. Resistance to BRAF inhibition in melanomas. N Engl J Med. 2011; 364: 772-74.
7. Edmunds SC, Cree IA, Di Nicolantonio F, Hungerford JL, Hurren JS, Kelsell DP. Absence of BRAF gene mutations in uveal melanomas in contrast to cutaneous melanomas. Br J Cancer. 2003; 88: 1403-5.
8. Maldonado JL, Fridlyand J, Patel H, Jain AN, Busam K, Kageshita T, Ono T, Albertson DG, Pinkel D, Bastian BC. Determinants of BRAF mutations in primary melanomas. J Natl Cancer Inst. 2003; 95: 1878-90.
9. Edwards RH, Ward MR, Wu H, Medina CA, Brose MS, Volpe P, Nussen-Lee S, Haupt HM, Martin AM, Herlyn M, Lessin SR, Weber BL. Absence of BRAF mutations in UV-protected mucosal melanomas. J Med Genet. 2004; 41: 270-2.
10. Sasaki Y, Niu C, Makino R, Kudo C, Sun C, Watanabe H, Matsunaga J, Takahashi K, Tagami H, Aiba S, Horii A. BRAF point mutations in primary melanoma show different prevalences by subtype. J Invest Dermatol. 2004; 123: 177-83.
11. Helmke BM, Mollenhauer J, Herold-Mende C, Benner A, Thome M, Gassler N, Wahl W, Lyer S, Poustka A, Otto HF, Deichmann M. BRAF mutations distinguish anorectal from cutaneous melanoma at the molecular level. Gastroenterology. 2004; 127: 1815-20.
12. Landi MT, Bauer J, Pfeiffer RM, Elder DE, Hulley B, Minghetti P, Calista D, Kanetsky PA, Pinkel D, Bastian BC. MC1R germline variants confer risk for BRAF-mutant melanoma. Science. 2006; 313: 521-22.
13. Bastian BC, LeBoit PE, Hamm H, Bröcker EB, Pinkel D. Chromosomal gains and losses in primary cutaneous melanomas detected by comparative genomic hybridization. Cancer Res. 1998; 58: 2170-75.
14. Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol. 2006; 24: 4340-46.
15. Beadling C, Jacobson-Dunlop E, Hodi FS, Le C, Warrick A, Patterson J, Town A, Harlow A, Cruz F 3rd, Azar S, Rubin BP, Muller S, West R, Heinrich MC, Corless CL. KIT gene mutations and copy number in melanoma subtypes. Clin Cancer Res. 2008; 14: 6821-28.
16. Namiki T, Tanemura A, Valencia JC, Coelho SG, Passeron T, Kawaguchi M, Vieira WD, Ishikawa M, Nishijima W, Izumo T, Kaneko Y, Katayama I, Yamaguchi Y, Yin L, Polley EC, Liu H and et al. AMP kinase-related kinase NUAK2 affects tumor growth, migration, and clinical outcome of human melanoma. Proc Natl Acad Sci USA. 2011; 108: 6597-602.
17. Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007; 8: 774-785.
18. Kato K, Ogura T, Kishimoto A, Minegishi Y, Nakajima N, Miyazaki M, Esumi H. Critical roles of AMP-activated protein kinase in constitutive tolerance of cancer cells to nutrient deprivation and tumor formation. Oncogene. 2002; 21: 6082-90.
19. Carling D, Aguan K, Woods A, Verhoeven AJ, Beri RK, Brennan CH, Sidebottom C, Davison MD, Scott J. Mammalian AMP-activated protein kinase is homologous to yeast and plant protein kinases involved in the regulation of carbon metabolism. J Biol Chem. 1994; 269: 11442-8.
20. Mitchelhill KI, Stapleton D, Gao G, House C, Michell B, Katsis F, Witters LA, Kemp BE. Mammalian AMP-activated protein kinase shares structural and functional homology with the catalytic domain of yeast Snf1 protein kinase. J Biol Chem. 1994; 269: 2361-4.
21. Stapleton D, Mitchelhill KI, Gao G, Widmer J, Michell BJ, Teh T, House CM, Fernandez CS, Cox T, Witters LA, Kemp BE. Mammalian AMP-activated protein kinase subfamily. J Biol Chem. 1996; 271: 611-4.
22. Heyer BS, Warsowe J, Solter D, Knowles BB, Ackerman SL. New member of the Snf1/AMPK kinase family, Melk, is expressed in the mouse egg and preimplantation embryo. Mol Reprod Dev. 1997; 47: 148-56.
23. Lefebvre DL, Bai Y, Shahmolky N, Sharma M, Poon R, Drucker DJ, Rosen CF. Identification and characterization of a novel sucrose-non-fermenting protein kinase/AMP-activated protein kinase-related protein kinase, SNARK. Biochem J. 2001; 355: 297-305.
24. Suzuki A, Kusakai G, Kishimoto A, Minegichi Y, Ogura T, Esumi H. Induction of cell-cell detachment during glucose starvation through F-actin conversion by SNARK, the fourth member of the AMP-activated protein kinase catalytic subunit family. Biochem Biophys Res Commun. 2003; 311: 156-61.
25. Suzuki A, Kusakai G, Kishimoto A, Lu J, Ogura T, Lavin MF, Esumi H. Identification of a novel protein kinase mediating Akt survival signaling to the ATM protein. J Biol Chem. 2003; 278: 48-53.
26. Lizcano JM, Göransson O, Toth R, Deak M, Morrice NA, Boudeau J, Hawley SA, Udd L, Mäkelä TP, Hardie DG, Alessi DR. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 2004; 23: 833-43
27. Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002; 298: 1912-34.
28. Woodard J, Platanias LC. AMP-activated kinase (AMPK)-generated signals in malignant melanoma cell growth and survival. Biochem Biophys Res Commun. 2010; 398: 135-9.
29. Kusakai G, Suzuki A, Ogura T, Miyamoto S, Ochiai A, Kaminishi M, Esumi H. ARK5 expression in colorectal cancer and its implications for tumor progression. Am J Pathol. 2004; 164: 987-95.
30. Luo Z, Zang M, Guo W. AMPK as a metabolic tumor suppressor: control of metabolism and cell growth. Future Oncol. 2010; 6: 457-70.
31. Vazquez-Martin A, Oliveras-Ferraros C, Lopez-Bonet E, Menendez JA. AMPK: Evidence for an energy-sensing cytokinetic tumor suppressor. Cell Cycle. 2009; 8: 3679-83.
32. Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009; 9: 563-75.
33. Suzuki A, Lu J, Kusakai G, Kishimoto A, Ogura T, Esumi H. ARK5 is a tumor invasion-associated factor downstream of Akt signaling. Mol Cell Biol. 2004; 24: 3526-35.
34. Tsuchihara K, Ogura T, Fujioka R, Fujii S, Kuga W, Saito M, Ochiya T, Ochiai A, Esumi H. Susceptibility of Snark-deficient mice to azoxymethane-induced colorectal tumorigenesis and the formation of aberrant crypt foci. Cancer Sci. 2008; 99: 677-82.
35. Becher R, Gibas Z, Karakousis C, Sandberg AA. Nonrandom chromosome changes in malignant melanoma. Cancer Res. 1983; 43: 5010-16.
36. Trent JM, Rosenfeld SB, Meyskens FL. Chromosome 6q involvement in human malignant melanoma. Cancer Genet Cytogenet. 1983; 9: 177-80.
37. Balaban G, Herlyn M, Guerry D 4th, Bartolo R, Koprowski H, Clark WH, Nowell PC. Cytogenetics of human malignant melanoma and premalignant lesions. Cancer Genet Cytogenet. 1984; 11: 429-39.
38. Parmiter AH, Balaban G, Herlyn M, Clark WH, Nowell PC. A t(1;19) chromosome translocation in three cases of human malignant melanoma. Cancer Res. 1986; 46: 1526-29.
39. Cowan JM, Halaban R, Lane AT, Francke U. The involvement of 6p in melanoma. Cancer Genet Cytogenet. 1986; 20: 255-61.
40. Nowell PC, Hungerford DA. A minute chromosome in human chronic granulocytic leukemia. Science. 1960; 132: 1497.
41. Rowley JD. Letter: A new consistent chromosomal abnormality in chronic myelogenous leukemia identified by quinacrine fluorescence and Giemsa staining. Nature. 1973; 243: 290-93.
42. de Klein A, van Kessel AG, Grosveld G, Bartram CR, Haqemeijer A, Bootsma D, Spurr NK, Heisterkamp N, Groffen J, Stephenson JR. A cellular oncogene is translocated to the Philadelphia chromosome in chronic myelocytic leukemia. Nature. 1982; 300: 765-67.
43. Groffen J, Stephenson JR, Heisterkamp N, de Klein A, Bartram CR, Grosveld G. Philadelphia chromosomal breakpoints are clustered within a limited region, bcr, on chromosome 22. Cell. 1984; 36: 93-9.
44. Shtivelman E, Lifshitz B, Gale RP, Canaani E. Fused transcript of abl and bcr genes in chronic myelogenous leukemia. Nature. 1985; 315: 550-54.
45. Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, Zimmermann J, Lydon NB. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med. 1996; 2: 561-66.
46. Druker BJ, Sawyers CL, Kantarjian H, Resta DJ, Reese SF, Ford JM, Capdeville R, Talpaz M. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med. 2001; 344: 1038-42.
47. Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, Lydon NB, Kantarjian H, Capdeville R, Ohno-Jones S, Sawyers CL. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001; 344: 1031-37.
48. Koretzky GA. The legacy of the Philadelphia chromosome. J Clin Invest. 2007; 117: 2030-32.
49. Thompson FH, Emerson J, Olson S, Weinstein R, Leavitt SA, Leong SP, Emerson S, Trent JM, Nelson MA, Salmon SE, Taetle R. Cytogenetics of 158 patients with regional or disseminated melanoma. Subset analysis of near-diploid and simple karyotypes. Cancer Genet Cytogenet. 1995; 83: 93-104.
50. Trent JM, Thompson FH, Meyskens FL. Identification of a recurring translocation site involving chromosome 6 in human malignant melanoma. Cancer Res. 1989; 49: 420-23.
51. Kallioniemi A, Kallioniemi OP, Sudar D, Rutovitz D, Gray JW, Waldman F, Pinkel D. Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science. 1992; 258: 818-21.
52. Pinkel D, Segraves R, Sudar D, Clark S, Poole I, Kowbel D, Collins C, Kuo WL, Chen C, Zhai Y, Dairkee SH, Ljung BM, Gray JW, Albertson DG. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat Genet. 1998; 20: 207-11.
53. Bastian BC, Olshen AB, LeBoit PE, Pinkel D. Classifying melanocytic tumors based on DNA copy number changes. Am J Pathol. 2003; 163: 1765-70.
54. Curtin JA, Fridlyand J, Kageshita T, Patel HN, Busam KJ, Kutzner H, Cho KH, Aiba S, Bröcker EB, LeBoit PE, Pinkel D, Bastian BC. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005; 353: 2135-47.
55. Ackerman AB, David KM. A unifying concept of malignant melanoma: biologic aspects. Hum Pathol. 1986; 17: 438-40.
56. Feibleman CE, Stoll H, Maize JC. Melanomas of the palm, sole, and nailbed: a clinicopathologic study. Cancer. 1980; 46: 2492-504.
57. Kato T, Suetake T, Tabata N, Takahashi K, Tagami H. Epidemiology and prognosis of plantar melanoma in 62 Japanese patients over a 28-year period. Int J Dermatol. 1999; 38: 515-19.
58. Jimbow K, Takahashi H, Miura S, Ikeda S, Kukita A. Biological behavior and natural course of acral malignant melanoma. Clinical and histologic features and prognosis of palmoplantar, subungual, and other acral malignant melanomas. Am J Dermatopathol. 1984; 6 Suppl: 43-53.
59. Bradford PT, Goldstein AM, McMaster ML, Tucker MA. Acral lentiginous melanoma: incidence and survival patterns in the United States, 1986-2005. Arch Dermatol. 2009; 145: 427-34.
60. Bastian BC, Kashani-Sabet M, Hamm H, Godfrey T, Moore DH 2nd, Bröcker EB, LeBoit PE, Pinkel D. Gene amplifications characterize acral melanoma and permit the detection of occult tumor cells in the surrounding skin. Cancer Res. 2000; 60: 1968-73.
61. Namiki T, Yanagawa S, Izumo T, Ishikawa M, Tachibana M, Kawakami Y, Yokozeki H, Nishioka K, Kaneko Y. Genomic alterations in primary cutaneous melanomas detected by metaphase comparative genomic hybridization with laser capture or manual microdissection: 6p gains may predict poor outcome. Cancer Genet Cytogenet. 2005; 157: 1-11.
62. Karlsson E, Waltersson MA, Bostner J, Pérez-Tenorio G, Olsson B, Hallbeck AL, Stål O. High-resolution genomic analysis of the 11q13 amplicon in breast cancers identifies synergy with 8p12 amplication, involving the mTOR targets S6K2 and 4EBP1. Genes Chromosomes Cancer. 2011;50: 775-87.
63. Sauter ER, Yeo UC, von Stemm A, Zhu W, Litwin S, Tichansky DS, Pistritto G, Nesbit M, Pinkel D, Herlyn M, Bastian BC. Cyclin D1 is a candidate oncogene in cutaneous melanoma. Cancer Res. 2002; 62: 3200-6.
64. Yamaura M, Takata M, Miyazaki A, Saida T. Specific dermoscopy patterns and amplifications of the cyclin D1 gene to define histopathologically unrecognizable early lesions of acral melanoma in situ. Arch Dermatol. 2005; 141: 1413-8.
65. Gast A, Scherer D, Chen B, Bloethner S, Melchert S, Sucker A, Hemminiki K, Schadendorf D, Kumar R. Somatic alterations in the melanoma genome: a high-resolution array-based comparative genomic hybridization study. Genes Chromosomes Cancer. 2010; 49: 733-45.
66. Takata M, Goto Y, Ichii N, Yamamura M, Murata H, Koga H, Fujimoto A, Saida T. Constitutive activation of the mitogen-activated protein kinase signaling pathway in acral melanomas. J Invest Dermatol. 2005; 125: 318-22.
67. Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol. 2006; 24: 4340-46.
68. Torres-Cabala CA, Wang WL, Trent J, Yang D, Chen S, Galbincea J, Kim KB, Woodman S, Davies M, Plaza JA, Nash JW, Prieto VG, Lazar AJ, Ivan D. Correlation between KIT expression and KIT mutation in melanoma: a study of 173 cases with emphasis on the acral-lentiginous/mucosal type. Mod Pathol. 2009; 22: 1446-56.
69. Kim KB, Eton O, Davis DW, Frazier ML, McConkey DJ, Diwan AH, Papadopoulos NE, Bedikian AY, Camacho LH, Ross MI, Cormier JN, Gershenwald JE, Lee JE, Mansfield PF, Billings LA, Ng CS, Charnsangavej C, Bar-Eli M, Johnson MM, Murgo AJ, Prieto VG. Phase II trial of imatinib mesylate in patients with metastatic melanoma. Br J Cancer. 2008; 99: 734-40.
70. Carvajal RD, Antonescu CR, Wolchok JD, Chapman PB, Roman RA, Teitcher J, Panageas KS, Busam KJ, Chmielowski B, Lutzky J, Pavlick AC, Fusco A, Cane L, Takebe N, Vemula S, Bouvier N, Bastain BC, Schwartz GK. KIT as a therapeutic target in metastatic melanoma. JAMA. 2011; 305: 2327-34.
71. Saldanha G, Potter L, Daforno P, Pringle JH. Cutaneous melanoma subtypes show different BRAF and NRAS mutation frequencies. Clin Cancer Res. 2006; 12: 4499-505.
72. Hawley SA, Davison M, Woods A, Davies SP, Beri RK, Carling D, Hardie DG. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J Biol Chem. 1996; 271: 27879-87.
73. Hurley RL, Anderson KA, Franzone JM, Kemp BE, Means AR, Witters LA. The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J Biol Chem. 2005; 280: 29060-66.
74. Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, Cantley LC. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci USA. 2004; 101: 3329-35.
75. Russell RR 3rd, Bergeron R, Shulman GI, Young LH. Translocation of myocardial GLUT-4 and increased glucose uptake through activation of AMPK by AICAR. Am J Physiol. 1999; 277: H643-9.
76. Luo Z, Zang M, Guo W. AMPK as a metabolic tumor suppressor: control of metabolism and cell growth. Future Oncol. 2010; 6: 457-70.
77. Mirouse V, Billaud M. The LKB1/AMPK polarity pathway. FEBS Lett. 2011; 585: 981-5.
78. Yamamoto H, Takashima S, Shintani Y, Yamazaki S, Seguchi O, Nakano A, Higo S, Kato H, Liao Y, Asano Y, Minamino T, Matsumura Y, Takeda H, Kitakaze M. Identification of a novel substrate for TNFalpha-induced kinase NUAK2. Biochem Biophys Res Commun. 2008; 365: 541-7.
79. Hoppe PE, Chau J, Flanagan KA, Reedy AR, Schriefer LA. Caenorhabditis elegans unc-82 encodes a serine/threonine kinase important for myosin filament organization in muscle during growth. Genetics. 2010; 184: 79-90.
80. Vallenius T, Vaahtomeri K, Kovac B, Osiceanu AM, Viljanen M, Mäkelä TP. An association between NUAK2 and MRIP reveals a novel mechanism for regulation of actin stress fibers. J Cell Sci. 2011; 124: 384-93.
81. Lefebvre DL, Rosen CF. Regulation of SNARK activity in response to cellular stresses. Biochim Biophys Acta. 2005; 1724: 71-85.
82. Koh HJ, Toyoda T, Fujii N, Jung MM, Rathod A, Middelbeek RJ, Lessard SJ, Treebak JT, Tsuchihara K, Esumi H, Richter EA, Wojtaszewski JF, Hirshman MF, Goodyear LJ. Sucrose nonfermenting AMPK-related kinase (SNARK) mediates contraction-stimulated glucose transport in mouse skeletal muscle. Proc Natl Acad Sci USA. 2010; 107: 15541-6.
83. Tsuchihara K, Ogura T, Fujioka R, Fujii S, Kuga W, Saito M, Ochiya T, Ochiai A, Esumi H. Susceptibility of Snark-deficient mice to azoxymethane-induced colorectal tumorigenesis and the formation of aberrant crypt foci. Cancer Sci. 2008; 99: 677-82.
84. Legembre P, Schickel R, Barnhart BC, Peter ME. Identification of SNF1/AMP kinase-related kinase as an NF-kappaB-regulated anti-apoptotic kinase involved in CD95-induced motility and invasiveness. J Biol Chem. 2004; 279: 46742-47.
85. Noonan FP, Dudek J, Merlino G, De Fabo EC. Animal models of melanoma: an HGF/SF transgenic mouse model may facilitate experimental access to UV initiating events. Pigment Cell Res. 2003; 16: 16-25.
86. Esteve-Puig R, Canals F, Colomé N, Merlino G, Recio A. Uncoupling of the LKB1-AMPKalpha energy sensor pathway by growth factors and oncogenic BRAF. PLoS One. 2009; 4: e4771.
87. Zheng B, Jeong JH, Asara JM, Yuan YY, Granter SR, Chin L, Cantley LC. Oncogenic B-RAF negatively regulates the tumor suppressor LKB1 to promote melanoma cell proliferation. Mol Cell. 2009; 33: 237-47.
88. Wong SS, Rajakulendran S. Peutz-Jeghers syndrome associated with primary malignant melanoma of the rectum. Br J Dermatol. 1996; 135: 439-42.
89. Guldberg P, thor Straten P, Ahrenkiel V, Seremet T, Kirkin AF, Zeuthen J. Somatic mutation of the Peutz-Jeghers syndrome gene, LKB1/STK11, in malignant melanoma. Oncogene. 1999; 18: 1777-80.
90. Mungai PT, Waypa GB, Jairaman A, Prakriya M, Dokic D, Ball MK, Schumacker PT. Hypoxia triggers AMPK activation through reactive oxygen species-mediated activation of calcium release-activated calcium channels. Mol Cell Biol. 2011;31: 3531-45.
91. Gusarova GA, Trejo HE, Dada LA, Briva A, Welch LC, Hamanaka RB, Mutlu GM, Chandel NS, Prakriya M, Sznajder JI. Hypoxia leads to Na,K-ATPase down-regulation via Ca2+ release-activated Ca2+ channels and AMPK activation. Mol Cell Biol. 2011;31: 3546-56.
92. Sealy RC, Hyde JS, Felix CC, Menon IA, Prota G, Swartz HM, Persad S, Haberman HF. Novel free radicals in synthetic and natural pheomelanins: distinction between dopa melanin and cysteinyldopa melanins by ESR spectroscopy. Proc Natl Acad Sci USA. 1982; 79: 2885-9.
93. Bedogni B, Welford SM, Cassarino DS, Nickoloff BJ, Giaccia AJ, Powell MB. The hypoxic microenvironment of the skin contributes to Akt-mediated melanocyte transformation. Cancer Cell. 2005; 8: 443-54.
94. Leontieva OV, Blaqosklonny MV. DNA damaging agents and p53 do not cause senescence in quiescent cells, while consecutive re-activation of mTOR is associated with conversion to senescence. Aging (Albany NY). 2010; 2: 924-35.
95. Korotchkina LG, Leontieva OV, Bukreeva EI, Demidenko ZN, Gudkov AV, Blaqosklonny MV. The choice between p53-induced senescence and quiescence is determined in part by the mTOR pathway. Aging (Albany NY). 2010; 2: 344-52.
96. Koomen JM, Smalley KS. Using quantitative proteomic analysis to understand genotype specific intrinsic drug resistance in melanoma. Oncotarget. 2011; 2: 329-35.
97. Beloueche-Babari M, Arunan V, Jackson LE, Perusinqhe N, Sharp SY, Workman P, Leach MO. Modulation of melanoma cell phospholipid metabolism in response to heat shock protein 90 inhibition. Oncotarget. 2010; 1: 185-97.
98. Ugurel S, Hildenbrand R, Zimpfer A, La Rosée P, Paschka P, Sucker A, Keikavoussi P, Becker JC, Rittgen W, Hochhaus A, Schadendorf D. Lack of clinical efficacy of imatinib in metastatic melanoma. Br J Cancer. 2005; 92: 1398-405.