
Introduction
Acute myeloid leukaemia (AML) is a malignancy of haemopoietic progenitor cells [1] and accounts for approximately 1% of all cancer deaths. At present the standard induction treatment for younger fitter patients consists of the antimetabolite cytarabine plus the anthracycline antibiotic daunorubicin [2, 3]. Depending on clinicopathological characteristics, patients who go into remission would commonly receive consolidation therapy with either high dose cytarabine or allogeneic stem cell transplant . Despite these intensive treatment strategies, significant numbers of patients relapse and only approximately 50% of younger fitter patients can be cured. The treatment outcomes are dependent on a variety of clinical and biological factors including cytogenetics, age and drug-resistance [2, 4-7].
A number of mechanisms have been suggested to contribute to drug-resistance in AML. These include, the targeted cells fail to undergo apoptosis in response to the chemotherapy agent, drugs failing to reach their intracellular targets or the removal by the ABC membrane transporter protein, P-glycoprotein (Pgp). Pgp is a efflux transporter, present within the cell’s plasma membrane, and its expression in AML has been reported to be relatively low, however an increase in its expression after drug treatment and also at the point of relapse, have been reported [8]. Moreover, Galmarini et al have shown that high levels of 5’-nucleotidase, which is involved in DNA repair and membrane transport, is related to the poor prognosis of AML patients [9]. These studies suggest that AML cells evolve to regulate pathways that provide protection against toxic chemotherapeutic agents.
Recently, we reported that haem oxygenase-1 (HO-1) has an important function in protecting human AML cells from TNF-induced apoptosis [10]. To date, three isoforms of haem oxygenase have been identified, HO-1, HO-2 and HO-3 [11]. HO-1, which is the most intriguing in terms of providing protection against cellular stresses, regulates cellular haem levels, and converts intracellular haem into carbon monoxide, free iron and biliverdin [11]. Biliverdin is further reduced into the potent antioxidant bilirubin [12, 13] by biliverdin reductase [14]. This metabolite possesses cytoprotective properties including anti-inflammatory, anti-oxidative and anti-apoptosis [15, 16]. HO-2 is constitutively expressed and HO-3 is not catalytically active and thought to be involved in oxygen-sensing. HO-1 belongs to the heat shock protein family (Hsp-32), thus its expression is triggered by a variety of stress-inducing stimuli including, UV irradiation, hyperthermia, inflammatory cytokines, bacterial endotoxins and heavy metals [17-21]. The regulation of HO-1 is under the control of signalling components [22, 23] and many transcription factors including nuclear factor-κB (NF-κB), NF-E2-related factor 2 (Nrf2) and activator protein-1 (AP-1)[24, 25].
Furthermore we have recently we showed that HO-1 is in fact down-regulated in AML by their constitutively active NF-κB activity present, and that inhibiting NF-κB brings HO-1 levels back to more normal levels, providing further secondary protection for AML cells against NF-κB inhibition. Here were undertaken to investigate the role of HO-1 in regulating cytoprotective responses to two common front-line chemotherapy agents, cytarabine and daunorubicin, which are currently widely used in treating patients presenting with AML.
Results
AML resistance to cytarabine and daunorubicin.
To understand the mechanisms of chemoresistance in AML cells to cytarabine and daunorubicin, we have examined the levels of apoptosis of primary AML samples and AML cell lines in response to varying concentrations of these drugs. The drug concentrations selected were based on previous studies [26, 27]. One µM of cytarabine is a clinically achievable concentration obtained in situations where a standard dose of cytarabine is administered [28]. Table 1 shows the relevant clinical data for the AML patient samples tested in these studies. Figure 1A shows apoptosis for both AML samples and AML cell lines in response to both cytarabine (0.5 µM and 1 µM) and daunorubicin (0.2 µM and 0.5 µM) in concentration-dependent manners. Figure 1B shows that cytarabine (0.5 µM), daunorubicin (0.2 µM) or a combination of both cytarabine and daunorubicin induce cell death by apoptosis as measured by annexin-V and PI staining. What is clear from these findings is that there is a range of sensitivities to the chemotherapeutics and that there are significant levels of living cells that remain resistant to these cytotoxic agents, even in combination.
| Number | Age | Gender | WHO diagnosis | Cytogenetics | % Blasts | Previous treatment |
| AML8 | 40 | male | Acute promyelocytic leukaemia with t(15;17)(q22:q12) PML-RARA | t(15;17) | 95* | 1999 DAT,DAT MACE,MiDAC [29] |
| AML9 | 49 | male | AML with maturation | normal | 80 | nil |
| AML10 | 84 | male | Acute monoblastic and monocytic leukaemia | not available | nil | |
| AML11 | 46 | female | AML with maturation | +4,+8, t(9;22) | 70 | nil |
| AML12 | 78 | male | AML with myelodysplasia related changes | not available | 85 | nil |
| AML13 | 27 | male | AML with t(8;21)(q22;q22) RUNX1-RUNX1T1 | t(8;21) | nil | |
| AML14 | 28 | female | Acute myelomonocytic leukaemia | normal | nil | |
| AML16 | 66 | female | therapy related myeloid neoplasm | complex | 85 | 1999 DAT,DAT MACE,MiDAC [29] |
| AML18 | 77 | male | AML with myelodysplasia related changes | complex | 95 | nil |
| AML19 | 31 | female | AML with minimal differentiation | normal | nil | |
| AML20 | 40 | male | Acute monoblastic and monocytic leukaemia | constitutional XYY only | 90% | nil |
Table 1: Characteristics of study patient samples. AML disease characteristics including WHO diagnosis and cytogenetics. Percent blast denotes % of AML blasts after purification using density gradient (* denotes % of blasts and promyelocytes). Previous treatments are as outlined [29]. Abbreviations used: AML, Acute myeloid leukaemia; WHO, world health organisation.
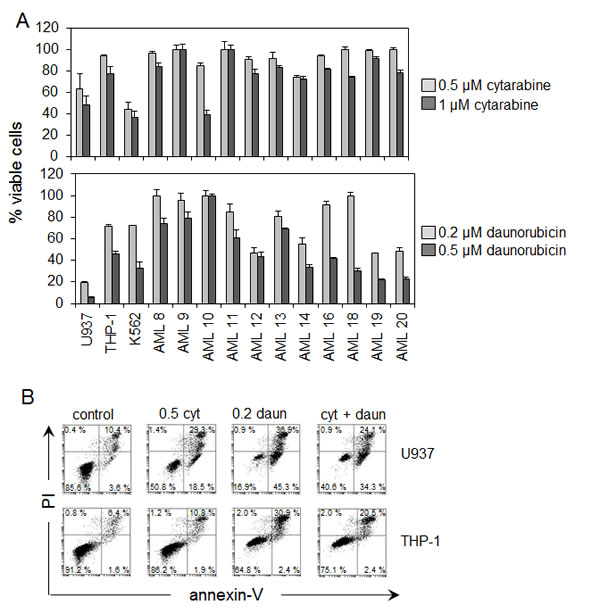
Figure 1: AML cell resistance to cytarabine and daunorubicin. (A) AML cells were treated with two doses of cytarabine (0.5 µM and 1 µM) and two doses of daunorubicin (0.2 µM and 0.5 µM) for 24 hours. Viable cell numbers were then devised using PI and annexin V staining followed by flow cytometry. Values indicate mean ± SD from three separate independent experiments. (B) U937 and THP-1 cells were treated with either cytarabine 0.5 µM, daunorubicin 0.2 µM or a combination of cytarabine 0.5 µM and daunorubicin 0.2 µM for 24 hours. Samples were stained with annexin V and PI. Flow cytometry analysis was used to determine the percentage of viable cells present within the sample population.
Cytarabine and daunorubicin induce HO-1 expression in AML cells.
Since we have previously shown that HO-1 protects AML cells from apoptotic stimuli, we wanted to determine if either cytarabine or daunorubicin could induce the expression of this cytoprotective gene in AML cells. Figure 2A shows that both cytarabine and daunorubicin induce HO-1 mRNA expression by 24 h of drug exposure in both AML cell lines and AML patient samples. Figure 2B also reiterates the above point in AML cell lines and shows the effect on HO-1 mRNA expression of 24 h drug exposure to cytarabine and daunorubicin alone or in combination. Interestingly, HO-1 mRNA expression is increased further when the cells are exposed to both chemotherapeutic agents together. Figure 2C shows a Western blot indicating that HO-1 protein was also induced by both cytarabine and daunorubicin after 24 h drug exposure. Thus these front-line chemotherapeutic agents are able to readily induce HO-1 in AML cells. Furthermore, at the concentrations used, daunorubicin is somewhat better at killing off U937 cells than is cytarabine (Figure 1), with daunorubicin only weakly inducing HO-1 in U937 cells compared to cytarabine’s induction (Figure 2C). This correlates perfectly with a role for HO-1 induction in drug-resistance in U937 cells. Likewise, it is notable that both agents can equally induce strong HO-1 expression in THP-1 cells, with a similar inability towards efficient cytotoxicity in those cells.
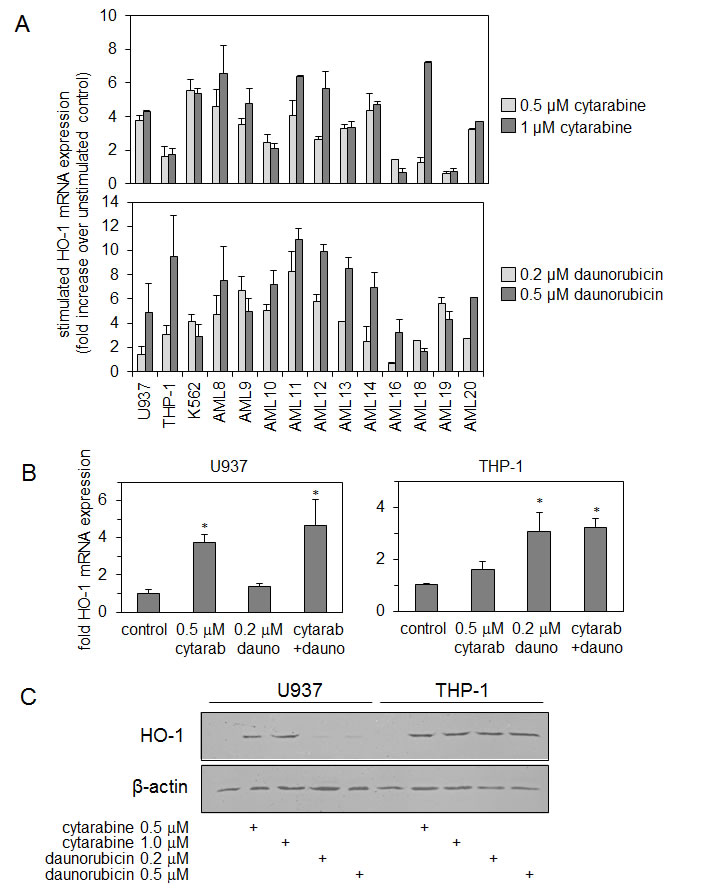
Figure 2: Cytarabine and daunorubicin induce HO-1 expression in AML cells. (A) AML cells were treated with cytarabine (0.5 µM or 1 µM) and daunorubicin (0.2 µM or 0.5 µM) for 24 hours. RNA was extracted from each sample and HO-1 mRNA was measured using real-time PCR. (B) U937 and THP-1 cells were treated with either cytarabine 0.5 µM, daunorubicin 0.2 µM or a combination of both cytarabine 0.5 µM and daunorubicin 0.2 µM for 24 hours. RNA was extracted from each sample and HO-1 expression was measured using real-time PCR. (C) U937 and THP-1 cells were treated with either cytarabine (0.5 µM or 1 µM) or daunorubicin (0.2 µM or 0.5 µM) for 24 hours. Whole cell protein extracts were collected and separated via SDS-PAGE and examined using Western immunoblotting analysis. HO-1 expression levels were examined. Membranes were reprobed with β-actin to ensure equal loading across the samples.
Silencing HO-1 increases AML cell apoptosis to cytarabine and daunorubicin.
We developed a lentivirus-based miRNA delivery system on a GFP backbone to target HO-1 mRNA knockdown and silence HO-1 expression in AML cells. Figure 3A shows that we can achieve > 95% transfection efficiency as measured by flow cytometry. Figure 3B shows that HO-1 mRNA expression in both THP-1 and U937 control and HO-1-silenced cells, with greater than 95% knock-down of endogenous HO-1 mRNA levels observed. Figure 3B also indicates HO-1 protein expression in THP-1 control and HO-1-silenced cells, with almost no detectable levels of HO-1 observed specifically with the HO-1 miRNA lentivirus. Next we wanted to determine if either cytarabine or daunorubicin induced HO-1 expression could protect AML cells from apoptosis. To do this we used lentiviral-based miRNA HO-1 knockdown cells (compared to control negative miRNA lentiviral knockdown cells) treated with either cytarabine or daunorubicin for 24 h. Figure 3Cand 3D show the apoptotic responses of control- and HO-1-silenced THP-1 and U937 cells after treatment with either cytarabine or daunorubicin. The HO-1-silenced THP-1 and U937 cells were more susceptible to apoptosis compared to the non-silenced cells. These findings indicate that HO-1 is playing a part in cellular protection mechanisms observed in apoptosis-resistance towards cytarabine or daunorubicin.
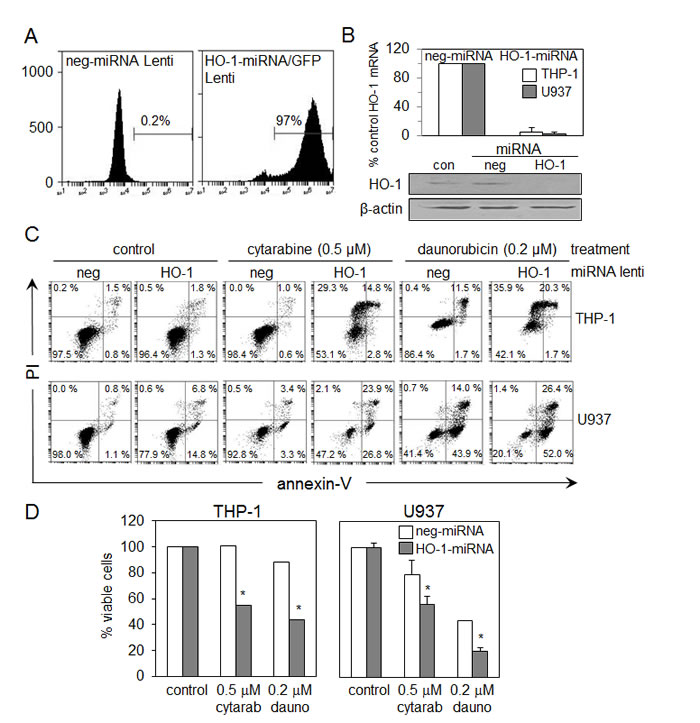
Figure 3: Silencing HO-1 increases AML cell apoptosis to cytarabine and daunorubicin (A) Flow cytometry was used to measure the transfection efficiency of the lentivirus based miRNA HO-1/GFP construct. (B) U937 and THP-1 cells were infected with a lentiviral based miRNA HO-1/GFP knockdown construct, whilst the control was infected with lentiviral based miRNA negative construct. RNA was extracted from each sample and HO-1 mRNA expression was measured using real-time PCR. (C) Whole cell protein extracts were collected and separated via SDS-PAGE and examined using Western immunoblotting analysis to investigate HO-1 expression. (D) Both control U937 and THP-1 cells and HO-1 silenced U937 and THP-1 cells were treated with cytarabine 0.5 µM and daunorubicin 0.2 µM for 24 hours. Cells were stained with annexin-V and PI, the percentage of viable cells was measured via flow cytometry.
ROS modulates the apoptotic potential of cytarabine and daunorubicin in AML cells.
We have previously shown in AML cells that H2DCFDA oxidation occurs in response to the proteasome inhibitor bortezomib and also in response to NF-ĸB inhibitor BAY 11-7082 [29, 30]. We have also shown that ROS induced by these drugs are responsible for upregulating HO-1 expression. Here we wanted to determine if cytarabine and daunorubicin increased HO-1 expression through a ROS-dependent mechanism. Figure 4A shows H2DCFDA oxidation in U937 and THP-1 cells in response to cytarabine and daunorubicin treatment over a 6 h time period. N-acetylcysteine (NAC) is an antioxidant scavenger which quenches ROS activity present within the cellular environment. NAC was used to determine if removing ROS would block cytarabine- and daunorubicin-induced HO-1 expression. Finally, we wanted to determine whether NAC could block apoptosis induced by cytarabine and daunorubicin. Figure 4B show that NAC effectively inhibits apoptosis in cytarabine- and daunorubicin-treated THP-1 and U937 cells, thus demonstrating that ROS are responsible for cytarabine- or daunorubicin-induced cell death of AML cells. Figure 4C shows a decrease in mRNA HO-1 expression in the presence of NAC compared to that of the control, further implicating ROS in the chemotherapeutic-induction of HO-1 and its consequent cytoprotection observed in AML.
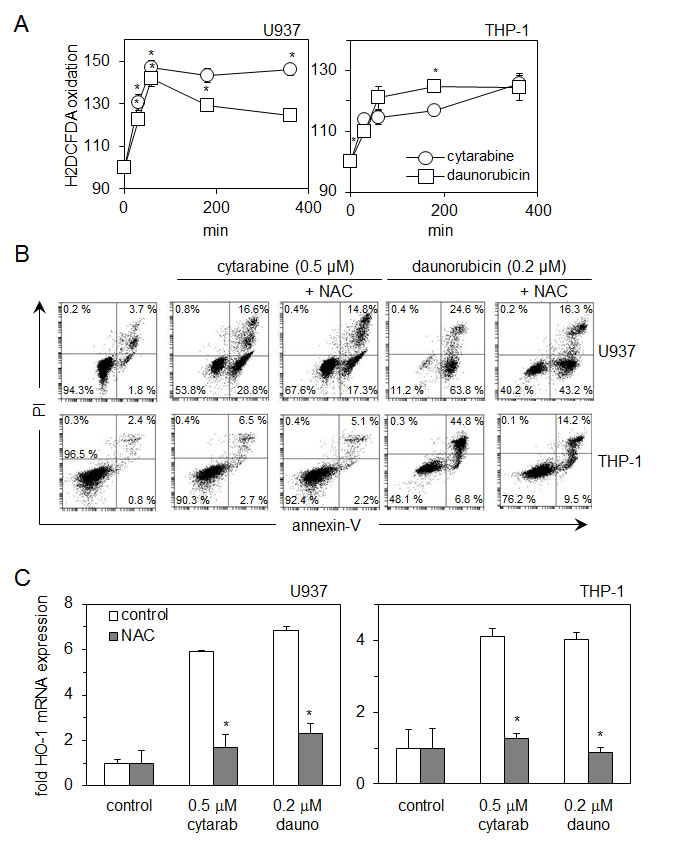
Figure 4: Reactive oxygen species (ROS) modulate the apoptotic potential of cytarabine and daunorubicin in AML cells. (A) U937 and THP-1 cells were treated with cytarabine 0.5 µM and daunorubicin 0.2 µM for up to 6 hours and then washed with PBS and incubated for 15 minutes with 10 µM of H2DCFDA. ROS production in response to drug exposure was examined using flow cytometry. (B) U937 and THP-1 cells were treated with cytarabine 0.5 µM and daunorubicin 0.2 µM for 24 hours with or without pre treatment with NAC (10 µM for daunorubicin treated cells and 5 µM for cytarabine treated cells) for 30 minutes. Cells were stained with annexin-V and PI, their response to NAC was examined using flow cytometry. (C) U937 and THP-1 cells were treated with either cytarabine 0.5 µM and NAC 5 µM or daunorubicin 0.2 µM and NAC 10µM for 24 hours. RNA was extracted and mRNA HO-1 expression was measured using real- time PCR.
Discussion
In this study we have investigated the role of HO-1 in protecting AML cells from apoptosis in response to cytarabine and daunorubicin. We have previously shown that HO-1 can protect AML cells from apoptosis in response to the proteosome inhibitor, bortezomib and the NF-κB inhibitor, BAY11-7082 [29, 30]. In the present work, we show that expression of HO-1 is induced in response to cytarabine, daunorubicin and a combination of both agents. Interestingly HO-1 mRNA expression is increased further when the cells are treated with a combination of the drugs which one would expect because of the presence of both agents acting via differing modes of action to induce apoptosis. Referring to Figure 1B, oddly the percentage of viable cells present within the sample population is actually between that of separate cytarabine and daunorubicin exposure. Furthermore silencing HO-1 expression in combination with either cytarabine or daunorubicin induces a greater apoptotic response in AML cells. Finally we show that both cytarabine and daunorubicin cause AML cells to generate ROS production and this in turn induces HO-1 expression, which subsequently reinforces any cellular resistance towards these drugs. A prior report has already shown ROS production in the presence of cytarabine in U937 cells [26]. These results provide novel insight into the role of HO-1 in protecting human AML cells from cytarabine and daunorubicin.
In a broad range of morphologic and cytogenetic subtypes of primary human AML cells and human AML cell lines, both cytarabine and daunorubicin consistently induce HO-1 expression by 24 h. We also examined the effects of daunorubicin and cytarabine on HO-1 expression at earlier time points finding no increase in HO-1 mRNA levels (not shown). This time lag between addition of drug and HO-1 induction in AML cells is interesting. We hypothesise that this is likely to be due to the complex nature of the HO-1 promoter and its regulation. Nuhn et al have reported that HO-1 expression is usually increased in human cancer cells and is also increased further by the addition of cytotoxic agents [31]. Subsequent silencing of HO-1 in these AML cell lines significantly decreases the percentage of viable AML cells present within the population treated with fixed doses of either cytarabine or daunorubicin. Therefore we suggest inhibiting HO-1 could potentially enhance AML cell response to these chemotherapeutic agents in vivo. Fang et al have determined the presence of a HO-1 inhibitor in vivo can enhance tumour responsiveness to chemotherapeutic agents [32].
Protection from apoptosis conferred to AML cells by HO-1 in the current study appears to be medicated through regulation of ROS produced in response to cytarabine and daunorubicin exposure. With regards to HO-1 and its ability to protect cells from undergoing apoptosis, there are a number of different proposed mechanisms. The first is by decreasing the levels of the pro-oxidant, haem [32, 33]; the second, by increasing the concentration of bilirubin, an antioxidant [32]; and the final mechanism is by increasing the levels of carbon monoxide, curiously generally thought to be an anti-apoptotic molecule [13, 34, 35]. Together, these mechanisms act either alone or in a combined way to protect cancer cells from apoptosis. What is intriguing about AML cells is that they seem to have evolved to manipulate this pathway to provide it with a growth advantage over normal non-cancerous control cells. AML (or any cancerous cell) probably doesn’t set out to be drug-resistant, but there is a varying population of cancer stem cells that are relatively able to cope with any cytotoxic insult they receive from cytarabine or daunorubicin. We show here that these resistance mechanisms heavily involve HO-1. A patient’s HO-1 response to chemotherapeutics will go a long way in determining their clinical outcome.
In our previous study we showed that basal levels of HO-1 are low compared to normal cells and that NF-κB activation reduced basal HO-1 expression. Furthermore, Nrf-2 activation could override this regulation [29]. In fact any number of transcription factors can regulate HO-1 expression [24, 25]. In this study we concentrated on the role of HO-1 expression in regulating chemotherapy-induced apoptosis but cannot preclude the involvement of other transcriptional elements.
In summary we report that silencing HO-1 significantly increases in vitro sensitivity of AML cells to cytarabine and daunorubicin, two front-line chemotherapy agents currently widely used to treat this type of leukaemia. Previously we have shown that HO-1 plays an important role in protecting AML cells from apoptotic stimuli and here we propose that this is likely to be clinically relevant and thus HO-1 warrants further investigation as a target in future therapeutic strategies.
Acknowledgements
SH and LZ performed the research; KMB, SAR and DJM designed the research study; SH and SAR analysed the data and all authors wrote the paper. This work was supported by a UEA Dean’s Studentship to S-A H.
Competing interests
The authors have no competing interests.
Materials and Methods
Materials
The AML-derived cell lines THP-1 and U937, were obtained from the European Collection of Cell Cultures, where cell identity is confirmed by cytogenetic analuysis. Cells were used in the laboratory for a maximum of 6 months post-revival from liquid nitrogen storage, to ensure integrity and retention of characteristics. Anti-human HO-1 antibody was purchased from R & D Systems (Abingdon, UK). All other antibodies were obtained from Abcam (Cambridge, UK). All primers were purchased from Invitrogen (Paisley, UK). Micro RNA (miRNA) vector was cloned on site. Annexin-V and propidium iodide (PI) were purchased from Abcam (Cambridge, UK). Cytarabine (dissolved in water, stored at -20 ºC as a 4.1 mM stock) and daunorubicin (dissolved in water, stored at -20 ºC as a 0.95 mM stock) were both purchased from Sigma-Aldrich (St Louis, MO). All other reagents were purchased from Sigma- Aldrich unless stated.
Cell Culture
Primary AML cells were obtained under local ethical approval (LREC ref 07/H0310/146). For primary cell isolation, heparinised blood was collected from volunteers and isolated by Histopaque (Sigma-Aldrich, St Louis, MO) density gradient centrifugation. U937 were cultured in a humidified atmosphere at 37ºC with 5% CO2 in RPMI-1640 from Invitrogen (Paisley, UK) supplemented with 10% foetal bovine serum (FBS)(Biosera, UK) and 1% L-glutamine incubated. 293FT cells were obtained from Invitrogen and were grown in DMEM high glucose (Invitrogen) supplemented with 10% FBS, 6 mM L-glutamine, 0.1 mM non-essential amino acids and 1 mM sodium pyruvate.
RNA extraction and real-time PCR
Total RNA was extracted from 1 x 106 cells using RNA lysis solution and a nucleic acid Prepstation both purchased from Applied Biosystems (Warrington, UK) following the manufactures instructions. Applied Biosystems RNA PCR core kit was used for reverse transcription. SYBR green technology (Roche) was used on cDNA produced via the reverse transcription of purified RNA. After pre-amplification (95 ºC for 2 min) the reactions were amplified for 45 cycles (95ºC for 15 s, then 60ºC for 10 s, followed by 72ºC for 10 s) on a Lightcycler 480 (Roche) as described [29]. HO-1 mRNA expression was standardised against GAPDH expression using the standard curve analysis method. Primer sequences were: GAPDH forward 5’-ACCAGCCTCAAGATCATCAGC-3’; GAPDH reverse 5’-TGCTAAGCAGTTGGTGGTGC-3’; HO-1 forward 5’-ATGGCCTC CCTGTACCACATC-3’ and HO-1 reverse 5’-TGTTGCGCTCAATCTCCTCCT-3’.
Western immunoblotting
Total protein was extracted from 1 x 106 cells using radioimmunoprecipitation assay (RIPA) buffer as described [29]. Protein was transferred onto nitrocellulose membrane, probed according to the antibody manufacturer’s guidelines and then examined using enhanced chemiluminescence (ECL) detection in a Li-Cor Odyssey IR imaging system.
Flow Cytometry
Flow cytometry for measuring apoptosis was performed on an Accuri C6 flow cytometer (Accurri). Samples were collected, centrifuged at 500 x g, 5 min then resupended and stained with Annexin-V and PI dye, followed by detection. N-acetylcysteine (NAC) antioxidant ROS-scavanger was used to assess the presence of ROS, whereby cells were treated with NAC with relevant chemotherapeutic agent for 24 h, prior to sample flow cytometry analysis.
Dichlorofluorescein Assay
A dichlorofluorescein (DCF) assay was used to determine cellular ROS generation [21]. Cells were washed with PBS and then incubated with 10 µM of 2’,7-dichlorodihydrofluorescein diacetate (H2DCFDA) for 15 min at 37 ºC and 5% CO2. Flow cytometry was used to measure the fluorescence intensity of H2DCFDA. The mean channel fluorescence of a treated sample is divided by that of the control and then multiplied by 100, thus obtaining the relative percentage change. The relative fluorescence intensity of H2DCFDA detected represents the steady state of ROS generation [21, 36, 37].
Virus construction and infection
MicroRNA (miRNA) sequence miRNA-HO-1-166 (5’-TCCTCATGAACTCAGCATTCT-3’) targeting human HO-1 was selected with Invitrogen Block-iT RNAi designer software (www.invitrogen.com/rnai). pcDNATM6.2-GW/EmGFP-miR-neg plasmid from Invitrogen was used as a source for negative control miRNA expression. miRNA-encoding viruses were produced with Block-iT Lentiviral Pol II miRNAi expression system (Invitrogen) in 293FT cells. Lentiviral stocks were concentrated using Lenti-XTM concentrator and titers were determined with Lenti-XTM qRT-PCR titration kit (both purchased from Clontech, Saint-Germain-en-Laye, France). For infection, cells were plated onto 12 well plates at 2.5 x 105 cells/well and infected with lentiviral stocks at an MOI of 10 in the presence of polybrene, then analysed by flow cytometry, RT-PCR and Western blotting 48 h post infection. Stably transduced cells were selected using Blasticidin selection marker (8 μg/ml) for 2 weeks before use.
Statistical analyses
Student’s T-test was used to assess the statistical significance compared to that of the control values. Results with a P < 0.05 were considered statistically significant and are indicated (*). Values represent the means ± SEMs from at least three independent experiments, or of representative blots/plots that are typical of at least three independent findings.
References
1. Estey E, Döhner H. Acute myeloid leukaemia. The Lancet. 2006; 368: 1894-1907.
2. Tallman MS, Gilliland DG, Rowe JM. Drug therapy for acute myeloid leukemia. Blood. 2005; 106: 1154-1163.
3. British National Formulary. www.bnf.org. 2011.
4. Grimwade D, Walker H, Oliver F, Wheatley K, Harrison C, Harrison G, Rees J, Hann I, Stevens R, Burnett A, Goldstone A. The importance of diagnostic cytogenetics on outcome in AML: analysis of 1,612 patients entered into the MRC AML 10 trial. Blood. 1998; 92: 2322-2333.
5. Dohner H, Estey EH, Amadori S, Appelbaum FR, Buchner T, Burnett AK, Dombret H, Fenaux P, Grimwade D, Larson RA, Lo-Coco F, Naoe T, Niederwieser D, Ossenkoppele GJ, Sanz MA, Sierra J et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010; 115: 453-474.
6. de Jonge HJM, Woolthuis CM, de Bont ESJM, Huls G. Paradoxical down-regulation of p16(INK4a) mRNA with advancing age in acute myeloid leukemia. Aging. 2009; 1: 949-953.
7. Abrams SL, Steelman LS, Shelton JG, Wong EWT, Chappell WH, Baesecke J, Stivala F, Donia M, Nicoletti F, Libra M, Martelli AM, McCubrey JA. The Raf/MEK/ERK pathway can govern drug resistance, apoptosis and sensitivity to targeted therapy. Cell Cycle. 2010; 9: 1781-1791.
8. Ross DD. Novel mechanisms of drug resistance in leukemia. Leukemia. 2000; 14: 467-743.
9. Galmarini CM, Graham K, Thomas X, Calvo F, Rousselot P, El Jafaari A, Cros E, Mackey JR, Dumontet C. Expression of high Km 5’-nucleotidase in leukemic blasts is an independent prognostic factor in adults with acute myeloid leukemia. Blood. 2001; 98: 1922-1926.
10. Rushworth SA, MacEwan DJ. HO-1 underlies resistance of AML cells to TNF-induced apoptosis. Blood. 2008; 111: 3793-3801.
11. Maines MD. Heme oxygenase: function, multiplicity, regulatory mechanisms, and clinical applications. FASEB J. 1988; 2: 2557-2568.
12. Stocker R, Yamamoto Y, McDonagh AF, Glazer AN, Ames BN. Bilirubin is an antioxidant of possible physiological importance. Science. 1987; 235: 1043-1046.
13. Barañano DE, Rao M, Ferris CD, Snyder SH. Biliverdin reductase: a major physiologic cytoprotectant. Proc Natl Acad Sci U S A. 2002; 99: 16093-16098.
14. Singleton JW, Laster L. Biliverdin reductase of guinea pig liver. J Biol Chem. 1965; 240: 4780-4789.
15. Otterbein LE, Soares MP, Yamashita K, Bach FH. Heme oxygenase-1: unleashing the protective properties of heme. Trends Immunol. 2003; 24: 449-455.
16. Miyazaki T, Kirino Y, Takeno M, Samukawa S, Hama M, Tanaka M, Yamaji S, Ueda A, Tomita N, Fujita H, Ishigatsubo Y. Expression of heme oxygenase-1 in human leukemic cells and its regulation by transcriptional repressor Bach1. Cancer Sci. 2010; 101: 1409-1416.
17. Pietsch EC, Chan JY, Torti FM, Torti SV. Nrf2 mediates the induction of ferritin H in response to xenobiotics and cancer chemopreventive dithiolethiones. J Biol Chem. 2003; 278: 2361-2369.
18. Ogborne RM, Rushworth SA, Charalambos CA, O’Connell MA. Haem oxygenase-1: a target for dietary antioxidants. Biochem Soc Trans. 2004; 32: 1003-1005.
19. Trekli MC, Riss G, Goralczyk R, Tyrrell RM. Beta-carotene suppresses UVA-induced HO1 gene expression in cultured FEK4. Free Radical Biol Med. 2003; 34: 456-464.
20. Ogborne RM, Rushworth SA, O’Connell MA. alpha-lipoic acid-induced heme oxygenase-1 expression is mediated by nuclear factor erythroid 2-related factor 2 and p38 mitogen-activated protein kinase in human monocytic cells. Arteriosclerosis Thrombosis Vasc Biol. 2005; 25: 2100-2105.
21. Chi L, Ke Y, Luo C, Gozal D, Liu R. Depletion of reduced glutathione enhances motor neuron degeneration in vitro and in vivo. Neurosci. 2007; 144: 991-1003.
22. Rushworth SA, Zaitseva L, Langa S, Bowles KM, MacEwan DJ. FLIP regulation of HO-1 and TNF signalling in human acute myeloid leukemia provides a unique secondary anti-apoptotic mechanism. Oncotarget. 2011; 1: 359-366.
23. Shirley S, Micheau O. The heme oxygenase-1 and c-FLIP in acute myeloid leukemias: two non-redundant but mutually exclusive cellular safeguards protecting cells against TNF-induced cell death? Oncotarget. 2010; 1: 317-319.
24. Kurata SI, Matsumoto M, Tsuji Y, Nakajima H. Lipopolysaccharide activates transcription of the heme oxygenase gene in mouse M1 cells through oxidative activation of nuclear factor κB. Eur J Biochem. 1996; 239: 566-571.
25. Camhi SL, Alam J, Wiegand GW, Chin BY, Choi AMK. Transcriptional activation of the H0-1 gene by lipopolysaccharide is mediated by 5’ distal enhancers: Role of reactive oxygen intermediates and AP-1. Am J Respir Cell Mol Biol. 1998; 18: 226-234.
26. Kanno S-I, Higurashi A, Watanabe Y, Shouji A, Asou K, Ishikawa M. Susceptibility to cytosine arabinoside (Ara-C)-induced cytotoxicity in human leukemia cell lines. Toxicol Lett. 2004; 152: 149-158.
27. Mitsuhashi M, Endo K, Obara K, Izutsu H, Ishida T, Chikatsu N, Shinagawa A. Ex vivo simulation of the action of antileukemia drugs by measuring apoptosis-related mRNA in blood. Clin Chem. 2008; 54: 673-681.
28. Capizzi RL, Yang JL, Cheng E, Bjornsson T, Sahasrabudhe D, Tan RS, Cheng YC. Alteration of the pharmacokinetics of high-dose ara-C by its metabolite, high ara-U in patients with acute leukemia. J Clin Oncol. 1983; 1: 763-771.
29. Rushworth SA, Bowles KM, Raninga P, MacEwan DJ. NF-κB-inhibited acute myeloid leukemia cells are rescued from apoptosis by heme oxygenase-1 induction. Cancer Res. 2010; 70: 2973-2983.
30. Rushworth SA, Bowles KM, MacEwan DJ. High basal nuclear levels of Nrf2 in acute myeloid leukemia reduces sensitivity to proteasome inhibitors. Cancer Res. 2011; 71: 1999-2009.
31. Nuhn P, Kunzli B, Hennig R, Mitkus T, Ramanauskas T, Nobiling R, Meuer S, Friess H, Berberat P. Heme oxygenase-1 and its metabolites affect pancreatic tumor growth in vivo. Mol Cancer. 2009; 8: 37.
32. Fang J, Sawa T, Akaike T, Greish K, Maeda H. Enhancement of chemotherapeutic response of tumor cells by a heme oxygenase inhibitor, pegylated zinc protoporphyrin. Intl J Cancer. 2004; 109: 1-8.
33. Jeney V, Balla J, Yachie A, Varga Z, Vercellotti GM, Eaton JW, Balla G. Pro-oxidant and cytotoxic effects of circulating heme. Blood. 2002; 100: 879-887.
34. Brouard S, Otterbein LE, Anrather J, Tobiasch E, Bach FH, Choi AMK, Soares MP. Carbon monoxide generated by heme oxygenase 1 suppresses endothelial cell apoptosis. J Exp Med. 2000; 192: 1015-1025.
35. Fang J, Akaike T, Maeda H. Antiapoptotic role of heme oxygenase (HO) and the potential of HO as a target in anticancer treatment. Apoptosis. 2004; 9: 27-35.
36. Rushworth SA, MacEwan DJ, O’Connell MA. Lipopolysaccharide-induced expression of NAD(P)H:quinone oxidoreductase 1 and heme oxygenase-1 protects against excessive inflammatory responses in human monocytes. J Immunol. 2008; 181: 6730-6737.
37. Ryan KA, Smith MF, Jr., Sanders MK, Ernst PB. Reactive oxygen and nitrogen species differentially regulate Toll-like receptor 4-mediated activation of NF-κB and interleukin-8 expression. Infect Immun. 2004; 72: 2123-2130.