
Introduction
Gliomas compose almost 60% of total human primary CNS malignancies [1, 2]. These tumors are recognized as a heterogeneous group of neoplasms that differ in location, morphological features, tendency for progression and response to therapy. The malignant Grade IV Glioblastoma Multiforme (GBM) is the most common, with an associated median survival of 14-16 months [2, 3]. Despite progress in research on the molecular aspects of malignant gliomas the prognosis of these brain tumors continues to be dismal. We believe that research on the ontogeny of GBMs, presents an opportunity for improving our understanding and treatment of this disease.
For decades, it was widely assumed that differentiated glia were the only cells capable of transformation as the adult brain was thought to be mitotically inactive [4] . The demonstration of functional neurogenesis in the adult CNS[5-8] provided new possibilities for the candidate cell of origin of CNS neoplasms. These progenitor populations reside in multiple regions of the adult brain, including the subventricular zone (SVZ) and the dentate gyrus within the hippocampus [5, 9-12] (Figure 1). The largest of these germinal regions in humans, the SVZ, has long be proposed as a source of gliomas, as many are either periventricular or contiguous with the subventricular zone [13-16]. Additionally, cells that express the NG2 proteoglycan, distributed throughout the brain have also been proposed as the cells origin of gliomas [17, 18]. In summary, the cell of origin maybe differentiated glial and/or neuro-glilal progenitor stem cells subjected to a variety of transforming molecular alterations.
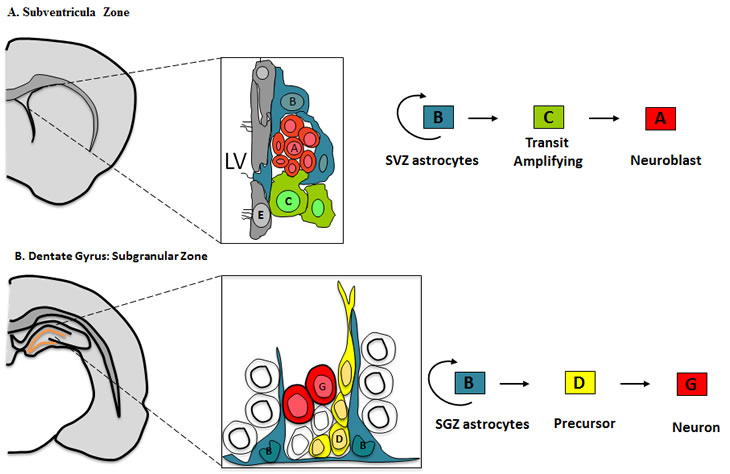
Figure 1: Cellular composition and cytoarchitecture of germinal regions within the adult brain
A) Frontal schema of the Subventricular zone in the adult mouse brain and the cell types from which it is composed. Multi-ciliated ependymal cells (E, Gray) line the lateral ventricle (LV). SVZ astrocytes (B, Blue) are primary progenitors which generate neuroblast (A, Red) through a rapidly dividing transit amplifying cell (C, Green). Occasionally Type B cells extend a short primary cilium into the ventricle.
B) Frontal schema of the Subgranular Zone within the dentate gyrus of the adult mouse brain. The cell types that compose the SGZ include, SGZ astrocytes (B, Blue), the in-vivo primary precursors of new granule neurons (G, Red) through an intermediate precursor Type D cell (D, Yellow).
Conditional mouse modeling combined with lineage tracing techniques makes it possible to target these distinct neurogenic regions in adults, as well as cell populations at different stages of their differentiation. These and other reagents have facilitated experiments towards a better understanding of the ontogeny of GBMs, which have underscored the relationship between gliomagenesis and brain development. A relationship further highlighted by the The Cancer Genome Atlas (TCGA) which sub-classified GBMs to four gene signature profiles (Classical, Neural, Proneural and Mesenchymal), which are characteristic of distinct stages, prognosis and patterns of differentiation [19, 20]. This reinforces the need to view the tumourigenic process of gliomas, in the context of normal brain development.
definition of CNS cell types and their regional diversity.
CNS development proceeds in a hierarchical manner; whereby, mature differentiated cells such as neurons and the varying glial subtypes (astrocytes, oligodendrocytes, ependymocytes) arise from CNS stem cells. These CNS stem cells arise from the pseudostratified neuroepithelium that lines the cerebral ventricles early in embryonic development and stains positive for Nestin [21]. By the onset of neurogenesis, these neuroephitelial cells are replaced by a more differentiated neural stem cell, the radial glia cell [22, 23]. Radial glia has a more glial like pathological profile with expression of BLBP (brain lipid binding protein)[24]. By adulthood, most CNS progenitors have disappeared, however, stem cells capable of giving rice to differentiated CNS cell types are retained in distinct anatomical regions, including the SVZ lining the lateral ventricles[9, 25] and the dentate gyrus within the hippocampus [11].
The subventricular zone (SVZ) is the largest germinal region, and it exhibits a high degree of organization, Figure 1A. It is composed of Type B cells, slowly dividing SVZ astrocytes that function as primary progenitors, giving rise to actively proliferating intermediate progenitors known as Type C cells [26]. Type C cells give rise to immature neuroblasts or Type A cells, which migrate in chains to the olfactory bulb, where they differentiate into distinct interneuronal populations[27]. Type B cells are marked by the expression of GFAP and they retain important properties of radial glia, thus it has been postulated that radial glia transforms directly into Type B cells[23-25].
Similarly, neural stem cells within the hippocampus seem to have a radial glial origin, Figure 1B. The Subgranular Zone (SGZ) within the dentate gyrus of the hippocampus, is composed of two types of dividing cells, astrocytes and darkly stained small cell with small basophilic nucleus [28]. Similar to the SVZ, radial astrocytes function as the primary precursors for new neurons through an intermediate progenitor, referred to as Type D cells [28].
A third population of CNS precursors in the adult brain, is a subpopulation of NG2 expressing cells shown to play a glial precursor role. The NG2 proteoglycan is believed to be an in vivo marker for oligodendrocytes progenitors found in the developing brain [17, 18, 29, 30]. However, a significant proportion of NG2-expressing cells remain in the adult CNS following the end of gliogenesis. These cells continue to proliferate and can give rise to neurons and macroglia, as opposed to the SVZ and SGZ progenitors described above. NG2 expressing cells are found throughout the adult brain [18], however, the heterogeneity of this population has not been adequately addressed in the adult CNS. While NG2 cells continue to divide in the adult CNS it is not clear what function they serve once myelination is complete. Future studies should elucidate the functional importance of NG2 in a variety of cell functions and shed light on the role NG2-expressing cells play in the intact and diseased CNS.
All of the neural stem and progenitor populations described above have different proliferative capacities and varying ability to generate astrocytes, oligodendrocytes and neurons that may reflect distinct patterns of gene expression. Thus regional differences in neuroglial precursor populations may dictate a significant component of CNS tumor gene expression and biology.
Implications for CNS tumorigenesis
All forms of cancer arise from the acquisition of genetic alteration, leading to several aberrant biological properties required for transformation. These biological properties include, sustained proliferation, evading growth suppression, resisting cell death, enabling replicative immortality, inducing angiogenesis and activating invasion and metastasis [31]. In the adult brain, existing normal neural stem cells and progenitor elements represent the path of least resistance to tumorigenesis. These cells already have the genetic machinery for promoting mitosis, to resist apoptosis and senescence [32], making them an attractive candidate for the cell of origin for gliomas. Nevertheless, any cell in the hierarchy with proliferative capacity could serve as a cell of origin in gliomas, if it acquires mutations that re-instigate self-renewal capacity and prevent differentiation to a post-mitotic state. The normal lineage hierarchy in the CNS can serve as a framework for understanding cells of origin in GBM’s, by comparing expression signature of normal cell populations with those of different tumor subtypes.
Comparative gene expression analysis in leukemias show that the different forms of leukemias appear to arise from distinct points in the hematopoietic lineage, that are susceptible to specific translocations. Furthermore, the biology behind this translocations-lineage stage mechanism might lie in the capacity of translocations to activate key leukemogenic programs [33, 34], reinforcing the idea that cancers arise from specific combinations of mutations and susceptible cell types. In the CNS this idea has been shown to hold true in ependymomas, which carry transcriptome, and DNA copy alterations that correlate with tumor location [35, 36]. Similarly gene expression profiles of medulloblastomas carve these cancers into clinically and molecular distinct subgroups [37]. Genomic profiling of GBMs has revealed patterns of molecular changes with tumor subclasses that harbor distinct underlying biology and clinical prognosis. One such study used correlative expression profiling to subdivide gliomas into groups based on similarities to known cell types. The resulting groups were termed Pro-neural, Proliferative and Mesenchymal [38]. A more recent analysis of one larger TCGA GBM data set, classified GBMs as Pro-neural, Neural, Classical and Mesenchymal. These subclasses are strongly associated with genomic abnormalities in PDGFR, IDH1/IDH2, EGFR or NF1 genes respectively [19, 20, 39]. However, the link between precursor cells to genomic classification of GBMs has not been firmly established. First, these analysis were on bulk GBM tumors, which show phenotypic plasticity or differentiation during neoplastic progression, Hence, the molecular signatures of bulk tumor cells may not precisely reflect the true cell of origin in normal tissue. To overcome this, rather than analyzing terminal stage tumors, this kind of studies should be carried out on tumors at different stages of their malignant progression.
There exists debate about the Cancer Stem Cell (CSC) hypothesis and the existence of BTIC in brain tumors. Unlike leukemias discussed above, there is no singular cell surface or genetic signature of the BTIC. Initial enthusiasm with CD133 has recently been questioned by studies demonstrating that alternations in mitochondrial function among glioma cells induce CD133 expression[40]. Conversely, replacement of dysfunctional mitochondrial genes can reverse CD133 expression[40]. These results suggest that CD133 expression in gliomas is triggered as a response to environmental stress, questioning the reliability of CD133 as a BTIC marker. Although the CSC hypothesis seems to be supportive of the hypothesis that brain tumors arise from the transformation of normal neural stem cells, these two concepts are distinct and in most instances the identity of the cell of origin may differ substantially from that of the BTIC.
Another but not mutually exclusive hypothesis is that some GBMs can arise from the transformation of terminally differentiated glia. Retroviral transduction of INK4a/Arf-/- mature astrocytes with a constitutively active mutant EGF receptor (EGFRvIII), prevalent in human GBMs, induces astrocyte de-differentiation and GBM formation[41]. A phenomenon, which is also observed, when GFAP positive cells are infected with a PDGF expressing retrovirus using the RCAS/tva system [42]. In addition, overexpression of the transcriptional factor c-myc, in astrocytes results in the down regulation of the astrocytic marker GFAP and upregulation of Nestin[panicker43, 44], suggestive of transformation. However one of the most relevant peaces of evidence comes from the demonstration, that adult fibroblast can be reprogrammed to a pluripotent stem cell state by transfection of a small number of transcription factors [45], suggesting that differentiated cells can be endowed stem cell like properties. Arguably, similar process might be relevant during brain tumor pathogenesis. This controversy can only be settled with in vivo studies to asses the propensity of cell populations to act as cell of origin. Mouse models of oncogenesis have been pivotal in this regard, and have been of great insight into the ontogeny of GBMs
Lessons from mouse models
To elucidate the ontogeny of gliomas conditional and somatic strategies coupled to lineage tracing techniques have come to the forefront, as summarized on Table 1. Conditional strategies were developed to allow for the control of gene expression in both a tissue and or time specific manner. For example, Tet/Tamoxifen-regulatable and Cre inducible genes, can allow for the control of the timing, duration and tissue compartment of gene expression or inactivation. In addition, several methods for somatic gene transfer have been developed using retroviral or adenoviral delivery of Cre recombinase. This approach, allows for a more accurate modeling of human cancers as most mutations are thought to occur somatically in a single cell or small group of specific cell types. All of the above mentioned strategies have been used to study the effects of different GBM relevant mutations and their cooperation at different developmental time-points. However, the most convincing data will come from experiments that clearly demonstrate the ability of a cell to get transformed and initiate tumor development. Towards this end, linage-tracing techniques are used to label and follow cells of known type in the CNS for changes. As reviewed below the use of these powerful techniques has shed some light into the cellular origins of GBMs.
| Table 1. Candidate cell of origin identified in GBMs by targeting distinct cellular compartments and or anatomical regions | ||||
| Mouse model | Genetic model | Promoter-Cre construct | Anatomical region | Cell of origin |
| Germline | ||||
| V12H-Ras activation | hGFAP-Cre | NA | SVZ Multipotent progenitor[47] | |
| p53, Nf1 inactivation | hGFAP-Cre | NA | SVZ Multipotent progenitor[49] | |
| p53,Pten,Nf1 inactivation | Nestin-CreERT2 | NA | Multipotent progenitor[50] | |
| Pten, Rb1, p53 inactivation | GFAP-Cre ERT2 | NA | Multipotent progenitor and Astrocytes[53] | |
| p53,Nf1 inactivation | GFAP-Cre | NA | Oligodendrocyte precursor[53] | |
| p53,Nf1 inactivation | NG2-Cre | NA | Oligodendrocyte precursor[53] | |
| Mutant p53 | GFAP-Cre | NA | Neural progenitor, Transit-amplifying cell[66] | |
| Somatic | ||||
| Nf1, p53, Pten inactivation | Adeno-Cre | SVZ, Striatum, Cortex | Multipotent progenitor[50] | |
| Rb, p53, Pten inactivation | Adeno-Cre | SVZ, Striatum | Multipotent progenitor, transit-amplifying cell[60] | |
| Kras, AKT activation | GFAP-tva | NA | Multipotent progenitor[62] | |
| Kras, AKT activation | Nestin-tva | NA | Multipotent progenitor[62] | |
| V12Hras, AKT activation, p53 inactivation | GFAP-Cre | SVZ, Cortex, Hippocampus | Multipotent progenitor[63] | |
| Ink4a/Arf, Pten, RCAS-PDGF | RCAS-Cre/ GFAP-tva | SVZ, striatum, cerebellum | Multipotent progenitors and astrocytes[65] | |
| Ink4a/Arf, Pten, RCAS-PDGF | RCAS-Cre/Nestin-tva | SVZ, striatum, cerebellum | Multipotent progenitors and astrocytes[65] | |
| Primary cultures | ||||
| P16Ink4a/P19ARFBmi1 inactivation, mutant EGFR | NA | Cortex, SVZ | Neural progenitor and astrocytes[67] | |
Germline strategies: Targeting defined cell populations at specific developmental time-points
The first germline derived mouse models for brain tumors, were models that over expressed viral oncogenes [46]. However it was soon realized that most are embryonically lethal. Overtime, genetic models evolved to the use of conditional strategies and genes where driven by cell type specific promoters [47, 48]. Using these strategies, brain tumor models have been developed by altering signaling pathways that are disrupted in human gliomas including Rb, Ras, AKT, Pten, NF1 amongst others.
To study the role of Ras signaling in gliomas, a mouse model carrying a constitutively active Ras (V12Ras) under the control of a human hGFAP promoter was developed [49] by our group. This model highlighted the importance of Ras gene dosage during tumor development, as high expression levels lead to the development of multifocal GBMs within two weeks after birth. As opposed to moderate levels of expression, which lead to 50% of the mice developing high-grade astrocytomas by three months of age [49, 50]. This model more accurately represents secondary GBMs, as these mice are born normal but develop low-grade astrocytomas by 3-8 weeks of age [49]. Upon micro-dissection, the low-grade gliomas harbored p53 mutations, similar to low grade astrocytomas in humans. Currently we are deciphering if they also harbor mutations in IDH, which are highly prevalent in human low-grade astrocytomas. At 12 weeks highly invasive GFAP-/Nestin + high-grade astrocytomas develop, a progression that is accompanied by additional changes including overexpression of EGR, MDMT, loss of Pten and p16 and p19 [50], all of which are known genetic markers associated with malignant human gliomas. The cell of origin in this model is uncertain, but likely from GFAP positive progenitors in the SVZ (unpublished data).
In a different model, Zhu et al. used the same hGFAP promoter to undertake Cre induced deletion of p53 and Nf1, showing that early inactivation of p53 gene cooperates with Nf1 loss to induce GBM development [51]. Similar to the Ras model, the earliest identifiable area of a tumor was confined to the SVZ. These two models suggest that either a specific cell type or the microenvironment in this region provide a favorable niche for the growth of early tumor cells [51].
However, the hGFAP promoter used in both models is expressed in both stem cells and white matter astrocytes. Thus whether the tumors arose from a stem or progenitor population or a differentiated astrocyte cannot be definitely determined. To address this, Alcantarallaguno et al. used a tamoxifen inducible Nestin-Cre to inactivate the tumor suppressor Pten, Nf1 and Tp53 at different developmental time points. During early embryogenesis, the Nestin promoter produced a broad pattern of expression similar to the hGFAP transgene [52]. However, induction during adulthood leads to recombination almost exclusively in the neural stem cell population of the SVZ and SGZ [52, 53]. In this model, Induction of recombination during early embryogenesis lead to the development of tumors at rates similar to the hGFAP-Cre; Tp53; Nf1 glioma model, with a median survival of 16 weeks. In contrast, tumors observed when recombination was induced at adulthood, had a median survival of 46 weeks [52]. This observation suggests that early progenitors seem to be more susceptible to transformation and at least in this model the cell of origin seems to reside in the neural stem/progenitor populations of the adult brain.
Recently, a tamoxifen inducible GFAP promoter was used to induce the deletion of Pten, Rb1 and Tp53 in astrocytes in a limited population of neural progenitors in the adult brain [54, 55]. This model showed selective cooperativity among tumor suppressors. TP53 deletion was required for the formation of high-grade astrocytomas induced by the deletion of Pten, Rb1 or combined Pten Rb1 deletions [55]. Many tumors were contiguous with regions rich in neural progenitor cells such as the SVZ, the rostral migratory stream and subgranular zone of the dentate gyrus. However, there were a significant 20% of gliomas that developed in sites independent of these proliferative niches. This suggests that astrocytomas induced by this set of mutations can develop outside proliferative niches, albeit at lower efficiency.
Despite all the insights from the models discussed above, the true cell of origin is still controversial. This mainly due to the fact that the cell of origin is difficult to identify by analyzing cells within terminal stage tumors, whose genetic identity could be concealed by the acquired plasticity. A recent study aimed at addressing this issue analyzed proliferative abnormalities in distinct lineages prior to malignancy [56]. Using a mouse model approach that allows simultaneous labeling and gene knockout in clones of somatic cells [57, 58], Liu et traced the growth of individual lineages that descended from a mutant neural stem cell lacking p53 and Nf1 expression. After recombination, a dramatic overexpansion and aberrant growth of oligodendrocytes progenitors (OPC), but not of neural stem cells or other lineages was observed. Further transcriptome and marker staining analysis confirmed upon tumor formation the OPC nature of the tumor cells. This demonstrates that in p53/Nf1 mutant driven gliomas, mutations may initially occur in either NSC or OPC, but only OPC provides the susceptible cellular context needed for transformation [56]. This highlights the concept that cells initially acquiring a mutation might not be the cell of origin, but rather its descendants who acquire additional transforming alterations.
Somatic strategies: Targeting defined anatomical regions in the CNS
Germinal regions such as the SVZ have long been proposed as more gliomagenic, as this region is more susceptible to viral and chemical oncogenesis [59-61]. In canine and rodent brains for example, avian sarcoma viral transformation [60]or systemic exposure to the carcinogen N-ethyl-N-nitrosourea, leads to tumor formation, preferentially in the proliferative SVZ rather than in non-proliferative regions of the brain [59]. This observation suggested that gliomas could originate from the neural stem cell population that resides in the SVZ; however, the genetic alterations necessary for transformation of these stem/progenitor cells are recently being unraveled.
Work by Alcantarallaguno et al. demonstrates that viral mediated Cre-recombinase targeting of the SVZ in an Nf1/p53 flox background induced astrocytoma formation with 100% penetrance [52]. Conversely, targeting regions such as the striatum did not lead to tumor formation. Targeting of the SVZ was carried out at two different developmental time-points, early postnatal and adulthood. Tumor suppressor inactivation in the SVZ of both early postnatal and adult stages induced astrocytoma formation, however, the early postnatal injected mice developed more and extensive gliomas compared to adult mice[52].
Using mice bearing conditional alleles of Rb, p53 and Pten Jacks et al showed that the initial combination of genetic mutations in the SVZ dictates the pathogenesis and phenotype of the tumors that develop. In these mice, recombination of Pten/p53 gave rise to gliomas, whereas deletion of Rb/p53 generated primitive neuroectodermal tumors (PNET) [62]. This model raises questions regarding the relationship between state of differentiation and the oncogenic growth signal. Is it that different populations of stem/progenitor cells are transformed by a specific set of mutations? Or is it that the different tumor types developed from the same “Tumor progenitor” but weather a glial or primitive neuroectodermal differentiation is observed is dependent on the genetic alteration. Further optimization of this techniques using Cre reporter mouse lines in combination with promoter specific Cre drivers will allow us to look at this questions.
Other retroviral-based approaches such as the RCAS system have allowed lineage tracing of specific cell populations through the process of tumorigenesis [63]. The virus infects mammalian cells that express the subgroup a receptor for the RCAS virus (tv-a), which is not encoded by the mammalian genome. This system has been used to transfer constitutively active mutant forms of Kras and AKT to both Nestin positive progenitor cells and GFAP positive astrocytes [64]. Although neither activated Kras nor AKT alone is sufficient to induce GBM formation, the combination is sufficient to induce high grade gliomas with histological features of human GBMs. Furthermore, the gliomas were found in the Nestin drive tv-a neural progenitors, but not in the differentiated GFAP-tv-a astrocytes [64], highlighting the importance of both cooperating mutations as well as the differentiation status of the cell of origin. Similar results were observed when a Cre loxP controlled Lentiviral vector was injected into specific anatomical regions to induce expression of activated H-Ras and AKT in GFAP+ astrocytes in the adult mice [65, 66]. In this model, high-grade gliomas developed only when the SVZ or hippocampus were targeted. Tumors were rarely detected when less proliferative areas such as the cortex were targeted [65].
In contrast to targeting the neural-glial progenitor population, a study used the RCAS system, with injection of the RCAS-PDGF + RCAS-Cre virus into various regions of a Nestin-tva; Ink4aARF null; Pten flox adult mice. These regions included the SVZ, left and right cerebral hemispheres and cerebellum, with development of gliomas in all of the areas targeted [67]. Similar results were also observed with a GFAP-tva model resulting in mainly mixed oligoastrocytomas. These mouse model studies collectively suggest that the cell of origin in gliomas depends on timing of transforming genetic alterations (embryonic or adult); the neuro-glial progenitor enriched regions in adults, but can also occur in differentiated astrocytes albeit with a lower efficiency.
DISCUSSION
Mouse models of gliomagenesis have been pivotal in determining how the process of differentiation is integrally related to the process of gliomagenesis and the maintenance of the neoplastic phenotype. They indicate that not only the nature of transforming genetic alteration, but also the differentiation status of the neuro-glial cell, modulates gliomagenesis. Of interest, the oncogenic stimuli can themselves alter the state of neuro-glial differentiation, to make them more susceptible to transformation.
However, the question of ontogeny of gliomas remains in its infancy despite these mouse models. One of the problems is that the models to date target, using broadly expressed promoters such as Nestin and GFAP. For example, targeting the SVZ with CMV, GFAP or Nestin promoters target the stem and progenitor cells, but also differentiated glia that resides in this region. Use of cell specific promoters to drive expression of transforming alterations in neuro-glial cells at different stage of the normal lineage hierarchies is crucial, towards our understanding of initiating events in gliomagenesis within a defined cellular compartment. Identifying the cell of origin of gliomas and the region in the brain where these cells reside would permit a more systematic analysis of the genetic lesions involved in tumor initiation and progression. Moreover, the gene signature of the cell of origin may elucidate key molecular pathways and driver mutations that could lead to the development of new therapeutic approaches to prevent and target early stage disease. Finally, the integration of developmental biology, genomics and cancer biology will lead to the production of more accurate models, where single cell genetic manipulations in unambiguously labeled cells, will allow a better understanding of the ontogeny of gliomas.
REFERENCES
1. Kleihues P, Soylemezoglu F, Schauble B, Scheithauer BW, Burger PC. Histopathology, classification, and grading of gliomas. Glia. 1995; 15:211-221.
2. Kleihues PSL. World health organization classification of tumors. cancer. 2000; 88:2887.
3. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005; 352:987-996.
4. Rakic P. DNA synthesis and cell division in the adult primate brain. Ann N Y Acad Sci. 1985; 457:193-211.
5. Altman J, Das GD. Autoradiographic and histological evidence of postnatal hippocampal neurogenesis in rats. J Comp Neurol. 1965; 124:319-335.
6. Altman J. Autoradiographic investigation of cell proliferation in the brains of rats and cats. Anat Rec. 1963; 145:573-591.
7. Sidman RL, Miale IL, Feder N. Cell proliferation and migration in the primitive ependymal zone: an autoradiographic study of histogenesis in the nervous system. Exp Neurol. 1959; 1:322-333.
8. Gross CG. Neurogenesis in the adult brain: death of a dogma. Nat Rev Neurosci. 2000; 1:67-73.
9. Reynolds BA, Weiss S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science. 1992; 255:1707-1710.
10. GD AJaD. Autoradiographic and histological evidence of postnatal hippocampal neurogenesis in rats. The journal of comparative neurology. 1965; 124:319-335.
11. Gage FH. Mammalian neural stem cells. Science. 2000; 287:1433-1438.
12. Kaplan MS, Bell DH. Mitotic neuroblasts in the 9-day-old and 11-month-old rodent hippocampus. J Neurosci. 1984; 4:1429-1441.
13. JW H. The subependymal plate and the geneisi of gliomas. The journal of pathology. 1975; 117.
14. Bohman L-E, Swanson KR, Moore JL, Rockne R, Mandigo C, Hankinson T, Assanah M, Canoll P, Bruce JN. Magnetic resonance imaging characteristics of glioblastoma multiforme: implications for understanding glioma ontogeny. Neurosurgery. 2010; 67:1319-1327; discussion 1327-1318.
15. Young GS, Macklin EA, Setayesh K, Lawson JD, Wen PY, Norden AD, Drappatz J, Kesari S. Longitudinal MRI evidence for decreased survival among periventricular glioblastoma. J Neurooncol. 2010.
16. Lim DA, Cha S, Mayo MC, Chen M-H, Keles E, VandenBerg S, Berger MS. Relationship of glioblastoma multiforme to neural stem cell regions predicts invasive and multifocal tumor phenotype. Neuro-oncology. 2007; 9:424-429.
17. Belachew S, Chittajallu R, Aguirre AA, Yuan X, Kirby M, Anderson S, Gallo V. Postnatal NG2 proteoglycan-expressing progenitor cells are intrinsically multipotent and generate functional neurons. J Cell Biol. 2003; 161:169-186.
18. Horner PJ, Thallmair M, Gage FH. Defining the NG2-expressing cell of the adult CNS. J Neurocytol. 2002; 31:469-480.
19. Brennan C, Momota H, Hambardzumyan D, Ozawa T, Tandon A, Pedraza A, Holland E. Glioblastoma subclasses can be defined by activity among signal transduction pathways and associated genomic alterations. PLoS ONE. 2009; 4:e7752.
20. Network CGAR. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008; 455:1061-1068.
21. Kriegstein A, Alvarez-Buylla A. The glial nature of embryonic and adult neural stem cells. Annu Rev Neurosci. 2009; 32:149-184.
22. Alvarez-Buylla A, Garcia-Verdugo JM, Tramontin AD. A unified hypothesis on the lineage of neural stem cells. Nat Rev Neurosci. 2001; 2:287-293.
23. Merkle FT, Tramontin AD, García-Verdugo JM, Alvarez-Buylla A. Radial glia give rise to adult neural stem cells in the subventricular zone. Proc Natl Acad Sci USA. 2004; 101:17528-17532.
24. Merkle FT, Alvarez-Buylla A. Neural stem cells in mammalian development. Curr Opin Cell Biol. 2006; 18:704-709.
25. Doetsch F, Caillé I, Lim DA, García-Verdugo JM, Alvarez-Buylla A. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell. 1999; 97:703-716.
26. Doetsch F, García-Verdugo JM, Alvarez-Buylla A. Cellular composition and three-dimensional organization of the subventricular germinal zone in the adult mammalian brain. J Neurosci. 1997; 17:5046-5061.
27. Doetsch F, Alvarez-Buylla A. Network of tangential pathways for neuronal migration in adult mammalian brain. Proc Natl Acad Sci USA. 1996; 93:14895-14900.
28. Seri B, García-Verdugo JM, Collado-Morente L, McEwen BS, Alvarez-Buylla A. Cell types, lineage, and architecture of the germinal zone in the adult dentate gyrus. J Comp Neurol. 2004; 478:359-378.
29. Liu A, Han YR, Li J, Sun D, Ouyang M, Plummer MR, Casaccia-Bonnefil P. The glial or neuronal fate choice of oligodendrocyte progenitors is modulated by their ability to acquire an epigenetic memory. J Neurosci. 2007; 27:7339-7343.
30. Nishiyama A, Komitova M, Suzuki R, Zhu X. Polydendrocytes (NG2 cells): multifunctional cells with lineage plasticity. Nat Rev Neurosci. 2009; 10:9-22.
31. Hanahan D, Weinberg Robert A. Hallmarks of Cancer: The Next Generation. Cell. 2011; 144:646-674.
32. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001; 414:105-111.
33. Chen W, Kumar AR, Hudson WA, Li Q, Wu B, Staggs RA, Lund EA, Sam TN, Kersey JH. Malignant transformation initiated by Mll-AF9: gene dosage and critical target cells. Cancer Cell. 2008; 13:432-440.
34. Barabé F, Kennedy JA, Hope KJ, Dick JE. Modeling the initiation and progression of human acute leukemia in mice. Science. 2007; 316:600-604.
35. Johnson RA, Wright KD, Poppleton H, Mohankumar KM, Finkelstein D, Pounds SB, Rand V, Leary SES, White E, Eden C, Hogg T, Northcott P, Mack S, Neale G, Wang Y-D, Coyle B et al. Cross-species genomics matches driver mutations and cell compartments to model ependymoma. Nature. 2010; 466:632-636.
36. Taylor MD, Poppleton H, Fuller C, Su X, Liu Y, Jensen P, Magdaleno S, Dalton J, Calabrese C, Board J, Macdonald T, Rutka J, Guha A, Gajjar A, Curran T, Gilbertson RJ. Radial glia cells are candidate stem cells of ependymoma. Cancer Cell. 2005; 8:323-335.
37. Northcott PA, Rutka JT, Taylor MD. Genomics of medulloblastoma: from Giemsa-banding to next-generation sequencing in 20 years. Neurosurg Focus. 2010; 28:E6.
38. Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, Misra A, Nigro JM, Colman H, Soroceanu L, Williams PM, Modrusan Z, Feuerstein BG, Aldape K. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006; 9:157-173.
39. Parsons DW. The evolving picture of the glioblastoma genome. Oncotarget. 2010; 1:237-238.
40. Griguer CE, Oliva CR, Gobin E, Marcorelles P, Benos DJ, Lancaster JR, Jr., Gillespie GY. CD133 is a marker of bioenergetic stress in human glioma. PLoS ONE. 2008; 3:e3655.
41. Bachoo RM, Maher EA, Ligon KL, Sharpless NE, Chan SS, You MJ, Tang Y, DeFrances J, Stover E, Weissleder R, Rowitch DH, Louis DN, DePinho RA. Epidermal growth factor receptor and Ink4a/Arf: convergent mechanisms governing terminal differentiation and transformation along the neural stem cell to astrocyte axis. Cancer Cell. 2002; 1:269-277.
42. Dai C, Celestino JC, Okada Y, Louis DN, Fuller GN, Holland EC. PDGF autocrine stimulation dedifferentiates cultured astrocytes and induces oligodendrogliomas and oligoastrocytomas from neural progenitors and astrocytes in vivo. Genes & Development. 2001; 15:1913-1925.
43. Hass R. Retrodifferentiation and cell death. Crit Rev Oncog. 1994; 5:359-371.
44. Panicker SP, Raychaudhuri B, Sharma P, Tipps R, Mazumdar T, Mal AK, Palomo JM, Vogelbaum MA, Haque SJ. p300- and Myc-mediated regulation of glioblastoma multiforme cell differentiation. Oncotarget. 2010; 1:289-303.
45. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006; 126:663-676.
46. Brinster RL, Chen HY, Messing A, Terry van Dyke, levine A and Palmiter RD. Transgenic mice harboring SV40 t antigen genes develop characteristic brain tumors. Cell. 1984; 37:367-379.
47. Jacks T Macleod KF. insight into cancer from transgenic mouse models. J Path. 1999; 187:43-60.
48. Talmadge JE, Singh RK, Fidler IJ, Raz A. Murine models to evaluate novel and conventional therapeutic strategies for cancer. Am J Pathol. 2007; 170:793-804.
49. Ding H, Roncari L, Shannon P, Wu X, Lau N, Karaskova J, Gutmann DH, Squire JA, Nagy A, Guha A. Astrocyte-specific expression of activated p21-ras results in malignant astrocytoma formation in a transgenic mouse model of human gliomas. Cancer Research. 2001; 61:3826-3836.
50. Shannon P, Sabha N, Lau N, Kamnasaran D, Gutmann DH, Guha A. Pathological and molecular progression of astrocytomas in a GFAP:12 V-Ha-Ras mouse astrocytoma model. Am J Pathol. 2005; 167:859-867.
51. Zhu Y, Guignard F, Zhao D, Liu L, Burns DK, Mason RP, Messing A, Parada LF. Early inactivation of p53 tumor suppressor gene cooperating with NF1 loss induces malignant astrocytoma. Cancer Cell. 2005; 8:119-130.
52. Alcantarallaguno S, Chen J, Kwon C, Jackson E, Li Y, Burns D, Alvarezbuylla A, Parada L. Malignant Astrocytomas Originate from Neural Stem/Progenitor Cells in a Somatic Tumor Suppressor Mouse Model. Cancer Cell. 2009; 15:45-56.
53. Chen J, Kwon CH, Lin L, Li Y, Parada LF. Inducible site-specific recombination in neural stem/progenitor cells. Genesis. 2009; 47:122-131.
54. Chow LM, Zhang J, Baker SJ. Inducible Cre recombinase activity in mouse mature astrocytes and adult neural precursor cells. Transgenic Res. 2008; 17:919-928.
55. Chow LML, Endersby R, Zhu X, Rankin S, Qu C, Zhang J, Broniscer A, Ellison DW, Baker SJ. Cooperativity within and among Pten, p53, and Rb pathways induces high-grade astrocytoma in adult brain. Cancer Cell. 2011; 19:305-316.
56. Liu C, Sage Jonathan C, Miller Michael R, Verhaak Roel GW, Hippenmeyer S, Vogel H, Foreman O, Bronson Roderick T, Nishiyama A, Luo L, Zong H. Mosaic Analysis with Double Markers Reveals Tumor Cell of Origin in Glioma. Cell. 2011:1-13.
57. Hippenmeyer S, Youn YH, Moon HM, Miyamichi K, Zong H, Wynshaw-Boris A, Luo L. Genetic mosaic dissection of Lis1 and Ndel1 in neuronal migration. Neuron. 2010; 68:695-709.
58. Zong H, Espinosa JS, Su HH, Muzumdar MD, Luo L. Mosaic analysis with double markers in mice. Cell. 2005; 121:479-492.
59. Vick NA LM, bigner DD. The role of the subependymal plate in glial tumorigenesis. Acta neuropathologica. 1977; 40:63-71.
60. Copeland DD VFaBD. The induction of intracranial neoplasms by the inoculation of avian sarcoma virus in perinatal and adult rats. Journal of neuropathology and experimental neurology. 1975; 34:340-358.
61. Copeland DD, Bigner DD. The role of the subependymal plate in avian sarcoma virus brain tumor induction: comparison of incipient tumors in neonatal and adult rats. Acta Neuropathol. 1977; 38:1-6.
62. Jacques T, Swales A, Brzozowski M, Henriquez N, Linehan J, Mirzadeh Z, O’ Malley C, Naumann H, Alvarez-Buylla A, Brandner S. Combinations of genetic mutations in the adult neural stem cell compartment determine brain tumour phenotypes. EMBO J. 2009.
63. Hughes SH. The RCAS vector system. Folia Biol (Praha). 2004; 50:107-119.
64. Holland EC, Celestino J, Dai C, Schaefer L, Sawaya RE, Fuller GN. Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat Genet. 2000; 25:55-57.
65. Marumoto T, Tashiro A, Friedmann-Morvinski D, Scadeng M, Soda Y, Gage FH, Verma IM. Development of a novel mouse glioma model using lentiviral vectors. Nat Med. 2009; 15:110-116.
66. Pfeifer A, Brandon EP, Kootstra N, Gage FH, Verma IM. Delivery of the Cre recombinase by a self-deleting lentiviral vector: efficient gene targeting in vivo. Proc Natl Acad Sci U S A. 2001; 98:11450-11455.
67. Hambardzumyan D, Amankulor NM, Helmy KY, Becher OJ, Holland EC. Modeling Adult Gliomas Using RCAS/t-va Technology. Transl Oncol. 2009; 2:89-95.