
INTRODUCTION
DNA sequence amplification is a phenomenon that occurs predictably at defined stages during normal development in Xenopus, Drosophila, Sciara and Tetrahymena [1–4]. A cell’s strategy of amplifying genes represents a means of satisfying a heavy demand for stage-specific proteins [1]. These amplifications affect specific DNA regions and appear during narrow windows of development [4]. In mammals, gene amplification appears to be absent in normal cells but commonly occurs in cancer cells. However, we recently published first evidence for gene amplifications during differentiation of human neural progenitor cells [5].
Recent publications on haploid embryonic stem cells reported an intact genome without amplifications and losses. However, comparative genomic hybridization (CGH) data in these studies showed genomic imbalances that were not further investigated due to the selected threshold for amplification detection [6]. Likewise genomic imbalances reported for stem cells and/or induced pluripotent stem cells have always been interpreted based on threshold settings that were consistent with the hypothesis of an intact genome. In addition these imbalances were found between tissue samples including brain, testis, liver and blood samples [7]. These tissues were known to contain stem cells and differentiating cells of varying stage. To explain the genomic imbalances authors frequently blame preparation conditions and the influence of early and late replication timing. As of now, there is, however, no final evidence about the origin of the imbalances that are observed throughout many studies.
Our results on human neural progenitor cells are indicative of amplification as physiological process during stages of differentiation [5]. To follow up on this finding, we set out to investigate the hypothesis that gene amplifications occur as a developmental process in different species. Interestingly double minutes (DMs) as cytogenetic manifestations of gene amplification were found in 1% of serum free mouse embryo (SFME) cells and an increased frequency of DMs was found in cells grown in medium containing fetal calf serum (FCS) [8]. SFME cells were a neural stem cell line consisting of neural progenitor cells that are capable of differentiating into astrocytes when grown with growth factor TGF-ß or fetal calf serum (FCS). SFME cells were routinely cultivated on fibronectin coated culture ware. Many studies however have shown, that cell surface interactions of neural stem cells to extracellular matrix proteins (e.g. fibronectin, laminin) were capable of inducing cell differentiation processes suggesting synergic effects of adhesion and growth factor signals [9]. Sphere growth was reported for SFME cells as unattached multicellular aggregates in the absence of fibronectin [10]. Here we analyzed mouse neural progenitor cells during differentiation using SFME sphere cells and primary mesencephalon E14 neurosphere cells. Since both, our previous human and the present mouse analyses, are performed with cells under in vitro differentiation conditions, we also investigate amplifications on mouse embryo tissue sections to provide in vivo evidence for gene amplification as a physiological process.
RESULTS
Amplification analysis in SFME cells
To identify early differentiation-associated amplifications we performed array-CGH analysis on SFME cells that were induced to differentiate using different conditions. Former studies showed an increased glial differentiation specific GFAP mRNA expression 24 h after TGF-ß addition and 8–16 h after FCS addition. Based on these observations we choose to analyze undifferentiated SFME cells grown as spheres, SFME cells grown for 12 h with 10% FCS, and SFME cells grown for 24 h with TGF-ß. As shown in Figure 1a–1c we found clear morphology changes between the treatments. The SFME cells were also analyzed by immune fluorescence (Figure 1d–1f). Undifferentiated SFME cells expressed the neural stem cell marker nestin. Out of the SFME cells that were grown 24 h with TGF-ß, 30% of cells did not show nestin expression but GFAP expression, 50% of cells showed simultaneous nestin and GFAP expression and 10% of cells showed only nestin expression. All SFME cells that were grown for 12 h with 10% FCS showed GFAP expression but no nestin expression.
The array-CGH experiments were done by a two-color approach and all analyzed samples were compared to mouse genomic DNA from Clontech. We established the following data analysis pipeline to determine amplifications: Following the array-CGH analysis, signal intensities were extracted from scanned images of each array using Roche NimbleGen NimbleScan v2.6 software. Intensity values for Cy3 and Cy5 were spatially corrected, normalized using Qspline normalization, and the log2 of the ratios from Cy3/Cy5 intensity values were calculated. To reduce size and noise of the data we applied a 10 x window-averaging step. To detect amplifications we used the dynamic segMNT algorithm that identifies segments by minimizing the squared error relative to the segment mean. To detect representative alterations and to minimize the identification of random alterations, we extracted segments with segment means above a 0.1 threshold and a size greater than 500 kb. Since we did not compare the SFME cells to the original parental strain (BALB/c) but to normal mouse cells DNA, we expected several CNVs (copy number variations) between the individual mouse samples. We therefore excluded chromosomal regions representing CNVs from our results across all investigated samples that were included in a list of CNVs published in 2010 [11]. For the identification of differentiation-associated amplifications we further excluded chromosomal regions that revealed a copy number gain with a comparable log2 ratio value in all samples.
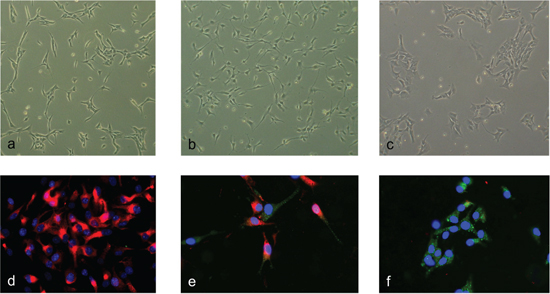
Figure 1: Morphology and marker expression changes upon differentiation induction. Undifferentiated SFME cells revealed a fibroblast-like morphology. (a) SFME cells differentiation-induced for 24 h by TGF-ß revealed filigree appendages. (b) SFME cells differentiation-induced for 12 h by FCS revealed a cobblestone-like morphology. (c) Immunofluorescence analysis was done with the neural stem cell marker nestin and glial marker GFAP. Undifferentiated SFME cells solely expressed nestin (red fluorescence). (d) TGF-ß differentiation induced SFME cells either expressed both nestin and GFAP, or GFAP only (green fluorescence). (e) FCS differentiation induced SFME cells expressed only GFAP. (f) Nuclei were counterstained with DAPI.
In total we detected 3 amplified regions in undifferentiated SFME sphere cells and 89 amplified chromosomal regions in 24 h-TGF-β-differentiation induced SFME cells. Amplifications were detected on all autosomal chromosomes with no specific clustering on a specific chromosome. In 12 h-FCS-differentiation induced SFME cells we detected only 13 amplified regions. Interestingly 2 out of 13 chromosome regions were solely detectable in FCS-differentiation induced cells and 11 of 13 chromosome regions were detectable under both differentiation induction conditions. The size of the amplified chromosome region varied between 250 kb–22 Mb. Amplified regions are summarized in Table 1. Representative array-CGH plots for mouse chromosomes 11, 9, 10 and 18 are shown in Figure 2a, 2b, 2c and 2d.
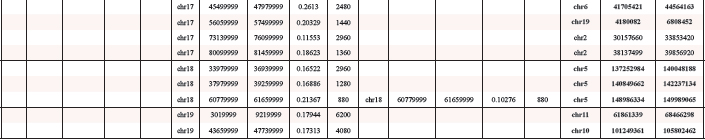
As an independent method of verifying amplification in single cells, we used fluorescence in situ hybridization (FISH). Representative BAC probes used for FISH analysis were selected for both the amplified and the control region. Corresponding gene names were only used for annotation of the BAC probes. Localization of probes used for FISH experiments are indicated in Figure 2. Two BAC probes were selected for two amplified chromosome regions on two different chromosomes with a log2 ratio value greater 0.24 (Table 1). We selected one region solely amplified in TGF-ß differentiation induced cells on chromosome 9 and one region amplified under both differentiation-induction conditions on chromosome 11. Both amplified chromosome regions were confirmed to be amplified using fluorescence in situ hybridization on individual cells. Examples of amplification of sequences on mouse chromosome 11q detected in 24 h-TGF-β–differentiation induced cells are illustrated in Figure 3a–3c. Amplifications were seen on interphase nuclei revealing several fluorescence signals for BAC RP23–235I12 (including C1QL1 gene in pink) and two fluorescence signals for the control probe BAC RP23–51E21 (including EGFR gene in green). An example of normal copy number fluorescence signals for BAC RP23–235I12 and the control probe BAC RP23–51E21 is shown in Figure 3d. Examples of sequence amplification on mouse chromosome 9q detected in 24 h-TGF-β–differentiation induced cells are illustrated in Figure 3e–3g. Amplifications were seen on interphase nuclei revealing several fluorescence signals for BAC probe RP23–4A11 (including S1PR5 gene in pink). Examples of normal copy number fluorescence signals for BAC RP23–4A11 are shown in Figure 3h.
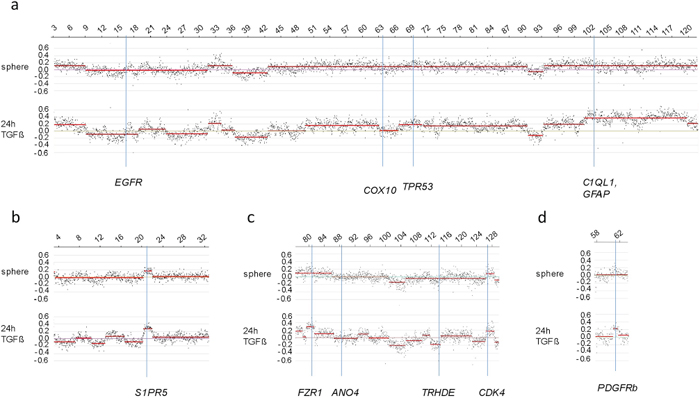
Figure 2: Relative copy number profiles on mouse chromosomes 11, 10, 9 and 18. CGH analysis of SFME cells grown as sphere or differentiation induced with TGF-β revealed multiple amplified and under-replicated regions on mouse chromosome 11 (a), mouse chromosome 9 (b), mouse chromosome 10 (c) and mouse chromosome 18 (d). Relative copy number is plotted at 40 kb resolution using a log2 scale. Localization of BAC clones used for FISH analysis were indicated including known genes: C1QL1, GFAP (RP23–235I12), TRP53 (RP23–150N14), EGFR (RP23–51E21), and COX10 (RP23–40P10) for chromosome 11; S1PR5 (RP23–4A11) for chromosome 9; FZR1 (RP23–421E11), CDK4 (RP23–432F11), ANO4 (RP23–279E23), and TRHDE (RP23–361I2) for chromosome 10; PDGFRb (RP23–143A24) for chromosome 18.
Table 1: Overview on amplified mouse chromosome regions
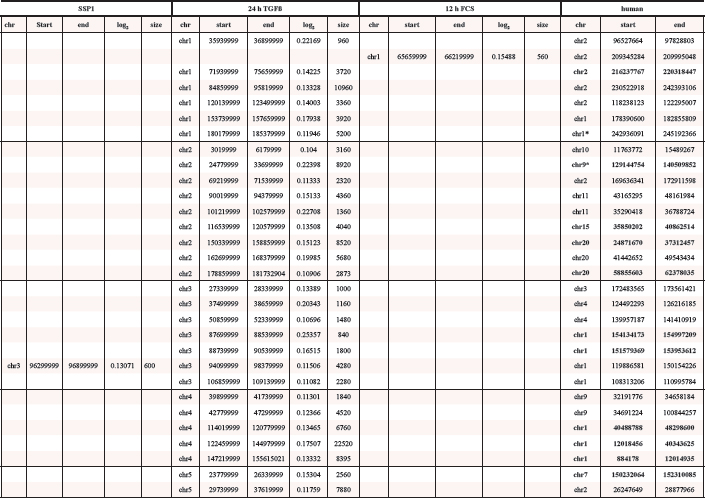
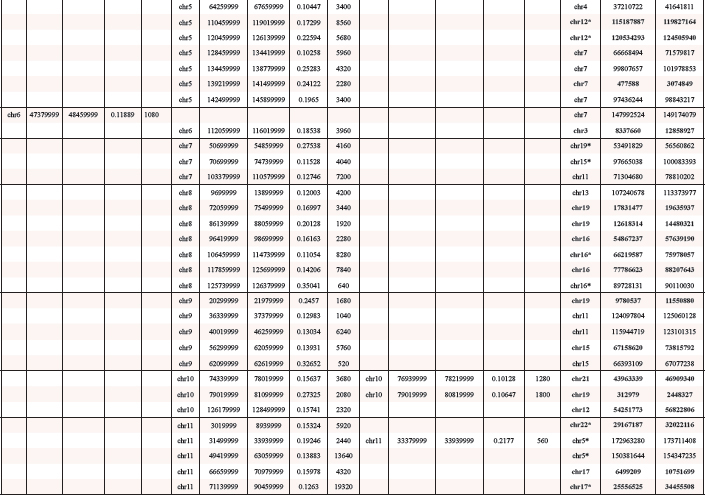
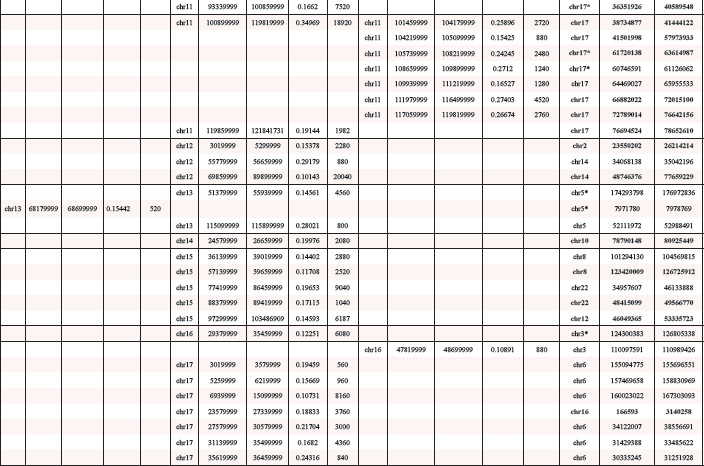
Start and end point of mouse amplified chromosome regions were according to NCBI37/mm9 and converted human sequences were according to NCBI36/HG18. Converted human sequences matching amplified human chromosome regions were displayed in bold. Human sequences marked by an asterisk were only partially converted. Sizes were displayed in kb.
Taken together, in differentiation induced SFME we determined a wavy pattern of genomic imbalances including amplifications that were confirmed in individual cells by fluorescence-in-situ hybridizations. Comparing our results with previously published results on array-CGH in haploid embryonic stem cells revealed noteworthy similarities. Representative chromosome plots for chromosome 11 from SFME array-CGH were presented in Figure 4a–4c. The pattern of imbalances present in TGF-ß-differentiation induced SFME cells is very similar to the pattern of imbalances at chromosome 11 in haploid embryonic stem cells published by Leeb and Wutz 2011 as presented in Figure 4d. This observation illustrates that far more stem cells use the mechanism of gene amplification during their development than hitherto believed.
Additional chromosome plots for chromosome 11 from array-CGH analysis at various time points and differentiation induction conditions were supplied in a Supplementary Figure S1a–S1d.
Analysis of amplified regions in mice
Previously, we reported 66 amplified chromosome regions after a 2 day long differentiation induction and 93 amplified chromosome regions after a 5 day differentiation induction in human neural progenitor cells [5]. We ask if and to what extend the human and the mouse amplifications overlap on the chromosomal level. Using UCSC-Genome Browser we converted mouse chromosome regions amplified in SFME sphere cells and FCS and/or TGF-β-differentiation induced SFME cells to the corresponding human chromosomal regions. Several regions failed to convert completely because sequences were split or duplicated. Out of 92 converted mouse sequences, 46 showed an overlap with amplified regions found in human neural progenitor cells after 5 day differentiation induction and 3 regions showed an overlap with amplified regions in human neural progenitor cells after 2 day differentiation induction. A detailed overview on this conversion is given in Table 1.
Figure 3: Gene amplifications on mouse chromosomes 11 and 9 on SFME cells. Interphase-FISH was used to confirm gene amplifications for C1QL1 on interphase nuclei from 24 h-TGF-β–differentiation induced SFME cells (a–c). C1QL1 specific BAC-probe (RP23–235I12) was labeled in pink and control BAC probe (RP23–51E21) (containing EGFR) from the same chromosome was labeled in green. Representative interphase nuclei without C1QL1 amplification from not-differentiated cells revealed comparable fluorescence signals for C1QL1 and EGFR control probe (d) Interphase-FISH was used to confirm gene amplifications for S1PR5 on interphase nuclei from 24 h TGF-β-differentiation induced cells (e–g). S1PR5 specific BAC-probe (RP23–4A11) was labeled in pink. Representative interphase nuclei from undifferentiated cells revealed single copy fluorescence signals for gene S1PR5 (h) Nuclei were counterstained with DAPI. Size calibration bar = 5 μm.
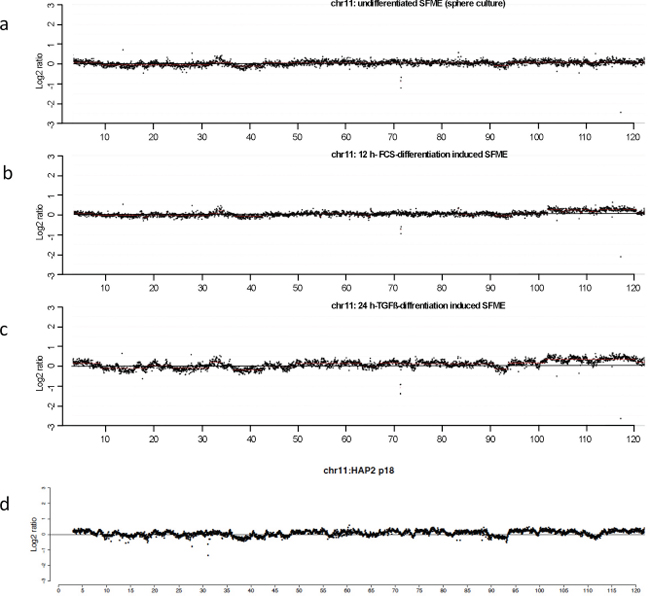
Figure 4: Comparative analysis of the ratio profiles from chromosome 11 indicating similarities and differences of the relative copy number. CGH results are displayed as representative chromosome 11 plots at 40 kb resolution using a log2 scale. Plots from undifferentiated sphere SFME cells revealed an almost smooth pattern of the ratio profile (a). Plot from 12 h-FCS differentiation induced SFME cells showed a similar smooth pattern of ratio profile with increased ratios for chromosome region 104–120 Mb (b). Plot from 24 h-TGF-ß differentiation induced SFME cells revealed a highly wavy pattern indicating multiple genomic imbalances with increased ratios for chromosome region 104–120 Mb (c). The wavy pattern visible in (c) revealed similarities to the wavy pattern of haploid embryonic stem cell line HAP2 recently published in Nature by Leeb and Wutz 2011 (Supplementary Figure) (d).
Gene amplification analysis on primary neural stem and progenitor cells
To exclude cell culture artifacts in the SFME neural stem cell line we analyzed gene amplifications in primary mesencephalon E14 neurosphere cells at passage P1 using FISH. For amplification confirmation we selected 6 amplified chromosome regions from four different chromosomes on the basis of log2 ratio value in array-CGH experiment. Representative BAC probes used for FISH analysis were selected for both the amplified and the control region. Localization of hybridization probes for amplified loci and control probes from the same chromosome are shown in Figure 2 and further information including the percentage of cells with amplification is summarized in Table 2. Undifferentiated neurosphere cells revealed normal copy number in 100 analyzed nuclei. We induced differentiation in primary mesencephalon E14 neurosphere cells for 2 or 3 days in differentiation medium containing small amounts of FCS as supplied by the manufacturer. Hybridization of FZR1 against neurosphere cells differentiated for 2 days (Figure 5a, 5b) and for 3 days (Figure 5c) revealed an increased number of fluorescence signals scattered over the whole nucleus in contrast to two fluorescence signals from the control probe ANO4. The increased number of fluorescence signals along with the two signals found for the control probe confirmed the amplification of FZR1. Gene amplifications of CDK4 after 3 days of differentiation induction are shown in Figure 5d. Gene amplifications of TRP53 (Figure 5e), C1QL1 (Figure 5f), S1PR5 (Figure 5g) and PDGFRb (Figure 5h) found in 3-day differentiation induced neurosphere cells confirmed our array-CGH results.
Simultaneous gene amplification and GFAP expression
Immune fluorescence was used to analyze the differentiation of neurosphere cells to glial lineage cells by simultaneous analysis of nestin and GFAP. As shown in Figure 6a neurosphere cells expressed nestin only. As shown in Figure 6b differentiation induced neurosphere cells expressed GFAP and showed a decreased nestin expression. To analyze a possible association between gene amplification and differentiation, we performed simultaneous FISH-analysis and immune fluorescence staining with an antibody against the astrocyte marker protein GFAP. Mesencephalon E14 neurosphere cells that were differentiation-induced for 2 days, showed amplifications for BAC RP23–421E11 containing FZR1 and BAC RP23–235I21 containing C1QL1 and GFAP. The differentiation-induced mesencephalon cells showed also GFAP protein expression (Figure 6c).
Multiple amplification analysis
We also investigated whether cells revealed multiple amplified chromosome regions per single cell. For co-hybridization experiments we labeled FZR1 from chromosome 10 with Alexa-594 in pink, TRP53 from chromosome 11 with Alexa-488 in green, and C1QL1 from a second amplified region on chromosome 11 with Alexa-555 in yellow. As shown in Figure 7a–7d a very variable pattern of hybridization signals was detectable. We found cells with normal copy number for all investigated probes (i) cells with amplification of either TRP53 (ii) or FZR1 (iii) amplification and cells with co-amplification of probes from chromosome 11 (iv) or co-amplification of all investigated probes (v and vi).
In vivo analysis of gene amplification
For in vivo analysis we selected a chromosome region that was amplified in mouse and human neural progenitor cells during in vitro differentiation. We selected TRP53 for further confirmation as the most prominent gene. Embryonic stage E11.5 was selected because we had preliminary evidence that amplifications occur during stages of brain development. Using FISH analysis we were able to confirm amplification of TRP53 on a cryosection from a mouse embryo at stage E11.5 (Figure 8a–8c). TRP53 amplifications were detectable in the developing brain, particularly in cells of the metencephalic part of the rhombencephalon and in cells of the cephalic mesenchyme (Figure 8d, 8e).
DISCUSSION
Gene amplification during differentiation was recently detected in human neural progenitor cells [5]. Here we report a similar complex amplification pattern in mouse neural progenitor cells during differentiation. Although gene amplifications were mainly an attribute of tumor cells and drug resistant cells in humans, there were many reports of programmed gene amplification during development of amphibians and flies. Interestingly polyploidization in mammalian cells gains more and more attention in respect to its physiological significance and distribution [12]. The reasons for polyploidy were manifold and sometimes achieved by multiple rounds of replication without entering mitosis (endocycles). The occurrence of endocycles is widespread in nature and points to an ancient and practical innovation [13]. In somatic follicle cells of Drosophila polyploid cells cease their genome replication and start to endoreplicate four specific genomic regions resulting in gene amplifications [14]. In the present study TGF-β–differentiation induced SFME cells revealed 89 amplified chromosome regions by array CGH analysis. Interestingly, FCS differentiation induced SFME cells show only 13 amplified chromosome regions. These results strongly argue against a simple artifact of DNA preparation or replication timing. In both scenarios one would expect genomic imbalances throughout the genome and not on specific chromosomes. As shown in other studies, a wavy CGH pattern was even found after increased protein digestion, further indicating that the wavy pattern that indicate genomic imbalances was not due to preparation effects [7]. In addition, it is very unlikely that early and late replication effects were responsible for the wavy pattern of amplifications in our experiments since we investigated a very heterogeneous cell population. This effect may apply to rapidly proliferating cells but not to a non-synchronized cell population during differentiation. Although induction of differentiation might have a synchronization effect we did not find an amplification pattern indicative of synchronization. The simultaneous analysis of multiple amplified regions revealed a very heterogeneous pattern of amplifications and co-amplifications that argues against synchronization. In a synchronized cell population one would expect a similar frequency for cells to be involved in amplification of a given gene. We found that the amplification frequency varies between 5% for TRP53 and 33% for C1QL1 and GFAP. One would also expect that cells with amplifications would have the same regions amplified and/or co-amplified. However we have cells with and without co-amplifications. One would further expect an almost similar intensity of the fluorescence signals for the amplified gene. Here, we have a great variation of fluorescence signal intensities between cells with an amplified gene.
Table 2: Details for chromosome regions selected for FISH-confirmation of amplification
Position (mm9) |
Position (HG18) |
log2 ratio value |
BAC |
Representative gene name |
Percentage of cells with amplification |
9 |
20299999–21979999 |
0.2457 |
RP23-4A11 |
S1PR5 |
15% |
10 |
79019999–81099999 |
0.2733 |
RP23-421E11 |
FZR1 |
10% |
10 |
126179999–128499999 |
0.1574 |
RP23-432F11 |
CDK4 |
14% |
11 |
66659999–70979999 |
0.1598 |
RP23-150N14 |
TRP53 |
5% |
11 |
100899999–119819999 |
0.3497 |
RP23-235I12 |
C1QL1 and GFAP |
33% |
18 |
60779999–61659999 |
0.2137 |
RP23-143A24 |
PDGFRb |
10% |
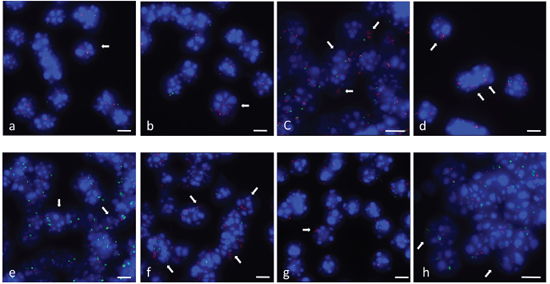
Figure 5: Gene amplifications in differentiation induced neurosphere cells. FISH was used to confirm gene amplifications of six gene loci on nuclei from differentiation induced mesencephalon E14 neurosphere cells. FZR1 (RP23–421E11) (pink) gene amplification was confirmed in 2-day differentiation induced cells and normal copy number of the control probe from the same chromosome ANO4 (RP23–279E23) (green) (a, b). FZR1 (pink) amplifications detected in 3-day differentiation induced cells frequently revealed an increase of fluorescence signals at only one of the corresponding loci (c). CDK4 (RP23-432F11) (pink) gene amplification in 3-day differentiation induced cells. Hybridization of the control gene (from the same chromosome) TRHDE (RP23–361I2) (green) revealed decreased fluorescence signal intensity (d). TRP53 (RP23–150N14) (green) gene amplification was confirmed in 3-day differentiation induced cells with normal copy number of control gene COX10 (RP23–40P10) (pink) from the same chromosome (e). C1QL1 (RP23–235I12) (pink) gene amplification was confirmed in 3-day differentiation induced cells with normal copy number of control gene COX10 (green) (f). S1PR5 (RP23–4A11) (pink) gene amplification in 3-day differentiation induced cells with normal copy number of control gene COX10 (green) (different chromosome) (g). PDGFRb (RP23–143A24) (green) gene amplification was confirmed in 3 day-differentiation induced cells with normal copy number of control gene COX10 (pink) (different chromosome) (h). Nuclei were counterstained with DAPI. Size calibration bar = 5 μm. Arrows point to examples of nuclei with amplifications.
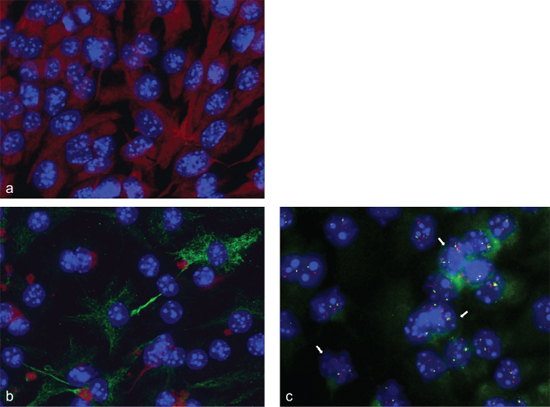
Figure 6: Immune fluorescence and FISH analysis. Neurosphere cells express nestin only (red fluorescence) (a); differentiation induced neurosphere cells express GFAP (green fluorescence) and showed a decreased nestin expression (red fluorescence) (b); Simultaneous fluorescence in situ hybridization and immune fluorescence analysis showed gene amplifications of FZR1 (RP23–421E11) (pink fluorescence) and GFAP/C1QL1 (RP23–235I12) (yellow fluorescence) in mesencephalon E14 neurosphere cells that were differentiation-induced for 2 d. These cells also expressed the GFAP protein (green fluorescence). GFAP expression was present in areas of the cells that developed appendages (c). Nuclei were counterstained with DAPI. Arrows point to examples of nuclei with amplifications.
As shown in Supplementary Figure S1 we found amplifications and an increased wavy pattern in SFME cells freshly seeded on fibronectin and cultivated for one day. Although still proliferating the SFME cells cultivated for 4 d revealed a more flattened pattern. This observation suggests an early influence mediated by fibronectin on differentiation-induced amplifications.
Interestingly, we found that 50% of the amplified mouse chromosome regions detected after TGF-β differentiation induction overlap with amplified chromosomal regions detectable in human neural progenitor cells upon differentiation induction for 2 and 5 days. The overlap of the amplified chromosome regions between both cell types points to a basic and conserved mechanism used by cells during differentiation. Out of the large number of genes that were affected by amplification, we would like to draw attention to the following groups of genes. Many amplified genes were involved in replication initiation or endocycling, including GINS genes, POLA2, PRIM1, CDH1, FZR, and genes from the CST (Cdc13-Stn1-Ten1) complex. Recently, a study reported that human CST complex is involved in replication restart [15]. Amplification of TP53 in human and mouse neural progenitor cells is consistent with previous observation that p53 expression might prevent neuronal terminal differentiation in neuroblasts [16].
There are several lines of evidence pointing to gene amplification as a mechanism to elevate the abundance of mRNAs and proteins in mammalian cells specifically during differentiation. Gurok et al 2004 investigated the mRNA expression pattern of mouse neurosphere cells differentiated by BDNF for 1, 2 and 4 days [17]. More than 50% of the up-regulated genes in the study by Gurok were localized in chromosome regions that we found amplified in our present study. The amplified regions included 3 genes that showed the highest up-regulation of expression including 6330403K07 RIKEN cDNA, complement component 3 and complement component 4 cDNAs. Regarding the protein level we showed that amplification of the chromosome region harboring the GFAP gene was associated with high GFAP expression detectable by immune fluorescence as early as 12 hours after FCS differentiation induction. Likewise, we found high GFAP expression in TGF-ß differentiation induced SFME cells. In mesencephalon neurosphere cells we confirmed GFAP amplification and GFAP expression using simultaneous FISH and immunefluorescence analysis after 2-day differentiation induction. Considering the biological effects of the amplifications, one has to bear in mind that array-CGH is likely to miss chromosome regions that are amplified during differentiation. Since the array-CGH data stem from a mixed population of cells, this method indentified only amplifications that are present in at least 5% of the cells. In addition, array-CGH analysis does not show if a given amplified domain has the same extend in the cells that harbor this amplification. Thus, the in vitro array-CGH analysis can only be a first step towards a complete picture of gene amplifications in differentiating cells. We used fluorescence-in-situ hybridization on differentiation induced SFME cells and mesencephanlon derived primary neuronal progenitor cells to validate our CGH-analysis. Six chromosome regions randomly selected from the amplified chromosome regions were confirmed in our in-situ hybridization experiments. The FISH experiments showed several cases with a sprinkled highly dense pattern of fluorescence signals, which is probably due to amplification on double minutes or even smaller units like episomes. As for the amplification mechanism, the process underlying the amplification in normal differentiating cells is likely different from the amplification mechanism assumed for mammalian tumor cells. The breakage-fusion-bridge-cycle model that may explain gene amplifications in tumor cells is not able to explain the tremendous increase in gene copy numbers observed in differentiating cells within 12–48 hours. The observed increase points to a mechanism independent from mitosis and appears more related to the endocycling and amplification mechanism described in Drosophila. A recent study reports amplified and under-replicated regions caused by repression of replication initiation and fork progression in Drosophila [18].
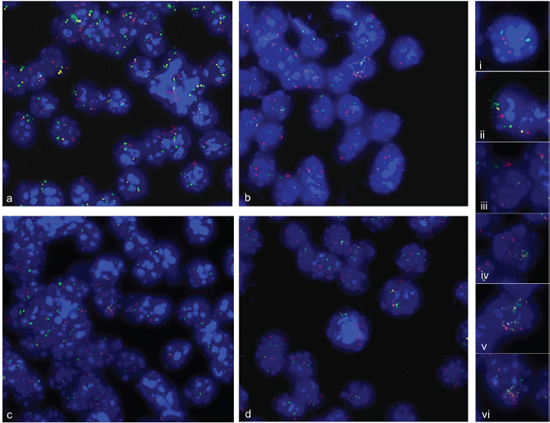
Figure 7: Multiple amplification analysis. Simultaneous fluorescence in situ hybridization of three genes from amplified chromosome regions on 2 d—differentiation induced neural progenitor cells. BAC probe C1QL1 (RP23–235I12) was labeled with Alexa-555 in yellow, BAC probe (RP23–150N14) TRP53 labeled with Alexa-488 in green, and BAC probe (RP23–421E11) FZR1 labeled with Alexa-594 in pink. An overview on hybridization results is shown in (a–d). Representative examples of fluorescence signal pattern were displayed as enlarged views of nuclei. We found normal copy number of C1QL1, TRP53 and FZR1 (i), TRP53 amplification (ii), FZR1 amplification (iii), C1QL1 and TRP53 co-amplification (iv), TRP53 and C1QL1 and FZR1 co-amplification (V, Vi). Nuclei were counterstained with DAPI.
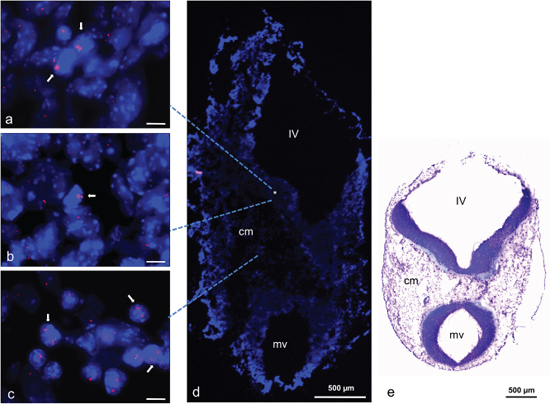
Figure 8: TRP53 amplification in mouse embryo stage E11.5. Amplification identified by FISH on transversal cryosection from mouse head region. High copy number of TRP53 is indicated by pink fluorescence signals (either enlarged signals and/or multiple signals) and normal copy number of COX10 by green signals. Both genes (BAC probes) were localized on mouse chromosome 11. Individual cells with amplifications were detected in multiple regions of the metencephalic part of the rhombencephalon (*) (a, b) and clustered cells with amplifications were detected in the cephalic mesenchyme (cm) (c). Nuclei were counterstained with DAPI. Size calibration bars in a–c = 5 μm. Arrows point to examples of nuclei with amplifications. Dashed lines point to areas where the pictures were taken from. For orientation, an enlarged DAPI stained overview of the transversal section is shown in (d) and a LFB/CV (Luxol Fast Blue und Cresyl Violet) histological stain from an analogical section is shown in (e). IV: fourth ventricle, mv: mesencephalic ventricle, cm: cephalic mesenchyme.
Our in vivo results on gene amplifications in a specific mouse embryonic stage provide further evidence that developmental gene amplification is not restricted to amphibians and flies. The tissue section contains a heterogeneous mixture of cells at different time points of the differentiation process including stem cells, progenitor cells and completely differentiated cells. We showed that gene amplification is present in specific cells of the developing mouse brain at stage E11.5 of embryonic development. We assume that amplification occurs in cells in the process of differentiation. Notably, fluorescence in situ hybridizations was done on tissue sections with non-repetitive hybridization probes. Under these conditions signal detection is complicated by the thickness of the section and the possibility that nuclei are cut in two. This may cause possible loss of one of the signals of the two gene copies. Higher numbers of gene copies as result of an amplification process are, however, likely to be detected by strong hybridization signals even under these experimental conditions. Independent of these technical issues, it remains to be seen to what extend gene amplifications may be found beyond the analyzed stage, genes and cell types. Published array-CGH data of previous studies on haploid embryonic stem cells, murine iPS or tissues containing differentiating cells show a striking similar wavy CGH pattern that we found in our experiments. As addressed above, the wavy pattern appears to result neither from short protein digestion nor simply from replication timing. Throughout our studies, samples from murine or human neural stem cells were processed the same way and amplifications were detectable at various time points during differentiation. We are aware that our threshold setting is below the setting used in other studies, but mixed populations of cells with only small number of cells harboring amplifications require this setting to allow for amplification detection. In conclusion we need a rethinking on the genomic stability of stem cells. It is very unlikely that amplification as a powerful mean to up-regulate genes was lost during evolution from amphibians and flies towards mammals and only reinvented by tumor cells. Stem cells during differentiation might represent the link to explain the appearance of gene amplifications during tumor development.
METHODS
Cell culture and differentiation
SFME cells (CRL-9392™) were obtained from ATCC as cryopreserved culture and were cultivated in DMEM:F12 Medium supplemented with bovine insulin (0.01 mg/ml), human transferrin (0.01 mg/ml), chemically defined lipids (1%), sodium selenite (10 nM) and mouse EGF (50 ng/ml). Cells were seeded on fibronectin coated cultureware and allowed to grow for 18 h prior to differentiation induction with TGF-β. SFME cells were differentiation induced using above supplemented ATCC DMEM:F12 Medium containing TGF-β (10 ng/ml) for 24 h or DMEM:F12 supplemented with FCS for 12 h.
For fibronectin control experiments cells were grown an additional 4 d and 1 d on fibronectin before DNA isolation. Cells cultured in the absence of fibronectin formed spheres and served as non-differentiated controls.
Mouse Ventral Mesencephalon E14 neurospheres were obtained as P1 cryopreserved neurospheres from STEMCELL Technologies. These neurospheres contain neural stem and progenitor cells. Cells were cultured in complete NeuroCultTM NSC proliferation medium supplemented with rhEGF (20 ng/ml). As undifferentiated controls those P1 neural stem and progenitor cells were cultivated on laminin coated glass slides for 2 d.
Differentiation was induced according to the manufacturer’s instructions using NeuroCultTM NSC differentiation medium without cytokines and with laminin or poly-D-lysine/laminin coated glass slides. Cells were differentiation induced for 2 d and 3 d. All experiments were done with neural stem and progenitor cells in P1 or P2. Cells differentiating for 3 d were cultured for 24 h in proliferation medium containing bFGF and EGF prior to differentiation induction. Culturing for 24 h in proliferation medium with bFGF and EGF is recommended by the manufacturers to stimulate progenitor cell proliferation.
Array preparation, hybridization and detection
Genomic DNA was isolated as described previously [5] and mouse genomic DNA from Clontech was used as control DNA. NimbleGen 3x720K mouse whole genome array hybridization was performed with the certified full service of NimbleGen provided by ImaGenes Berlin, Germany. Array data were deposited in GEO (GSE35523).
Fluorescence in situ hybridization
BAC clones were taken from the RP-11 (http://www.chori.org/bacpac/) libraries of the Welcome Trust Sanger Institute and available from ImaGenes GmbH, Germany.
BAC probes for C1QL1 and S1PR5 were directly labeled using the High Prime Labeling System (Roche Molecular Biochemicals, Germany). BAC-DNA (1 μg) was labeled with Cyanine-3-dCTP (Cy3) (pink fluorescence signals) according to the manufacturer’s instructions. Additional BAC probes were either labeled with Alexa-488-dCTP (green fluorescence signals) or with Alexa-594-dCTP (pink fluorescence signals) using the FISHTag DNA labeling Kit according to the manufacturer’s instructions. For multiple amplification analysis three BAC probes were labeled with Alexa-555-dCTP, with Alexa-488-dCTP and with Alexa-594-dCTP.
Differentially labeled probe DNAs (60 ng) were precipitated in the presence of mouse Cot-1 DNA. Samples were resuspended in hybridization mix (50% formamide, 2xSSPE, 10% dextrane sulphate and 4% SDS).
Fluorescence in situ hybridization using SFME cells
SFME interphase nuclei were fixed using Carnoy’s fixative (methanol ice acetic acid). Nuclei were dropped on clean glass slides. Slides were RNase treated (100 μg/ml RNaseA in 2 × SSC) for 30 minutes at 37°C and pepsin treated (0.005% in 0.01 M HCl at 37°C) for 10 minutes. Postfixation was performed using 1% formaldehyde/1x PBS for 10 minutes at room temperature.
Labeled BAC probes were applied to the slides and denatured for 2 min at 80°C. Hybridization was carried out in a humid chamber at 37°C for 16 h. Post hybridization washes were performed in 50% formamide/2 × SSPE (4 × 5 minutes; 45°C) followed by 0.1 × SSPE (3 × 5 minutes) at 60°C. Nuclei were counterstained with DAPI (4’,6’-Diamidino-2-phenylindole) (1 μg/ml in PBS) for 4 minutes and mounted with VectaShield mounting medium (Vector Laboratories, Orton Southgate, England) for microscopic analysis.
Fluorescence in situ hybridization on neural stem and progenitor cells
Differentiating neural stem and progenitor cells (primary mesencephalon E14 neurosphere cells) on coated glass slides were fixed in ice-cold methanol for 20 minutes. Slides were washed in PBS for 5 minutes and treated with 0.02% Tween-20 for 5 minutes. Slides were treated with RNaseA for 45 minutes, digested with pepsin and fixed with formaldehyde/1xPBS for 10 minutes. Hybridization and post hybridization washes were as described above.
Fluorescence in situ hybridization on mouse embryo cryosections
Transverse cryosections (10 μm) from mouse brain at embryonic stage E11.5 were treated with Carnoy’s fixative for 15 min at 4°C, pepsin digested and fixed 4% paraformaldehyde in diethylpyrocarbonate-treated PBS for 10 minutes.
Probes were labeled as described above, applied to the section and denatured for 5 min at 80°C. Hybridization was for 2–3 days at 37°C. Posthybridization washes and DAPI staining were as described above.
Fluorescence in situ hybridization with simultaneous immune fluorescence
Differentiating neural stem and progenitor cells (primary mesencephalon E14 neurosphere cells) on coated glass slides were fixed in ice-cold methanol for 20 minutes. Slides were washed in PBS for 5 minutes and treated with 0.2% Tween-20 for 5 minutes. Postfixation was done by 1% formaldehyde/1x PBS for 10 minutes at room temperature. Slides were blocked with goat serum and incubated for 1 h with chicken antibody polyclonal to GFAP (ab4674, Abcam) and detected using an Alexa-488 coupled secondary antibody. Finally, slides were dehydrated by an ascending ethanol series (70%/80%/96%) and air-dried. Hybridization and post hybridization washes were as described above.
Immune fluorescence
SFME cells were cultivated on fibronectin-coated glass coverslips. Subsequently, cells were differentiation induced as described above. Both induced cells and untreated controls were methanol fixed and treated with 0.2% Tween-20 for 2 minutes. Coverslips were blocked with goat serum and incubated for 1 h with chicken antibody polyclonal to GFAP (ab4674, Abcam) and rabbit antibody polyclonal to nestin (ab27952, Abcam). Detection was done with an Alexa-488 coupled secondary antibody against chicken and Alex-594 coupled secondary antibody against rabbit.
Differentiating neural stem and progenitor cells (primary mesencephalon E14 neurosphere cells) grown on coated glass slides were fixed in ice-cold methanol for 20 minutes. Slides were washed in PBS for 5 minutes and treated with 0.2% Tween-20 for 2 minutes. Subsequently, slides were blocked with goat serum and incubated for 1 h with chicken antibody polyclonal to GFAP (ab4674, Abcam) and rabbit antibody polyclonal to nestin. Detection was done with an Alexa-488 coupled secondary antibody against chicken and Alex-594 coupled secondary antibody against rabbit.
Microscope imaging and analysis
Fluorescence images were captured with an Olympus AX70 microscope using ISIS software from Metasystems. For an overview of DAPI stained cryosections, images were taken at an Olympus BX61 microscope. For histological staining, cryosections of 10 μm thickness were fixed in acetone at –20°C for 10 min and stained in luxol fast blue solution at 56°C overnight, followed by cresyl violet staining. Histological images were taken at an Olympus BX51 TF microscope.
ACKNOWLEDGMENTS
The “Deutsche Forschungsgemeinschaft” funded this study (Fi644/2–1; Fi644/2–2).
REFERENCES
1. Bostock CJ. Mechanisms of DNA sequence amplification and their evolutionary consequences. Philos Trans R Soc Lond B Biol Sci. 1986; 312:261–273.
2. Gall JG, Pardue ML. Formation and detection of RNA-DNA hybrid molecules in cytological preparations. Proc Natl Acad Sci U S A. 1969; 63:378–383.
3. Spradling AC, Mahowald AP. Amplification of genes for chorion proteins during oogenesis in Drosophila melanogaster. Proc Natl Acad Sci U S A. 1980; 77:1096–1100.
4. Tower J. Developmental gene amplification and origin regulation. Ann Rev Genetics. 2004; 38:273–304.
5. Fischer U, Keller A, Voss M, Backes C, Welter C, Meese E. Genome-wide gene amplification during differentiation of neural progenitor cells in vitro. PloS One. 2012; 7:e37422.
6. Leeb M, Wutz A. Derivation of haploid embryonic stem cells from mouse embryos. Nature. 2011; 479:131–134.
7. van Heesch S, Mokry M, Boskova V, Junker W, Mehon R, Toonen P, de Bruijn E, Shull JD, Aitman TJ, Cuppen E, Guryev V. Systematic biases in DNA copy number originate from isolation procedures. Gen Biol. 2013; 14:R33.
8. Rawson C, Loo D, Helmrich A, Ernst T, Natsuno T, Merrill G, Barnes D. Serum inhibition of proliferation of serum-free mouse embryo cells. Exp Cell Res. 1991; 192:271–277.
9. Nakajima M, Ishimuro T, Kato K, Ko IK, Hirata I, Arima Y, Iwata H. Combinatorial protein display for the cell-based screening of biomaterials that direct neural stem cell differentiation. Biomaterials. 2007; 28:1048–1060.
10. Loo D, Rawson C, Helmrich A, Barnes D. Serum-free mouse embryo cells: growth responses in vitro. J Cell Physiol. 1989; 139:484–491.
11. Agam A, Yalcin B, Bhomra A, Cubin M, Webber C, Holmes C, Flint J, Mott R. Elusive copy number variation in the mouse genome. PloS One. 2010; 5:e12839.
12. Pandit SK, Westendorp B, de Bruin A. Physiological significance of polyploidization in mammalian cells. Trends Cell Biol. 2013; 23:556–566.
13. Edgar BA, Zielke N, Gutierrez C. Endocycles: a recurrent evolutionary innovation for post-mitotic cell growth. Nat Rev Mol Cell Biol. 2014; 15:197–210.
14. Calvi BR, Lilly MA, Spradling AC. Cell cycle control of chorion gene amplification. Genes Dev. 1998; 12:734–744.
15. Stewart JA, Wang F, Chaiken MF, Kasbek C, Chastain PD 2nd, Wright WE, Price CM. Human CST promotes telomere duplex replication and general replication restart after fork stalling. The EMBO journal. 2012; 31:3537–3549.
16. Ferreira A, Kosik KS. Accelerated neuronal differentiation induced by p53 suppression. J Cell Sci. 1996; 109:1509–1516.
17. Gurok U, Steinhoff C, Lipkowitz B, Ropers HH, Scharff C, Nuber UA. Gene expression changes in the course of neural progenitor cell differentiation. J Neurosci. 2004; 24:5982–6002.
18. Sher N, Bell GW, Li S, Nordman J, Eng T, Eaton ML, Macalpine DM, Orr-Weaver TL. Developmental control of gene copy number by repression of replication initiation and fork progression. Gen Res. 2012; 22:64–75.