
Introduction
Signal transduction cascades downstream of epidermal growth factor (EGF) receptor (EGFR) isoforms (e.g., EGFR & HER2) have been associated with breast cancer development and resistance to anticancer agents [1]. Among the signaling pathways downstream of these receptors, the Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways have been shown to regulate apoptosis and their deregulation is often implicated in malignant transformation [2-9]. Indeed, the PI3K p110 catalytic subunit gene (PIK3CA) is one of the most frequently mutated genes in breast cancer [10-11]. Oncogenic mutations often elicit a permanent cell cycle withdrawal, termed oncogene-induced-senescence (OIS), to eliminate pre-malignant cells before they acquire additional mutations [12].
Phosphatidylinositol (PI) (3,4)P2 and PI(3,4,5)P3 are produced by class 1A PI3Ks and recruit phosphoinositide dependent kinase-1 (PDK1) as well as Akt isoforms to the plasma membrane by interacting with their pleckstrin homology (PH) domains [13-17]. Colocalization of PDK1 with Akt proteins at the plasma membrane causes PDK1 to phosphorylate Akt proteins at a threonine residue (T308) [18-19] and a serine residue (S473). Activation of PDK1 and Akt by class 1A PI3Ks is negatively regulated by phosphatase and tensin homologue deleted on chromosome ten (PTEN) [4,6,8,16,20,21]. PTEN removes phosphate groups from PI(3,4)P2 and PI(3,4,5)P3 added by PI3K as well as from tyrosine phosphorylated proteins including focal adhesion kinase (FAK) and Shc [4,16,20,22]. PTEN also has important roles in the nucleus and can regulate genome stability [6, 21]. In some cases, there are complex interactions with the p53, PI3K/PTEN/Akt/mTOR pathways which determine whether senescence, quiescence or autophagy occurs in response to DNA damaging drugs [12, 15, 23-49].
Diverse mechanisms regulate PTEN expression [7, 50-53], including gene deletion, alterations in mRNA splicing, subcellular localization, epigenetic repression and protein:protein interactions [51]. There are numerous microRNAs (miRNAs) which target the PTEN gene to inhibit its expression [4,6,50,54,55]. Furthermore there is a pseudo PTEN gene [6] which serves as a decoy to bind and neutralize some of these miRNAs. PTEN interacts with p53 to influence key points in the regulation of cellular proliferation. Loss of these tumor suppressor interactions can lead to cancer or in some cases cellular senescence [12,53,56].
PTEN mutations occur in breast cancer at varying frequencies (5-21%). While PTEN is deleted in certain cancers, loss of heterozygosity (LOH) is a more common genetic event (30%) leading to decreased PTEN expression [50,57]. Additionally, PTEN expression can be decreased by promoter methylation [50]. In one study, 26% of primary breast cancers had low PTEN levels which correlated with lymph node metastases and poor prognoses [58]. Disruption of PTEN activity by various mechanisms frequently results in Akt activation.
Elevated Akt activity can have pleiotropic effects on cell growth, which include activation/inactivation of transcription factors controlling pivotal gene expression, inactivation of pro-apoptotic molecules by their phosphorylation and subsequent proteasomal degradation, or by regulating the efficiency of translation of mRNAs involved in growth. Akt can regulate translation directly by activating mammalian target of rapamycin (mTOR) or indirectly by inhibiting TSC2. Active mTOR then can phosphorylate ribosomal S6 kinase (p70S6K), which in turn regulates the efficiency of translation of certain mRNAs and also functions in a negative feedback loop to control Akt activity [7,16,59-61]. The activities of Akt and p70S6K along with other molecules are essential for the formation of the eIF4F translation complex. The eIF4F complex is necessary for the translation of mRNAs containing long 5’UTRs which are highly-structured and have an elevated G+C content. These “weak” mRNAs often encode proteins involved in oncogenesis and survival such as c-Myc, Mcl-1, cyclin-D, VEGF and survivin. Furthermore p70S6K has important roles in autophagy and cellular senescence [62-65].
Akt, mTOR and p70S6K activation have been associated with a more severe prognosis in breast and other cancers [66-73]. Targeting the PI3K/PTEN/Akt/mTOR pathway may prove effective therapy in a variety of cancers [9,16,74-76]. Indeed some studies have evaluated the effectiveness of targeting mTOR in PTEN-negative cells [69]. Elevated active Akt expression has been associated with both chemo- and hormonal resistance in breast cancer [67-68,77]. Cells expressing activated Akt may be more sensitive to mTOR inhibitors such as rapamycin and may increase the efficacy in combination with chemo- and hormonal-based therapies [6,69]. A distinct advantage of targeting mTOR with rapamycin is that it has been used for many years to treat organ transplant patients. Rapamycin is now being examined in treatment of certain cancers and in the prevention of aging and other diseases including AIDS [7,8,78]. Previously it was determined that mutated forms of Akt and PTEN can induce chemotherapeutic- and hormonal-based drug resistance in breast cancer [5,67,77]. PTEN mutants that eliminate the lipid phosphatase activity result in activated Akt expression and drug resistance, which can be reversed by the mTOR inhibitor rapamycin [5].
In addition to deregulation of the PI3K/mTOR pathway, the Ras/Raf/MEK/ERK pathway plays a critical role in cellular transformation and drug resistance. Growth factor/cytokine/mitogen activation recruits a Src homology 2 (Shc) domain containing adaptor protein to the C-terminus of the receptor [6-8,60]. Shc in turn recruits the growth factor receptor-bound protein 2 (Grb2) protein and the son of sevenless (SOS) homolog protein, resulting in the loading of membrane-bound Ras with GTP [6-8,60]. Additionally, Ras can be activated by growth factor receptor tyrosine kinases (RTK), such as insulin receptor (IR), and insulin-like growth factor one receptor (IGF1-R), via intermediates like insulin receptor substrate (IRS) proteins that bind Grb2 [6-8,60,79]. Ras:GTP can recruit Raf to the membrane where it becomes activated, likely via a Src-family tyrosine kinase [6-8,60,80,81]. Ras and Raf are members of multi-gene families and there are three Ras members (KRAS, NRAS and HRAS) and three Raf members (BRAF, CRAF (a.k.a Raf-1) and ARAF) [6-8,60]. Raf is responsible for serine/threonine phosphorylation of mitogen-extracellular activated protein kinase kinase-1 (MEK1) [6-8,60]. MEK1 phosphorylates extracellular signal-regulated kinase 1 and 2 (ERK1/2) at specific threonine and tyrosine residues [6-8,60]. Activated ERK1 and ERK2 serine/threonine kinases phosphorylate and activate a variety of substrates, including p90 ribosomal six kinase (p90Rsk1) [6-8,60,82,83]. ERK1/2 has many downstream (>60) and even upstream substrates. The Ras/Raf/MEK/ERK pathway plays pivotal roles in: differentiation, chemotherapeutic drug resistance, cellular senescence and metastasis [2,84-94]. Thus suppression of MEK and ERK activities will have profound effects on cell growth. While Ras is frequently mutated in human cancers overall, it is rarely mutated in breast cancer. However, this pathway can be activated by multiple other mechanisms including: mutations in upstream receptors, mutations in pathway components, subcellular localization and changes in the level of expression of pathway constituents and regulators [82-83,95-98].
We have previously demonstrated that activated HRAS, CRAF, AKT1 and mutated phosphatase-inactive PTEN genes confer drug resistance to breast cancer cells [2,3,5,99,100]. Cellular senescence is clearly a very important component in regulating cancer development and responding to DNA-damaging chemotherapeutics as well as anti-cancer dietary considerations [12,101-108]. However, it remains unclear as to how drug-resistant cells bypass the induction of cellular senescence.
In the following study, we examined the effects of activated PI3K/PTEN/Akt/mTOR and Ras/Raf/MEK/ERK pathways on the induction of cellular senescence in response to chemo-/hormonal therapy. Our results suggest that deregulation of PI3K/PTEN/Akt/mTOR, and to a lesser extent the Ras/Raf/MEK/ERK pathway, decreases drug-induced cellular senescence in response to chemo-/hormonal therapy.
Results
Doxorubicin and Tamoxifen Induce Cellular Senescence in MCF-7 Breast Cancer Cells
We first examined the ability of doxorubicin and 4 hydroxy-tamoxifen (4HT) to induce senescence in p53 wild type (WT) and estrogen receptor (ER) positive MCF-7 breast cancer cells [2,3,5,99,100]. Cells were plated in 6 well plates containing an etched, gridded coverslip on the bottom of the well at 5 x 106 cells/well. The MCF-7 cells adhered to the etched coverslips and essentially grew as colonies (Figure 1). After 6 days, the cells on the coverslips were processed for β-galactosidase staining as described [109]. Both doxorubicin (10-100 nM) and 4HT (50-1000 nM) induced senescence in MCF-7 cells in a dose-dependent fashion (Figure 1). At high concentrations of 4HT (1000 nM), MCF-7 cells did not grow and form colonies. Thus in subsequent experiments, lower doses of 4HT were used. Quantitation of the effects of doxorubicin and 4HT on MCF-7 cells is presented in Figures 2 and 4.
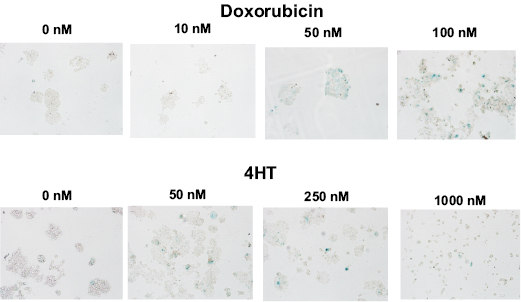
Figure 1: Effects of Doxorubicin and Tamoxifen (4HT) on the Induction of Cellular Senescence in MCF-7 Breast Cancer Cells. Top Panels, MCF-7 cells were treated with increasing concentration of doxorubicin for 6 days and then stained for senescence associated (SA) β-galactosidase (SA-β-gal). Bottom Panels, MCF-7 cells were treated with increasing concentration of 4HT for 6 days and then stained for SA-β-gal.
Effects of Doxorubicin on the Induction of Cellular Senescence in Doxorubicin-Resistant MCF-7 Cells
We also isolated MCF-7 cells with increased resistance to chemotherapeutic drugs by culturing the cells in medium containing 25 nM doxorubicin. These cells were named MCF7/DoxR. The MCF7/DoxR cells were approximately 5.7-fold more resistant to doxorubicin than MCF-7 cells as determined by MTT analysis.
The ability of MCF-7 and MCF7/DoxR cells to undergo cellular senescence was quantified from β-galactosidase-positive cells in the presence of doxorubicin (10, 50 and 100nM) for 6 days (Figure 2, Panels A and B). While MCF7/DoxR cells displayed lower levels of senescence compared to MCF-7 cells at 10 and 50 nM, similar levels of senescence was achieved at 100 nM, suggesting that drug-resistance cells have a diminished ability to arrest (Figure 2, Panels A and B). Photomicrographs of the induction of cellular senescence in MCF-7 and the doxorubicin-resistant cells in response to doxorubicin treatment are presented in Figure 3.
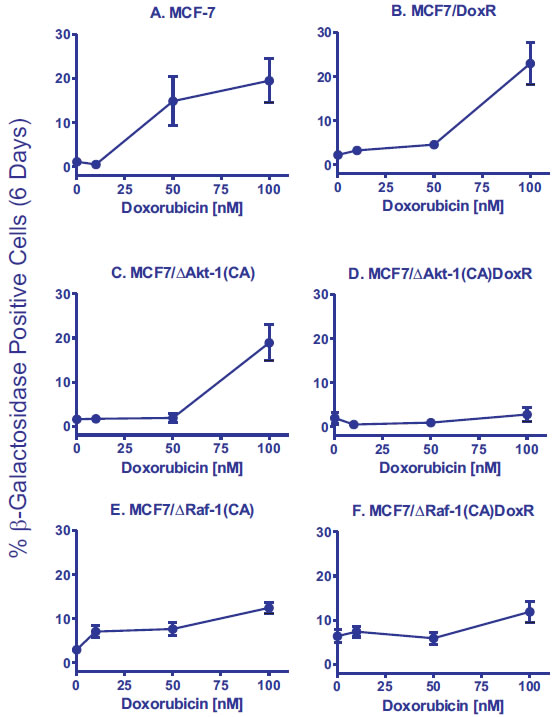
Figure 2: Effects of Activated Akt-1, Raf-1 and Selection for Doxorubicin-Resistance on the Induction of Cellular Senescence in Response to Different Concentrations of Doxorubicin in MCF-7 Derivatives. Quantification of the induction of cellular senescence in response to doxorubicin treatment. 10 individual images were taken of different areas of the cover slip for each cell line, for a total of 1,000-3,000 cells per condition. The total number of SA-β-gal positive and negative cells was determined on each image, and the percentages of SA-β-gal positive from each culture condition and cell type were then determined and averaged.
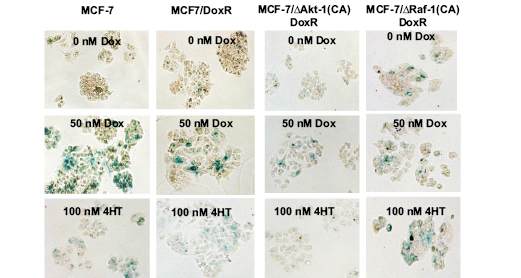
Figure 3: Effects of Doxorubicin and 4HT on the Induction of Cellular Senescence in MCF-7 Cells Containing Activated Akt-1, Raf-1 or Selected for Doxorubicin-Resistance. Photomicrographs of wild type MCF-7 and doxorubicin-resistant ∆Akt-1(CA) or ∆Raf-1(CA) cells after staining for SA-β-gal. Top row: untreated cells, middle row: cells treated with 50 nM doxorubicin, bottom row: cells treated with 100 nM 4HT.
Ectopic Akt-1 Expression Promotes Resistance of MCF-7 Cells to Induction of Senescence after Doxorubicin Treatment
Since the PI3K/PTEN/Akt/mTOR pathway is frequently altered in breast cancer and associated with drug resistance, we used a retrovirus to stably-transfect MCF-7 cells with a constitutively-active Akt-1 gene [ ∆Akt-1(CA)]. Expression of activated Akt-1 alone was sufficient to increase the IC50 for doxorubicin by 2-fold in MCF7/∆Akt-1(CA) [100]. Similar to MCF7/DoxR cells, expression of ∆Akt-1 reduced senescence 10-fold compared to MCF-7 cells when the cells were treated with 50 nM doxorubicin (Figure 2, Panels A and C).
We also isolated MCF7/∆Akt-1(CA) cells with increased resistance to chemotherapeutic drugs by culturing the cells in medium containing 25 nM doxorubicin. These cells were named MCF7/∆Akt-1(CA)DoxR. The MCF7/∆Akt-1(CA)DoxR were 5-fold more frequently recovered from MCF7/∆Akt-1(CA) than the MCF7/DoxR were from MCF-7 cell line. The MCF7/∆Akt-1(CA)DoxR cells were approximately 6.7-fold more resistant to doxorubicin than control MCF-7 cells [100].
Interestingly, constitutive activation of Akt-1 in the drug resistant cells [MCF7/ ∆Akt-1(CA)DoxR] resulted in the complete abrogation of drug-induced senescence at all doses examined (Panel D), suggesting an interplay between active Akt-1 and other resistance mechanisms acquired by the MCF7/DoxR cells. We have observed that MCF7/DoxR cells overexpress activated ERK, but not activated Akt when cultured in medium containing doxorubicin. In addition, we have seen that MCF7/∆Akt-1(CA) also overexpress activated ERK, suggesting the presence of both active pathways may serve to prevent drug-induced senescence.
Effects of Ectopic Raf-1 Expression on the Induction of Senescence
Since we observed elevated activated ERK1/2 expression in MCF7/DoxR cells, the activity of the Raf/MEK/ERK cascade was manipulated in MCF-7 cells in order to determine whether signals transduced by this pathway control the induction of drug-induced senescence. MCF-7 cells were stably transfected with a constitutively-active Raf-1 gene [Raf-1(CA)] using a retrovirus [2,3]. Expression of constitutively-active Raf-1 increased resistance to doxorubicin by 9-fold compared to parental MCF-7 cells [2,3].
Consistent with prior reports, activated Raf-1 expression increased baseline cellular senescence in the absence of stress by 3-fold, indicating that MCF-7 cells were undergoing oncogene-induced senescence (OIS) (Figure 2, Panels A and E). MCF7/∆Raf-1(CA) cells treated with 10 nM doxorubicin showed a 2.4-fold increase in cellular senescence, whereas similar treatments did not induce cellular senescence in either MCF-7 or MCF7/∆Akt-1(CA) cells (Figure 2, Panels A, C and E). Interestingly, increasing concentrations of doxorubicin in MCF7/∆Raf-1(CA) cells failed to induce senescence to as high levels as observed in MCF-7 or unselected MCF7/Akt-1(CA) cells.
We also isolated MCF7/∆Raf-1(CA)DoxR cells with increased resistance to chemotherapeutic drugs by culturing the cells in medium containing 25 nM doxorubicin. These cells are approximately 5-fold more resistant to doxorubicin than MCF7/∆Raf-1(CA) cells. The MCF7/∆Raf-1(CA)DoxR cells had a 5.8-fold higher baseline level of cellular senescence than MCF-7 cells (Figure 2, Panels A and F) and a 2.1-fold higher baseline level of cellular senescence than MCF7/∆Raf-1(CA) cells (Figure 2, Panels E & F). However, these MCF7/∆Raf-1(CA)DoxR cells displayed a similar induction of senescence in response to doxorubicin treatment as MCF7/∆Raf-1(CA) cells (Figure 2, Panels E & F).
Active Akt-1 Prevents the Induction of Cellular Senescence Following Tamoxifen Treatment
Since chemotherapies are commonly administered in combination with hormonal therapy in ER-positive breast cancers, we sought to determine if drug resistance and oncogenic signaling through Akt-1 or Raf-1 effects induction of cellular senescence in response to 4-hydroxy tamoxifen (4HT). MCF-7 or MCF7/DoxR cells expressing constitutively-active Akt-1 or Raf-1 were treated with 10, 50 and 100 nM 4HT for six days then stained for β-galactosidase activity (Figure 4). Photomicrographs of the induction of cellular senescence in MCF-7 and the doxorubicin-resistant cells in response to 4HT or doxorubicin treatment are presented in Figure 3.
Similar levels of cellular senescence were observed in MCF-7 and MCF7/DoxR (Panel A) after 4HT treatment, suggesting that mechanisms of drug resistance are independent of senescence inducted by 4HT. Interestingly, ∆Akt-1(CA) expression did not prevent cellular senescence induction by 4HT treatment in the unselected MCF7/∆Akt-1(CA) cells (Panel B), but potently suppressed senescence in doxorubicin-resistant MCF7/∆Akt-1(CA)DoxR cells (Panel B). In contrast, Raf-1 activation in doxorubicin-resistant MCF7/∆Raf-1(CA)DoxR cells did not suppress senescence by 4HT treatment (Panel C). Together, these results suggest that presence activated PI3K/PTEN/Akt/mTOR pathway in the presence of acquired drug resistant Raf/MEK/ERK pathway activation is sufficient to block drug-induced senescence to both doxorubicin and 4HT.
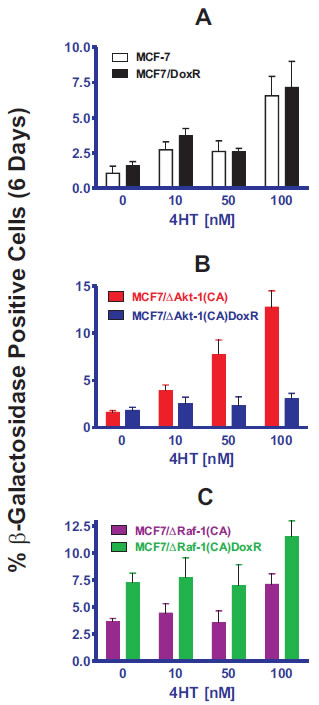
Figure 4: Effects of Activated Akt-1, Raf-1 and Selection for Doxorubicin-Resistance on the Induction of Cellular Senescence in Response to Different Concentrations of Tamoxifen in MCF-7 Derivatives. Quantification of the induction of cellular senescence in response to 4HT treatment. 10 individual images were taken of different areas of the cover slip for each cell line, for a total of 1,000-3,000 cells per condition. The total number of SA-β-gal positive and negative cells was determined on each image, and the percentages of SA-β-gal positive cells from each culture condition and cell type were then determined and averaged.
Discussion
In our studies, we determined the effects of the chemotherapeutic drug doxorubicin and the hormonal drug tamoxifen (4HT) on the induction of cellular senescence in MCF-7 and derivative cell lines which varied in their levels of activated Akt-1 or Raf-1 expression. Moreover, we also examined the induction of cellular senescence in cells which were selected for drug resistance by prolonged culture in medium containing 25 nM doxorubicin to determine whether drug resistance would alter the induction of senescence after either doxorubicin or 4HT treatment. An advantage of our investigation was that all cells had the same basic genetic background (MCF-7) and differed only in the levels of activated Akt-1 or Raf-1, or their resistance to doxorubicin.
A lower level of senescence was observed after 50 nM doxorubicin treatment of MCF7/DoxR than in MCF-7 cells. However, when the cells were treated with 100 nM doxorubicin, similar levels of cellular senescence were observed. These results demonstrated that the doxorubicin-resistant MCF7/DoxR wells were more resistant to the induction of senescence after doxorubicin treatment than unselected MCF-7 cells. However, the MCF7/DoxR doxorubicin-resistant phenotype did not prevent the induction of senescence in response to 4HT treatment. This indicates that acquired resistance to DNA damage-induced senescence does not protect against senescence induced by hormone blockade, and thus the two are likely to occur through independent signaling cascades.
While previous studies have shown that overexpression of either activated Akt-1 or Raf-1 results in resistance to doxorubicin [2,3,99,100], in our current work activated Akt-1 and Raf-1 expression have different effects on the induction of senescence after doxorubicin treatment. Overexpression of activated Akt-1 resulted in decreased induction of senescence compared with MCF-7 cells after 50 nM doxorubicin treatment. Drug-resistant MCF7/∆Akt-1[CA]DoxR cells were even more resistant to the induction of senescence than either MCF7/∆Akt-1[CA], MCF7/DoxR or MCF-7 cells (Figure 2). In contrast, a higher background level of cellular senescence was observed in untreated MCF7/∆Raf-1(CA) and MCF7/∆Raf-1(CA)DoxR cells compared to MCF-7 cells. However, the levels of cellular senescence observed after 100 nM doxorubicin treatment of MCF7/∆Raf-1(CA) and MCF7/∆Raf-1(CA)DoxR cells were not as high as observed in MCF-7 cells, suggesting that while there were higher levels of constitutive senescence in MCF7/∆Raf-1(CA) and MCF7/∆Raf-1(CA)DoxR cells, they were blocked in their ability to undergo as much senescence as MCF-7 cells.
MCF-7 cells contain WT p53 and doxorubicin induces p53 in MCF-7 cells [99]. Lower levels of p53 and downstream p21Cip-1 were induced in doxorubicin-resistant MCF-7 cells which also contain activated Akt-1[99]. p53 plays key roles in the regulation of cellular senescence [38-40,43-46,110-120], DNA damage response [121-123], cell cycle progression [124-143], chromosome dynamics [144-146], apoptosis [147-151], sensitivity to chemotherapy [152-154], radiotherapy [107,155] tumorigenesis and metastasis [53,156-158], metabolism [159-161], cellular redox [41,162], hypoxia [61,75,136,163,164], autophagy [49,165,166] and aging [167] as well as other important biological processes. Often some of the purported functions of p53 are overlapping and even sometimes contradictory. p53 is regulated by other transcription factors [168,169], the proteins ATM [170], MDM2 and MDMX [174-180] and other interacting proteins [181-187] and miRNAs [171-173]. Small molecule inhibitors to proteins such as MDM2 which negatively regulate p53 have been developed [56,188-190]. Akt-1 can phosphorylate MDM-2 which can promote p53 destabilization. Decreased p53 and p21Cip-1 levels may result in less cellular senescence in response to doxorubicin in the cells containing activated Akt-1 than in wild type MCF-7 cells.
The target genes of p53 and other members of the p53 family are clearly important in regulating many diverse processes [191-200]. p21Cip1 can activate pRb which can regulate entry into replicative senescence [12,201]. p53 also suppresses mTOR [38,40,44], however Akt can suppress the effects of p53 documenting the complexity of these pathways in how they regulate senescence induction [202-204]. miRNAs have also been shown to regulate critical genes involved in senescence such as pRb, p53, and PTEN [51,171-173,190,205]. Integrating these complex and often redundant interactions into a model by which p53 regulates cellular senescence in MCF-7 cells remains an ongoing process.
Recently it has been demonstrated by Blagosklonny and colleagues that the mTOR complex plays a critical role in regulating senescence and quiescence [30-32,38,40,206]. Inhibition of mTOR by rapamycin treatment in the presence of Nutlin-3A, will prevent the induction of senescence and result in p53-mediated quiescence [38, 40]. Interestingly, they also observed that inhibiting mTOR with rapamycin resulted in increased activation of “upstream” Akt [40]. This induction of Akt could have resulted from a shift in the equilibrium between mTORC1 and mTORC2, leading to higher levels of mTORC2 which resulted in Akt activation that prevented the induction of senescence by a mTORC1-independent mechanism [59].
Other studies by Blagosklonny’s group demonstrated that knock down of TSC2, a negative upstream regulator of mTOR, could convert quiescent cells into senescent cells in the presence of Nutlin-3A [38]. Nutlin-3A is a MDM2 inhibitor, which results in p53 induction (accumulation). Inhibiting TSC2 resulted in increased mTORC1 activity which switched the cells from the quiescent into the senescent phenotype.
The effects of Akt on the induction of cellular senescence remain controversial. Often abnormal oncogene expression or tumor suppressor gene deletion (PTEN, NF1 or RB) has been associated with “oncogene induced senescence” (OIS) which can be viewed as a protective mechanism to prevent tumor development [80,101]. However, recently it has been observed that Akt activation can suppress the senescence induced by mutant Ras signaling to promote tumorigenesis [207]. In this system, mutations at PI3K/Akt accelerated tumorigenesis potentially by bypassing the senescence induced by the activated Ras.
Activating mutations at oncogenes in the EGFR/Ras/PI3K/Akt pathway do not always result in OIS. AKT1, FGFR3, PIK3CA, KRAS, HRAS, and EGFR mutations have been found in some benign Seborrheic keratosis tumors which are not associated with increased cellular senescence. Interesting mutations in tumor suppressor genes TSC1 and PTEN were not observed in this study [208].
Inhibition of Akt activity can also have effects resulting in the induction of senescence. Pharmacological suppression of Akt activity in combination with vitamin D3 can synergistically inhibit growth and result in the induction of senescence in prostate cancer cells [209]. This research demonstrates that quenching the effects of Akt can result in the induction of senescence. Expression of an inducible Akt gene suppressed temozolomide-induced Chk2 activation and G2 arrest and senescence in a glioma cell line model [210]. These studies indicate the protective effects of Akt with regards to certain chemotherapeutic drug treatments and support our findings that constitutive activation of Akt-1 can prevent the induction of senescence in MCF-7 cells after doxorubicin treatment.
Previously we have determined that there are higher levels of activated Akt-1 in the drug resistant cell line MCF7/ΔAkt-1ER*(Myr+)DoxR compared with MCF7/ΔAkt-1ER*(Myr+) cells which had not been selected for doxorubicin- or hormonal-resistance [99]. Growth in medium containing either doxorubicin or 4HT selects for ∆Akt-1(CA)-infected cells, as higher levels of Akt-1 expression provides a selective growth advantage in the presence of these drugs [99].
We have also shown that introduction of mutated forms of PTEN into MCF-7 cells which lacked lipid and protein phosphatase activity also suppressed the ability of doxorubicin to induce cellular senescence [5]. These cells which express the PTEN mutants were also more drug resistant than MCF-7 cells and also overexpressed activated Akt [5]. The PI3K/PTEN/Akt/mTOR and Raf/MEK/ERK pathways differed in their abilities to modulate the induction of cellular senescence in response to doxorubicin treatment in breast cancer cells as activated Akt-1 impeded senescence while activated Raf-1 did not, similar to the results of the present study.
Doxorubicin induces the Raf/MEK/ERK pathway in MCF-7 cells and higher levels of active ERK are detected in MCF7/DoxR cells than in drug sensitive MCF-7 cells. ERK can in some cases phosphorylate p53 (or additional proteins) and alter its activity. However, this increased ERK expression does not appear to prevent the induction of cellular senescence. In fact, a higher as a higher baseline level of cellular senescence was detected in MCF7/∆Raf-1(CA) cells. Thus it can be argued that there is probably an additional modification(s) besides increased ERK expression in MCF7/DoxR which results in the decreased induction of senescence after doxorubicin treatment. While both MCF7/∆Akt-1(CA) and MCF7/∆Raf-1(CA) cells display increased resistance to doxorubicin compared with MCF-7 cells [2,3,100], they varied in terms of induction of senescence after doxorubicin treatment. Thus the genetic mechanisms responsible for chemotherapeutic drug resistance and senescence induction are different.
The ability of 4HT to induce cellular senescence in breast cancer is not well documented. 4HT likely induces reactive oxygen species (ROS) which in turn cause DNA damage and subsequently cellular senescence [211-213]. Similar levels of cellular senescence were observed in MCF-7 and MCF7/DoxR cells when they were cultured in medium containing 4HT. Thus the genetic modifications that occur in MCF7/DoxR cells which allow them to grow in medium containing 25 nM doxorubicin are not sufficient to prevent the induction of senescence induced by 4HT.
Induction of cellular senescence in response to 4HT differed in these models which expressed activated Akt-1(CA) or Raf-1(CA). In the MCF7/∆Akt-1(CA) cells that had not been selected in doxorubicin, there was more cellular senescence than in the MCF-7 cells in response to 4HT treatment (compare in Figure 4 panels A and B). In contrast, with the MCF7/Akt-1(CA)DoxR cells that had been selected in doxorubicin, less cellular senescence in response to 4HT treatment was observed than in all the other cell lines examined. These results suggest that doxorubicin-selected drug resistant cells which overexpress activated Akt-1 are resistant to the induction of senescence upon 4HT as well as doxorubicin treatment. As stated previously, the doxorubicin-resistant cells express higher levels of activated Akt-1 than cells which have not been selected in doxorubicin. This higher level of Akt-1 confers increased resistance to 4HT [99,100].
Activated Raf-1 did not appear to suppress the induction of cellular senescence in response to 4HT treatment. Although in general, relatively constant levels of cellular senescence were observed in MCF7/∆Raf-1(CA) and MCF7/∆Raf-1(CA)DoxR cells in response to 4HT treatment as was seen after doxorubicin treatment of these cells. Taken together with the results observed with MCF7/DoxR cells, which express high levels of activated ERK, it is likely that increased levels of the Raf/MEK/ERK pathway does not inhibit the induction of cellular senescence induced by either 4HT or doxorubicin.
In some cases, the Ras/Raf/MEK/ERK pathway may induce cellular senescence [27,214-218]. Ras can activate both the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways [6-8,59,60]. Other investigators have demonstrated that sustained Ras activation in endothelial cells can result in autonomous growth and senescence bypass and alter the differentiation status [219]. However, this altered phenotype was demonstrated to be regulated by the PI3K/Akt pathway.
These results have clinical significance as the PI3K/PTEN/Akt/mTOR pathway is often activated in breast cancer by mutations at PIK3CA and dysregulation of PTEN. Thus this pathway and downstream substrates such as the transcription factor Twist are critically involved in breast cancer and are targets for improved therapy [220-222]. Furthermore, drug resistance frequently develops in breast cancer after chemo- or hormonal-based therapies. Akt is frequently activated by upstream PIK3CA or PTEN mutations or gene silencing. PTEN can be mutated or silenced by various mechanisms in human cancer. Mutations occur which either delete the PTEN gene or alter its activity. These mutations which result in activated Akt expression may have effects on the induction of cellular senescence in response to chemotherapeutic drugs currently used to treat breast cancer patients. Our studies also indicate that just because a cell is drug resistant, that does not mean that it will be resistant to the induction of cellular senescence by a chemotherapeutic drug. Doxorubicin induced cell cycle arrest in the G2/M phase in both MCF7/∆Raf-1 and MCF7/Akt-1 cells, yet these cells differed in the extent of cellular senescence after doxorubicin treatment. Clearly cell cycle regulatory proteins play key roles in regulation of cellular senescence [223]. Other studies have shown that certain proteins such as p21Cip-1 are required in some cells for the induction of senescence but not for cell cycle arrest in response to HDAC inhibitors such as sodium butyrate [224]. Together these studies point to the complexities of drug resistance and cellular senescence.
Materials and Methods
Cell Culture
MCF-7 cells were obtained from the American Type Culture Collection (ATCC) (Manassas, VA). Cell culture medium for MCF-7 cells consisted of Roswell Park Memorial Institute-1640 (RPMI 1640) medium (Invitrogen, Carlsbad, CA) supplemented with 10% (v/v) heat inactivated fetal bovine serum (FBS) as described [2].
Transfection of MCF-7 cells with Akt-1 and Raf-1 Constructs
5 x 105 MCF-7 cells were plated into 6-well cell culture plates (BD Biosciences, Mountainview, CA) and one day later infected with a retrovirus containing the various plasmid DNAs as described [3,5,99,100,109]. The nomenclature of the transfected cells is MCF7/ followed by the name of the introduced plasmid DNA. Stably transfected/infected cells were isolated by culture in medium containing 2 mg/ml G418 (Geneticin, Invitrogen).
Cellular Senescence Assay
Senescent cells were identified by a senescence associated (SA) β-galactosidase (SA-β-gal) assay as described [109].
Acknowledgements
This work was supported in part by a grant from the US National Institutes of Health to JAM (R01CA098195).
References
1. Shelton JG, Steelman LS, Abrams SL, Bertrand FE, Franklin RA, McMahon M, McCubrey JA. The epidermal growth factor receptor gene family as a target for therapeutic intervention in numerous cancers: what’s genetics got to do with it? Expert Opin Ther Targets. 2005;9:1009-1030.
2. Weinstein-Oppenheimer CR, Henriquez-Roldan CF, Davis JM, Navolanic PM, Saleh OA, Steelman LS, Franklin RA, Robinson PJ, McMahon M, McCubrey JA. Role of the Raf signal transduction cascade in the in vitro resistance to the anticancer drug doxorubicin. Clin Cancer Res. 2001;7:2898-2907.
3. Davis JM, Navolanic PM, Weinstein-Oppenheimer CR, Steelman LS, Hu W, Konopleva M, Blagosklonny MV, McCubrey JA. Raf-1 and Bcl-2 induce distinct and common pathways that contribute to breast cancer drug resistance. Clin Cancer Res. 2003; 9:1161-1170.
4. Steelman LS, Bertrand FE, McCubrey JA. The complexity of PTEN: mutation, marker and potential target for therapeutic intervention. Expert Opin Ther Targets. 2004;8:537-550.
5. Steelman LS, Navolanic PN, Sokolosky M, Taylor JR, Lehmann BD, Chappell WH, Abrams SL, Wong EW, Stadelman K, Terrian DM, Leslie N, Martelli AM, Stivala F, Libra M, Franklin RA, McCubrey JA. Suppression of PTEN function increases breast cancer chemotherapeutic drug resistance while conferring sensitivity of mTOR inhibitors. Oncogene. 2008;27:4086-4095.
6. McCubrey JA, Steelman LS, Kempf CR, Chappell W, Abrams SL, Stivala F, Malaponte G, Nicoletti F, Libra M, Bäsecke J, Maksimovic-Ivanic D, Mijatovic S, Montalto G, Cervello M, Cocco L, Martelli AM. Therapeutic Resistance Resulting from Mutations in Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR Signaling Pathways. J Cell Physiol. 2011; doi: 10.1002/jcp.22647.
7. Steelman LS, Chappell WH, Abrams SL, Kempf CR, Long J, Laidler P, Mijatovic S, Maksimovic-Ivanic D, Stivala F, Mazzarino MC, Donia M, Fagone P, Malaponte G, Nicoletti F, Libra M, Milella M, Tafuri A, Bonati A, Bäsecke J, Cocco L, Evangelisti C, Martelli AM, Montalto G, Cervello M, McCubrey JA. Roles of the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways in controlling growth and sensitivity to therapy-implications for cancer and aging. Aging. 2011;3:192-222.
8. Chappell WH, Steelman LS, Long JM, Kempf CR, Abrams SL, Franklin RA, Basecke J, Stivala F, Donia M, Fagone P, Malaponte G, Mazzarino MC, Nicoletti F, Libra M, Maksimovic-Ivanic D, Miatovic S, Montalto G, Cervello M, Laidler P, Milella M, Tafuri A, Bonati A, Evangelisti C, Cocco L, Martelli AM, McCubrey JA. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR inhibitors: rational and importance to inhibiting these pathways in human health. Oncotarget. 2011;2:135-164.
9. Xu R, Spencer VA, Groesser DL, Bissell MJ. Laminin regulates PI3K basal localization and activation to sustain STAT5 activation. Cell Cycle. 2010;9:4315-4322.
10. Ligresti G, Militello L, Steelman LS, Cavallaro A, Basile F, Nicoletti F, Stivala F, McCubrey JA, Libra M. PIK3CA mutations in human solid tumors. Cell Cycle. 2009:8:1352-1358.
11. Zhao L, Vogt PK. Hot-spot mutations in p110alpha of phosphatidylinositol 3-kinase (pI3K): differential interactions with the regulatory subunit p85 and with RAS. Cell Cycle. 2010;9:596-600.
12. Haferkamp S, Tran SL, Becker TM, Scurr LL, Kefford RF, Rizos H. The relative contributions of the p53 and pRb pathways in oncogene-induced melanocyte senescence. Aging. 2009;1:542-556.
13 Franke TF, Kaplan DR, Cantley LC, Toker A. Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science. 1997;275:665-668.
14. Frech M, Andjelkovic M, Ingley E, Reddy KK, Falck JR, Hemmings. High affinity binding of inositol phosphates and phosphoinositides to the pleckstrin homology domain of RAC/protein kinase B and their influence on kinase activity. J Biol Chem. 1997;272:8474-8881.
15. Brauer PM, Tyner AL. RAKing in AKT: a tumor suppressor function for the intracellular tyrosine kinase FRK. Cell Cycle. 2009;8:2728-2732.
16. Martelli AM, Evangelisti C, Chiarini F, Grimaldi C, Cappellini A, Ognibene A, McCubrey JA. The emerging role of the phosphatiylinositol 3-kinase/Akt/mammalian target of rapamycin signaling network in normal myelopoiesis and leukemogensis. Biochim Biophys Act. 2010;1803:991-1002.
17. Saudemont A, Colucci F. PI3K signaling in lymphocyte migration. Cell Cycle. 2009;8:3307-3310.
18. Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol. 1997;7:261-269.
19. Stokoe D, Stephens LR, Copeland T, Gaffney PR, Reese CB, Painter GF, Holmes AB, McCormick F, Hawkins PT. Dual role of phosphatidylinositol-3,4,5-trisphosphate in the activation of protein kinase B. Science. 1997;277:567-570.
20. Palomero T, Dominguez M, Ferrando AA. The role of the PTEN/AKT pathway in NOTCH1-induced leukemia. Cell Cycle. 2008;7:965-970.
21. Gupta A, Yang Q, Pandita RK, Hunt CR, Xiang T, Misri S, Zeng S, Pagan J, Jeffery J, Puc J, Kumar R, Feng Z, Powell SN, Bhat A, Yaguchi T, Wadhwa R, Kaul SC, Parsons R, Khanna KK, Pandita TK. Cell cycle checkpoint defects contribute to genomic instability in PTEN deficient cells independent of DNA DSB repair. Cell Cycle. 2009;8:2198-2210.
22. Zheng Y, Lu Z. Paradoxical roles of FAK in tumor cell migration and metastasis.
Cell Cycle. 2009; 8: 3474-3479.
23. Gonzalez E, McGraw TE. The Akt kinases: isoform specificity in metabolism and cancer. Cell Cycle. 2009;8:2502-2508.
24. Narasimhan SD, Mukhopadhyay A, Tissenbaum HA. InAKTivation of insulin/IGF-1 signaling by dephosphorylation. Cell Cycle. 2009;8:3878-3884.
25. Masuda K, Marasa B, Martindale JL, Halushka MK, Gorospe M. Tissue- and age-dependent expression of RNA-binding proteins that influence mRNA turnover and translation Aging. 2009;1:681-698.
26. Hart JR, Vogt PK. Phosphorylation of AKT: a mutational analysis. Oncotarget. 2011;2: 467-476.
27. Blagosklonny MV. Revisiting the antagonistic pleiotropy theory of aging: TOR-driven program and quasi-program. Cell Cycle. 2010;9:3151-3156.
28. Marciniak-Czochra A, Stiehl T, Wagner W. Modeling of replicative senescence in hematopoietic development Aging. 2009;1:723-732.
29. Harikumar KB, Aggarwal BB. Resveratrol: a multitargeted agent for age-associated chronic diseases. Cell Cycle. 2008;7:1020-1035.
30. Demidenko ZN, Blagosklonny MV. At concentrations that inhibit mTOR, resveratrol suppresses cellular senescence. Cell Cycle. 2009; 8:1901-1904.
31. Demidenko ZN, Shtutman M, Blagosklonny MV. Pharmacologic inhibition of MEK and PI-3K converges on the mTOR/S6 pathway to decelerate cellular senescence. Cell Cycle. 2009;8:1896-1900.
32. Demidenko ZN, Zubova SG, Bukreeva EI, Pospelov VA, Pospelova TV, Blagosklonny MV. Rapamycin decelerates cellular senescence. Cell Cycle. 2009;8:1888-1895.
33. Blagosklonny MV. Aging-suppressants: Cellular senescence (hyperactivation) and its pharmacologic deceleration. Cell Cycle. 2009;8:1883-1887.
34. Yang WL, Wu CY, Wu J, Lin HK. Regulation of Akt signaling activation by ubiquitination. Cell Cycle. 2010;9:487-497.
35. Lal MA, Bae D, Camilli TC, Patierno SR, Ceryak S. AKT1 mediates bypass of the G1/S checkpoint after genotoxic stress in normal human cells. Cell Cycle. 2009;8:1589-1602.
36. Jin J, Wang GL, Timchenko L, Timchenko NA. GSK3beta and aging liver. Aging. 2009;1:582-585.
37. Gan B, DePinho RA. mTORC1 signaling governs hematopoietic stem cell quiescence. Cell Cycle. 2009;8:1003-1006.
38. Korotchkina LG, Leontieva OV, Bukreeva EI, Demidenko ZN, Gudkov AV, Blagosklonny MV. The choice between p53-induced senescence and quiescence is determined in part by the mTOR pathway. Aging. 2010;2:344-352.
39. Huang B, Vassilev LT. Reduced transcriptional activity in the p53 pathway of senescent cells revealed by the MDM2 antagonist nutlin-3. Aging. 2009;1:845-854.
40. Leontieva OV, Blagosklonny MV. DNA damaging agents and p53 do not cause senescence in quiescent cells, while consecutive re-activation of mTOR is associated with conversion to senescence. Aging. 2010;2:924-935.
41. Vigneron A, Vousden KH. p53, ROS and senescence in the control of aging. Aging. 2010;2:471-474.
42. Hodny Z, Hubackova S, Bartek J. Cytokines shape chemotherapy-induced and ‘bystander’ senescence. Aging. 2010;2:375-376.
43. Lane DP, Verma C, Fang CC. The p53 inducing drug dosage may determine quiescence or senescence. Aging. 2010;2:748.
44. Galluzzi L, Kepp O, Kroemer G. TP53 and MTOR crosstalk to regulate cellular senescence. Aging. 2010;2:535-537.
45. Mallette FA, Calabrese V, Ilangumaran S, Ferbeyre G. SOCS1, a novel interaction partner of p53 controlling oncogene-induced senescence. Aging. 2010;2:445-452.
46. Schug, TT. mTOR favors senescence over quiescence in p53-arrested cells. Aging. 2010;2:327-328.
47. Narita M. Quality and quantity control of proteins in senescence Aging. 2010;2:311–314.
48. Kandouz M, Haidara K, Zhao J, Brisson ML, Batist G. The EphB2 tumor suppressor induces autophagic cell death via concomitant activation of the ERK1/2 and PI3K pathways. Cell Cycle. 2010;9:398-407.
49. Galluzzi L, Morselli E, Kepp O, Maiuri MC, Kroemer G. Defective autophagy control by the p53 rheostat in cancer. Cell Cycle. 2010;9:250-255.
50. Leslie NR, Foti M. Not in my genes: non-genomic loss of PTEN function in cancer. Trends in Pharmacological Sciences. 2011;32:131-140.
51. Diaz-Meco MT, Abu-Baker S. The Par-4/PTEN connection in tumor suppression. Cell Cycle. 2009;8:2518-2522.
52. Shan W, Liu J. Epithelial ovarian cancer: focus on genetics and animal models. Cell Cycle. 2009;8:731-735.
53. Poon JS, Eves R, Mak AS. Both lipid- and protein-phosphatase activities of PTEN contribute to the p53-PTEN anti-invasion pathway. Cell Cycle. 2010;9:4450-4454.
54. Sayed D, Abdellatif M. AKT-ing via microRNA. Cell Cycle. 2010;9:3213-3217.
55. Shalgi R, Brosh R, Oren M, Pilpel Y, Rotter V. Coupling transcriptional and post-transcriptional miRNA regulation in the control of cell fate. Aging. 2009;1:762-770.
56. Cipriano R, Patton JT, Mayo LD, Jackson MW. Inactivation of p53 signaling by p73 or PTEN ablation results in a transformed phenotype that remains susceptible to Nutlin-3 mediated apoptosis. Cell Cycle. 2010;9:1373-1379.
57. Singh B, Ittmann MM, Krolewski JJ. Sporadic breast cancers exhibit loss of heterozygosity on chromosome segment 10q23 close to the Cowden disease locus. Genes Chrom Cancer. 1998;21:166-171.
58. Tsutsui S, Inoue H, Yasuda K, Suzuki K, Higashi H, Era S, Mori M. Reduced expression of PTEN protein and its prognostic implications in invasive ductal carcinoma of the breast. Oncology. 2005;68:398-404.
59. Martelli AM, Evangelisti C, Chappell W, Abrams SL, Bäsecke J, Stivala F, Donia M, Fagone P, Nicoletti F, Libra M, Ruvolo V, Ruvolo P, Kempf CR, Steelman LS, McCubrey JA. Targeting the translational apparatus to improve leukemia therapy: roles of the PI3K/PTEN/Akt/mTOR pathway. Leukemia. 2011;25:1064-1079 .
60. Steelman LS, Franklin RA, Abrams SL, Chappell W, Kempf CR, Bäsecke J, Stivala F, Donia M, Fagone P, Nicoletti F, Libra M, Ruvolo P, Ruvolo V, Evangelisti C, Martelli AM, McCubrey JA. Roles of the Ras/Raf/MEK/ERK pathway in leukemia therapy. Leukemia. 2011;25:1080-1094.
61. Rodriguez-Jimenez FJ, Moreno-Manzano V, Mateos-Gregorio P, Royo I, Erceg S, Murguia JR, Sanchez-Puelles JM. FM19G11: A new modulator of HIF that links mTOR activation with the DNA damage checkpoint pathways. Cell Cycle. 2010;9:2803-2813.
62. Armour SM, Baur JA, Hsieh SN, Land-Bracha A, Thomas SM, Sinclair DA. Inhibition of mammalian S6 kinase by resveratrol suppresses autophagy. Aging. 2009;1:515-528.
63. Shahbazian D, Parsyan A, Petroulakis E, Hershey J, Sonenberg N. eIF4B controls survival and proliferation and is regulated by proto-oncogenic signaling pathways. Cell Cycle. 2010;9:4106-4109.
64. Tamburini J, Green AS, Chapuis N, Bardet V, Lacombe C, Mayeux P, Bouscary D. Targeting translation in acute myeloid leukemia: a new paradigm for therapy? Cell Cycle. 2009;8:3893-3899.
65. Krymskaya VP, Goncharova EA. PI3K/mTORC1 activation in hamartoma syndromes: therapeutic prospects. Cell Cycle. 2009;8:403-413.
66. Bose S, Crane A, Hibshoosh H, Mansukhani M, Sandweis L, Parsons R. Reduced expression of PTEN correlates with breast cancer progression. Hu Pathol. 2002;33:405-409.
67. Clark AS, West K, Streicher S, Dennis PA. Constitutive and inducible Akt activity promotes resistance to chemotherapy, trastuzumab, or tamoxifen in breast cancer cells. Mol Cancer Ther. 2002;1:707-717.
68. Tokunaga E, Kimura Y, Mashino K, Oki E, Oki E, Kataoka A, Ohno S, Morita M, Kakeji Y, Baba H, Maehara Y. Activation of PI3K/Akt signaling and hormone resistance in breast cancer. Breast Cancer. 2006;13:137-144.
69. deGraffenried LA, Friedrichs WE, Russell DH, Donzis EJ, Middleton AK, Silva JM, Roth RA, Hidalgo M. Inhibition of mTOR activity restores tamoxifen response in breast cancer cells with aberrant Akt Activity. Clin Cancer Res. 2004;10:8059-8067.
70. Nagata Y, Lan KH, Zhou X, Tan M, Esteva FJ, Sahin AA, Klos KS, LI P, Monia BP, Nguyen NT, Hortobagyi GN, Hung MC, YuD. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004;6:117-127.
71. Lin HJ, Hsieh FC, Song H, Lin J. Elevated phosphorylation and activation of PDK-1/AKT pathway in human breast cancer. Br J Cancer. 2005;12:1372-1381.
72. Klos KS, Wyszomierski SL, Sun M, Tan M, Zhou X, Li P, Yang W, Yin G, Hittelman WN, Yu D. ErbB2 increases vascular endothelial growth factor protein synthesis via activation of mammalian target of rapamycin/p70S6K leading to increased angiogenesis and spontaneous metastasis of human breast cancer cells. Cancer Res. 2006;66:2028-2037.
73. Yim EK, Siwko S, Lin SY. Exploring Rak tyrosine kinase function in breast cancer. Cell Cycle. 2009;8:2360-2364.
74.Vazquez-Martin A, Oliveras-Ferraros C, Menendez JA. The antidiabetic drug metformin suppresses HER2 (erbB-2) oncoprotein overexpression via inhibition of the mTOR effector p70S6K1 in human breast carcinoma cells. Cell Cycle. 2009;8:88-96.
75. Matthew EM, Hart LS, Astrinidis A, Navaraj A, Dolloff NG, Dicker DT, Henske EP, El-Deiry WS. The p53 target Plk2 interacts with TSC proteins impacting mTOR signaling, tumor growth and chemosensitivity under hypoxic conditions. Cell Cycle. 2009;8:4168-4175.
76. Kim E, Guan KL. RAG GTPases in nutrient-mediated TOR signaling pathway.
Cell Cycle. 2009;8:1014-1018.
77. Faridi J, Wang L, Endemann G, Roth RA. Expression of constitutively active Akt-3 in MCF-7 breast cancer cells reverses the estrogen and tamoxifen responsivity of these cells in vivo. Clin Cancer Res. 2003;9:2933-2939.
78. Donia M, McCubrey JA, Bendtzen K, Nicoletti F. Potential use of rapamycin in HIV infection. British Journal of Clinical Pharmacology. 2010;70:784-793.
79. Hayashi K, Shibata K, Morita T, Iwasaki K, Watanabe M, Sobue K. Insulin receptor substrate-1/SHP-2 interaction, a phenotype-dependent switching machinery of insulin-like growth factor-I signaling in vascular smooth muscle cells. J Biol Chem. 2004;279:40807-40818.
80. Dimauro T. David G. Ras-induced senescence and its physiological relevance in cancer. Current Cancer Drug Targets. 2010;10:869-876.
81. Marais R, Light Y, Paterson HF, Marshall CJ. Ras recruits Raf-1 to the plasma membrane for activation by tyrosine phosphorylation. EMBO J. 1995;14:3136-3145.
82. Lefloch R, Pouyssegur J, Lenormand P. Total ERK1/2 activity regulates cell proliferation. Cell Cycle. 2009;8:705-711.
83. Mebratu Y, Tesfaigzi Y. How ERK1/2 Activation Controls Cell ... Cell Death : Is Subcellular Localization the Answer? Cell Cycle. 2009;8:1168-1175.
84. Steelman LS, Abrams SL, Shelton JG, Chappell WH, Bäsecke J, Stivala F, Donia M, Nicoletti F, Libra M, Martelli AM, McCubrey JA. Dominant roles of the Raf/MEK/ERK pathway in cell cycle progression, prevention of apoptosis and sensitivity to chemotherapeutic drugs. Cell Cycle. 2010;9:1629-1638.
85. Abrams SL, Steelman LS, Shelton JG, Wong ET, Chappell WH, Bäsecke J, Stivala F, Donia M, Nicoletti F, Libra M, Martelli AM, McCubrey JA. The Raf/MEK/ERK pathway can govern drug resistance, apoptosis and sensitivity to targeted therapy. Cell Cycle. 2010; 9:1781-1791.
86. Abrams SL, Steelman LS, Shelton JG, Chappell WH, Bäsecke J, Stivala F, Donia M, Nicoletti F, Libra M, Martelli AM, McCubrey JA. Enhancing therapeutic efficacy by targeting non-oncogene addicted cells with combinations of signal transduction inhibitors and chemotherapy. Cell Cycle. 2010;9:1839-1846.
87. Campaner S, Doni M, Verrecchia A, Faga G, Bianchi L, Amati B. Myc, Cdk2 and cellular senescence: Old players, new game. Cell Cycle. 2010;9:3655-3661.
88. Wood MD, Sanchez Y. Deregulated Ras signaling compromises DNA damage checkpoint recovery in S. cerevisiae. Cell Cycle. 2010;9:3353-3363.
89. Chawla R, Procknow JA, Tantravahi RV, Khurana JS, Litvin J, Reddy EP. Cooperativity of Cdk4R24C and Ras in melanoma development. Cell Cycle. 2010;9:3305-3314.
90. Demarchi F, Cataldo F, Bertoli C, Schneider C. DNA damage response links calpain to cellular senescence. Cell Cycle. 2010;9:755-760.
91. Evdokimova V, Tognon C, Ng T, Sorensen PH. Reduced proliferation and enhanced migration: two sides of the same coin? Molecular mechanisms of metastatic progression by YB-1. Cell Cycle. 2009;8:2901-2906.
92. Wang S, Koromilas AE. Stat1 is an inhibitor of Ras-MAPK signaling and Rho small GTPase expression with implications in the transcriptional signature of Ras transformed cells. Cell Cycle. 2009;8:2070-2079.
93. Safina AF, Varga AE, Bianchi A, Zheng Q, Kunnev D, Liang P, Bakin AV. Ras alters epithelial-mesenchymal transition in response to TGFbeta by reducing actin fibers and cell-matrix adhesion. Cell Cycle. 2009;8:284-298.
94. Yasuda T, Kurosaki T. Regulation of lymphocyte fate by Ras/ERK signals. Cell Cycle. 2008;7:3634-3640.
95. Murugan AK, Dong J, Xie J, Xing M. MEK1 mutations, but not ERK2 mutations, occur in melanomas and colon carcinomas, but none in thyroid carcinomas. Cell Cycle. 2009;8:2122-2124.
96. De La O JP, Murtaugh LC. Notch and Kras in pancreatic cancer: at the crossroads of mutation, differentiation and signaling. Cell Cycle. 2009;8:1860-1864.
97. Zaravinos A, Kanellou P, Baritaki S, Bonavida B, Spandidos DA. BRAF and RKIP are significantly decreased in cutaneous squamous cell carcinoma. Cell Cycle. 2009;8:1402-1408.
98. Chang B, Liu G, Yang G, Mercado-Uribe I, Huang M, Liu J. REDD1 is required for RAS-mediated transformation of human ovarian epithelial cells. Cell Cycle. 2009;8:780-786.
99. Steelman LS, Navolanic P, Chappell WH, Abrams SL, Wong EWT, Martelli AM, Cocco L, Stivala F, Libra M, Nicoletti F, Drobot LB, Franklin RA and McCubrey JA. Involvement of Akt and mTOR in chemotherapeutic- and hormonal-based drug resistance and response to radiation in breast cancer cells. Cell Cycle 2011; In Press.
100. Sokolosky ML, Stadelman KM, Chappell WH, Abrams SL, Martelli AM, Stivala F, Libra M, Nicoletti F, Drobot LB, Franklin RA, Steelman LS, McCubrey JA. Involvement of Akt-1 and mTOR in Sensitivity of Breast Cancer to Targeted Therapy. Oncotarget 2011; 2:538-550.
101. Smit MA, Peeper DS. Epithelial-mesenchymal transition and senescence: two cancer-related processes are crossing paths. Aging. 2010;2:735-741.
102. Jensen DK, Lal A. Micromanaging senescence. Aging. 2010;2:322-323.
103. Wagner W, Bork S, Lepperdinger G, Joussen S, Ma N, Strunk D, Koch C. How to track cellular aging of mesenchymal stromal cells? Aging. 2010;2:224-230.
104. Demaria M, Giorgi C, Lebiedzinska M, Esposito G, D’Angeli L, Bartoli A, Gough DJ, Turkson J, Levy DE, Watson CJ, Wieckowski MR, Provero P, Pinton P, Poli V. A STAT3-mediated metabolic switch is involved in tumour transformation and STAT3 addiction. Aging. 2010;2:823-842.
105. Hubackova S, Novakova Z, Krejcikova K, Kosar M, Dobrovolna J, Duskova P, Hanzlikova H, Vancurova M, Barath P, Bartek J, Hodny Z. Regulation of the PML tumor suppressor in drug-induced senescence of human normal and cancer cells by JAK/STAT-mediated signaling. Cell Cycle. 2010;9:3085-3099.
106. Wang C, Maddick M, Miwa S, Jurk D, Czapiewski R, Saretzki G, Langie SA, Godschalk RW, Cameron K, von Zglinicki T. Adult-onset, short-term dietary restriction reduces cell senescence in mice. Aging. 2010;2:555-566.
107. Kirkland JL. Perspectives on cellular senescence and short term dietary restriction in adults. Aging. 2010;2:542-544.
108. Kuhlow D, Florian S, von Figura G, Weimer S, Schulz N, Petzke KJ, Zarse K, Pfeiffer AF, Rudolph KL, Ristow M. Telomerase deficiency impairs glucose metabolism and insulin secretion. Aging. 2010;2:650-658.
109. Lehmann BD, McCubrey JA, Jefferson HS, Paine MS, Chappell WH, Terrian DM. A dominant role for p53-dependent cellular senescence in radiosensitization of human prostate cancer cells. Cell Cycle. 2007;6:595-605.
110. Leontieva OV, Gudkov AV, Blagosklonny MV. Weak p53 permits senescence during cell cycle arrest. Cell Cycle. 2010;9:4323-4327.
111. Jee HJ, Kim AJ, Song N, Kim HJ, Kim M, Koh H, Yun J. Nek6 overexpression antagonizes p53-induced senescence in human cancer cells. Cell Cycle. 2010; 9: 4703-4710.
112. Akakura S, Nochajski P, Gao L, Sotomayor P, Matsui S, Gelman IH. Rb-dependent cellular senescence, multinucleation and susceptibility to oncogenic transformation through PKC scaffolding by SSeCKS/AKAP12. Cell Cycle. 2010;9:4656-4665.
113. Schmidt S, Essmann F, Cirtea IC, Kuck F, Thakur HC, Singh M, Kletke A, Janicke RU, Wiek C, Hanenberg H, Ahmadian MR, Schulze-Osthoff K, Nurnberg B, Piekorz RP. The centrosome and mitotic spindle apparatus in cancer and senescence. Cell Cycle. 2010;9:4469-4473.
114. Menendez S, Camus S, Izpisua Belmonte JC. p53: guardian of reprogramming. Cell Cycle. 2010;9:3887-3891.
115. Apontes P, Leontieva OV, Demidenko ZN, Li F, Blagosklonny MV. Exploring long-term protection of normal human fibroblast and epithelial cells from chemotherapy in cell culture. Oncotarget. 2011;2:222-233.
116. Serrano M. Shifting senescence into quiescence by turning up p53. Cell Cycle. 2010;9:4256-4257.
117. Tavana O, Benjamin CL, Puebla-Osorio N, Sang M, Ullrich SE, Ananthaswamy HN, Zhu C. Absence of p53-dependent apoptosis leads to UV radiation hypersensitivity, enhanced immunosuppression and cellular senescence. Cell Cycle. 2010;9:3328-3336.
118. Jung-Hynes B, Ahmad N. Role of p53 in the anti-proliferative effects of Sirt1 inhibition in prostate cancer cells. Cell Cycle. 2009;8:1478-1483.
119. Chen YL, Chen YJ, Tsai WH, Ko YC, Chen JY, Lin SF. The Epstein-Barr virus replication and transcription activator, Rta/BRLF1, induces cellular senescence in epithelial cells. Cell Cycle. 2009;8:58-65.
120. Mele DA, Bista P, Baez DV, Huber BT. Dipeptidyl peptidase 2 is an essential survival factor in the regulation of cell quiescence. Cell Cycle. 2009;8:2425-2434.
121. Gillis LD, Leidal AM, Hill R, Lee PW. p21Cip1/WAF1 mediates cyclin B1 degradation in response to DNA damage. Cell Cycle. 2009;8:253-256.
122. Perucca P, Cazzalini O, Madine M, Savio M, Laskey RA, Vannini V, Prosperi E, Stivala LA. Loss of p21 CDKN1A impairs entry to quiescence and activates a DNA damage response in normal fibroblasts induced to quiescence. Cell Cycle. 2009;8:105-114.
123. McAndrew CW, Gastwirt RF, Donoghue DJ. The atypical CDK activator Spy1 regulates the intrinsic DNA damage response and is dependent upon p53 to inhibit apoptosis. Cell Cycle. 2009;8:66-75.
124. Larrieu D, Ythier D, Brambilla C, Pedeux R. ING2 controls the G1 to S-phase transition by regulating p21 expression. Cell Cycle. 2010;9:3984-3990.
125. Chien WW, Domenech C, Catallo R, Salles G, French M. S-phase lengthening induced by p16(INK4a) overexpression in malignant cells with wild-type pRb and p53. Cell Cycle. 2010;9:3286-3296.
126. Santoro R, Blandino G. p53: The pivot between cell cycle arrest and senescence. Cell Cycle. 2010;9:4262-4263.
127. Browne G, Sayan AE, Tulchinsky E. ZEB proteins link cell motility with cell cycle control and cell survival in cancer. Cell Cycle. 2010;9:886-891.
128. Nevis KR, Cordeiro-Stone M, Cook JG. Origin licensing and p53 status regulate Cdk2 activity during G(1). Cell Cycle. 2009;8:1952-1963.
129. Vivo M, Di Costanzo A, Fortugno P, Pollice A, Calabro V, La Mantia G. Downregulation of DeltaNp63alpha in keratinocytes by p14ARF-mediated SUMO-conjugation and degradation. Cell Cycle. 2009;8:3537-3543.
130. Zhuang Z, Lu J, Lonser R, Kovach JS. Enhancement of cancer chemotherapy by simultaneously altering cell cycle progression and DNA-damage defenses through global modification of the serine/threonine phospho-proteome. Cell Cycle. 2009;8:3303-3306.
131. Hohla F, Buchholz S, Schally AV, Seitz S, Rick FG, Szalontay L, Varga JL, Zarandi M, Halmos G, Vidaurre I, Krishan A, Kurtoglu M, Chandna S, Aigner E, Datz C. GHRH antagonist causes DNA damage leading to p21 mediated cell cycle arrest and apoptosis in human colon cancer cells. Cell Cycle. 2009;8:3149-3156.
132. Varmark H, Sparks CA, Nordberg JJ, Koppetsch BS, Theurkauf WE. DNA damage-induced cell death is enhanced by progression through mitosis. Cell Cycle. 2009;8:2951-2963.
133. Stubbert LJ, Spronck JC, Hamill JD, McKay BC. Ultraviolet light induces the sustained unscheduled expression of cyclin E in the absence of functional p53. Cell Cycle. 2009;8:2995-3002.
134. Fan J, Wray J, Meng X, Shen Z. BCCIP is required for the nuclear localization of the p21 protein. Cell Cycle. 2009;8:3019-3024.
135. Meng S, Arbit T, Veeriah S, Mellinghoff IK, Fang F, Vivanco I, Rohle D, Chan TA. 14-3-3sigma and p21 synergize to determine DNA damage response following Chk2 inhibition. Cell Cycle. 2009;8:2238-2246.
136. Fasullo M, Burch AD, Britton A. Hypoxia enhances the replication of oncolytic herpes simplex virus in p53- breast cancer cells. Cell Cycle. 2009;8:2194-2197.
137. Curry J, Angove H, Fazal L, Lyons J, Reule M, Thompson N, Wallis N. Aurora B kinase inhibition in mitosis: strategies for optimizing the use of aurora kinase inhibitors such as AT9283. Cell Cycle. 2009;8:1921-1929.
138. Chen B, Simpson DA, Zhou Y, Mitra A, Mitchell DL, Cordeiro-Stone M, Kaufmann WK. Human papilloma virus type16 E6 deregulates CHK1 and sensitizes human fibroblasts to environmental carcinogens independently of its effect on p53. Cell Cycle. 2009;8:1775-
139. Acquaviva C, Chevrier V, Chauvin JP, Fournier G, Birnbaum D, Rosnet O. The centrosomal FOP protein is required for cell cycle progression and survival. Cell Cycle. 2009;8:1217-1227.
140. Jung P, Hermeking H. The c-MYC-AP4-p21 cascade. Cell Cycle. 2009; 8: 982-989.
141. Hallstrom TC, Nevins JR. Balancing the decision of cell proliferation and cell fate. Cell Cycle. 2009;8:532-535.
142. Kreis NN, Sommer K, Sanhaji M, Kramer A, Matthess Y, Kaufmann M, Strebhardt K, Yuan J. Long-term downregulation of Polo-like kinase 1 increases the cyclin-dependent kinase inhibitor p21(WAF1/CIP1). Cell Cycle. 2009;8:460-472.
143. Liu X. Plk1, a negative regulator of p21? Cell Cycle. 2009;8:336-337.
144. Bakhanashvili M, Hizi A, Rahav G. The interaction of p53 with 3’-terminal mismatched DNA. Cell Cycle. 2010;9:1380-1389.
145. Li Z, Hann SR. The Myc-nucleophosmin-ARF network: a complex web unveiled. Cell Cycle. 2009;8:2703-2707.
146. Vernole P, Neale MH, Barcaroli D, Munarriz E, Knight RA, Tomasini R, Mak TW, Melino G, De Laurenzi V. TAp73alpha binds the kinetochore proteins Bub1 and Bub3 resulting in polyploidy. Cell Cycle. 2009;8:421-429.
147. Schlereth K, Charles JP, Bretz AC, Stiewe T. Life or death: p53-induced apoptosis requires DNA binding cooperativity. Cell Cycle. 2010;9:4068-4076.
148. Labi V, Villunger A. PUMA-mediated tumor suppression: A tale of two stories. Cell Cycle. 2010;9:4269-4275.
149. O’Prey J, Crighton D, Martin AG, Vousden KH, Fearnhead HO, Ryan KM. p53-mediated induction of Noxa and p53AIP1 requires NFkappaB. Cell Cycle. 2010;9:947-952.
150. Chipuk JE, Green DR. PUMA cooperates with direct activator proteins to promote mitochondrial outer membrane permeabilization and apoptosis. Cell Cycle. 2009;8:2692-2696.
151. Senovilla L, Vitale I, Galluzzi L, Vivet S, Joza N, Younes AB, Rello-Varona S, Castedo M, Kroemer G. p53 represses the polyploidization of primary mammary epithelial cells by activating apoptosis. Cell Cycle. 2009;8:1380-1385.
152. Vaseva AV, Marchenko ND, Moll UM. The transcription-independent mitochondrial p53 program is a major contributor to nutlin-induced apoptosis in tumor cells. Cell Cycle. 2009;8:1711-1719.
153. Steelman LS, Martelli AM, Nicoletti F, McCubrey JA. Exploiting p53 status to enhance effectiveness of chemotherapy by lowering associated toxicity. Oncotarget. 2011;2:109-112.
154. Pajic M, Kersbergen A, van Diepen F, Pfauth A, Jonkers J, Borst P, Rottenberg S. Tumor-initiating cells are not enriched in cisplatin-surviving BRCA1;p53-deficient mammary tumor cells in vivo. Cell Cycle. 2010;9:3780-3791.
155. Tao Y, Leteur C, Yang C, Zhang P, Castedo M, Pierre A, Golsteyn RM, Bourhis J, Kroemer G, Deutsch E. Radiosensitization by Chir-124, a selective CHK1 inhibitor: effects of p53 and cell cycle checkpoints. Cell Cycle. 2009;8:1196-1205.
156. Bao W, Chen M, Zhao X, Kumar R, Spinnler C, Thullberg M, Issaeva N, Selivanova G, Stromblad S. PRIMA-1Met/APR-246 induces wild-type p53-dependent suppression of malignant melanoma tumor growth in 3D culture and in vivo. Cell Cycle. 2011;10:301-307.
157. Ho L, Alman B. Protecting the hedgerow: p53 and hedgehog pathway interactions. Cell Cycle. 2010;9:506-511.
158. Dominguez-Brauer C, Brauer PM, Chen YJ, Pimkina J, Raychaudhuri P. Tumor suppression by ARF: gatekeeper and caretaker. Cell Cycle. 2010;9:86-89.
159. Ide T, Chu K, Aaronson SA, Lee SW. GAMT joins the p53 network: branching into metabolism. Cell Cycle. 2010;9:1706-1710.
160. Vazquez-Martin A, Oliveras-Ferraros C, Lopez-Bonet E, Menendez JA. AMPK: Evidence for an energy-sensing cytokinetic tumor suppressor. Cell Cycle. 2009;8:3679-3683.
161. Senovilla L, Vitale I, Galluzzi L, Vivet S, Joza N, Younes AB, Rello-Varona S, Castedo M, Kroemer G. Growth controls connect: interactions between c-myc and the tuberous sclerosis complex-mTOR pathway. Cell Cycle. 2009;8:1344-1351.
162. Bykov VJ, Lambert JM, Hainaut P, Wiman KG. Mutant p53 rescue and modulation of p53 redox state. Cell Cycle. 2009;8:2509-2517.
163. Nardinocchi L, Puca R, Givol D, D’Orazi G. HIPK2-a therapeutical target to be (re)activated for tumor suppression: role in p53 activation and HIF-1 inhibition. Cell Cycle. 2010;9:1270-1275.
164. Eitel JA, Bijangi-Vishehsaraei K, Saadatzadeh MR, Bhavsar JR, Murphy MP, Pollok KE, Mayo LD. PTEN and p53 are required for hypoxia induced expression of maspin in glioblastoma cells. Cell Cycle. 2009;8:896-901.
165. O’Prey J, Skommer J, Wilkinson S, Ryan KM. Analysis of DRAM-related proteins reveals evolutionarily conserved and divergent roles in the control of autophagy. Cell Cycle. 2009;8:2260-2265.
166. Maiuri MC, Malik SA, Morselli E, Kepp O, Criollo A, Mouchel PL, Carnuccio R, Kroemer G. Stimulation of autophagy by the p53 target gene Sestrin2. Cell Cycle. 2009;8:1571-1576.
167. Effects of p21 deletion in mouse models of premature aging. Cell Cycle. 2009;8:2002-2004.
168. Kasper LH, Thomas MC, Zambetti GP, Brindle PK. Double null cells reveal that CBP and p300 are dispensable for p53 targets p21 and Mdm2 but variably required for target genes of other signaling pathways. Cell Cycle. 2011;10:212-221.
169. Kim, DH, Rho K, Kim S. A theoretical model for p53 dynamics: identifying optimal therapeutic strategy for its activation and stabilization. Cell Cycle. 2009;8:3707-3716.
170. Cheng Q, Chen J. Mechanism of p53 stabilization by ATM after DNA damage. Cell Cycle. 2010;9:472-478.
171. Herrera-Merchan A, Cerrato C, Luengo G, Dominguez O, Piris MA, Serrano M, Gonzalez S. miR-33-mediated downregulation of p53 controls hematopoietic stem cell self-renewal. Cell Cycle. 2010;9:3277-3285.
172. Scheel AH, Beyer U, Agami R, Dobbelstein M. Immunofluorescence-based screening identifies germ cell associated microRNA 302 as an antagonist to p63 expression. Cell Cycle. 2009;8:1426-1432.
173. Yamakuchi M, Lowenstein CJ. MiR-34, SIRT1 and p53: the feedback loop. Cell Cycle. 2009;8:712-715.
174. Li B, Cheng Q, Li Z, Chen J. p53 inactivation by MDM2 and MDMX negative feedback loops in testicular germ cell tumors. Cell Cycle. 2010;9:1411-1420.
175. Gillotin S, Yap D, Lu X. Mutation at Ser392 specifically sensitizes mutant p53H175 to mdm2-mediated degradation. Cell Cycle. 2010;9:1390-1398.
176. Zdzalik M, Pustelny K, Kedracka-Krok S, Huben K, Pecak A, Wladyka B, Jankowski S, Dubin A, Potempa J, Dubin G. Interaction of regulators Mdm2 and Mdmx with transcription factors p53, p63 and p73. Cell Cycle. 2010;9:4584-4591.
177. Lo D, Lu H. Nucleostemin: Another nucleolar “Twister” of the p53-MDM2 loop. Cell Cycle. 2010;9:3227-3232.
178. Lane DP, Cheok CF, Brown C, Madhumalar A, Ghadessy FJ, Verma C. Mdm2 and p53 are highly conserved from placozoans to man. Cell Cycle. 2010;9:540-547.
179. Lau J, Lew QJ, Diribarne G, Michels AA, Dey A, Bensaude O, Lane DP, Chao SH. Ubiquitination of HEXIM1 by HDM2. Cell Cycle. 2009;8:2247-2254.
180. Czarna A, Popowicz GM, Pecak A, Wolf S, Dubin G, Holak TA. High affinity interaction of the p53 peptide-analogue with human Mdm2 and Mdmx. Cell Cycle. 2009;8:1176-1184.
181. Llanos S, Serrano M. Depletion of ribosomal protein L37 occurs in response to DNA damage and activates p53 through the L11/MDM2 pathway. Cell Cycle. 2010;9:4005-4012.
182. Jain AK, Barton MC. Regulation of p53:TRIM24 enters the RING. Cell Cycle. 2009;8:3668-3674.
183. Hedstrom E, Eriksson S, Zawacka-Pankau J, Arner ES, Selivanova G. p53-dependent inhibition of TrxR1 contributes to the tumor-specific induction of apoptosis by RITA. Cell Cycle. 2009;8:3576-3583.
184. Reinhardt HC, Jiang H, Hemann MT, Yaffe MB. Exploiting synthetic lethal interactions for targeted cancer therapy. Cell Cycle. 2009;8:3112-3119.
185. Farnebo M. Wrap53, a novel regulator of p53. Cell Cycle. 2009;8:2343-2346.
186. Kampa KM, Bonin M, Lopez CD. New insights into the expanding complexity of the tumor suppressor ASPP2. Cell Cycle. 2009;8:2871-2876.
187. Madhumalar A, Jun LH, Lane DP, Verma CS. Dimerization of the core domain of the p53 family: a computational study. Cell Cycle. 2009;8:137-148.
188. Steelman LS, McCubrey JA. Intriguing novel abilities of Nutlin-3A: induction of cellular quiescence as opposed to cellular senescence--implications for chemotherapy. Cell Cycle. 2009;8:3634-3635.
189. Blundell TL, Bolanos-Garcia, VM. Targeting protein interactions of p53 for therapeutic intervention: success in a frustrated landscape. Cell Cycle. 2009;8:3631-3632.
190. Korotchkina LG, Demidenko ZN, Gudkov AV, Blagosklonny MV. Cellular quiescence caused by the Mdm2 inhibitor nutlin-3A. Cell Cycle. 2009;8:3777-3781.
191. Wang B, Xiao Z, Ko HL, Ren EC. The p53 response element and transcriptional repression. Cell Cycle. 2010;9:870-879.
192. Zawacka-Pankau J, Kostecka A, Sznarkowska A, Hedstrom E, Kawiak A. p73 tumor suppressor protein: a close relative of p53 not only in structure but also in anti-cancer approach? Cell Cycle. 2010;9:720-728.
193. Ahn J, Poyurovsky MV, Baptiste N, Beckerman R, Cain C, Mattia M, McKinney K, Zhou J, Zupnick A, Gottifredi V, Prives C. Dissection of the sequence-specific DNA binding and exonuclease activities reveals a superactive yet apoptotically impaired mutant p53 protein. Cell Cycle. 2009;8:1603-1615.
194. Graupner V, Schulze-Osthoff K, Essmann F, Janicke RU. Functional characterization of p53beta and p53gamma, two isoforms of the tumor suppressor p53. Cell Cycle. 2009;8:1238-1248.
195. Di Costanzo A, Festa L, Duverger O, Vivo M, Guerrini L, La Mantia G, Morasso MI, Calabro V. Homeodomain protein Dlx3 induces phosphorylation-dependent p63 degradation. Cell Cycle. 2009;81185-1195.
196. Chen D, Yoon JB, Gu W. Reactivating the ARF-p53 axis in AML cells by targeting ULF. Cell Cycle. 2010;9:2946-2951.
197. Drewelus I, Gopfert C, Hippel C, Dickmanns A, Damianitsch K, Pieler T, Dobbelstein M. p63 antagonizes Wnt-induced transcription. Cell Cycle. 2010;9:580-587.
198. Leonova KI, Shneyder J, Antoch MP, Toshkov IA, Novototskaya LR, Komarov PG, Komarova EA, Gudkov AV. A small molecule inhibitor of p53 stimulates amplification of hematopoietic stem cells but does not promote tumor development in mice. Cell Cycle. 2010;9:1434-1443.
199. Baker DJ, van Deursen JM. Chromosome missegregation causes colon cancer by APC loss of heterozygosity. Cell Cycle. 2010;9:1711-1716.
200. Schoppy DW, Ruzankina Y, Brown EJ. Removing all obstacles: a critical role for p53 in promoting tissue renewal. Cell Cycle. 2010;9:1313-1319.
201. Wei W, Herbig U, Wei S, Dutriaux A, Sedivy JM. Loss of retinoblastoma but not p16 function allows bypass of replicative senescence in human fibroblasts. EMBO Rep. 2003;4:1061-1066.
202. Ogawara Y, Kishishita S, Obata T, Isazawa Y, Suzuki T, Tanaka K, Masuyama N, Gotoh Y. Akt enhances Mdm2-mediated ubiquitination and degradation of p53. J Biol Chem. 2002;277:21843-21850.
203. Jeong SJ, Pise-Masison CA, Radonovich MF, Park HU, Brady JN. Activated AKT regulates NF-kappaB activation, p53 inhibition and cell survival in HTLV-1-transformed cells. Oncogene. 2005;24:6719-6728.
204. Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci U S A. 2005;102:8204-8209.
205. Noonan EJ, Place RF, Basak S, Pookot D, Li LC. miR-449a causes Rb-dependent cell cycle arrest and senescence in prostate cancer cells. Oncotarget. 2010;1:349-358.
206. Demidenko ZN, Korotchkina LG, Gudkov AV, Blagosklonny MV. Paradoxical suppression of cellular senescence by p53. Proc Natl Acad Sci U S A. 2010;107:9660-9664.
207. Kennedy AL, Morton JP, Manoharan I, Nelson DM, Jamieson NB, Pawlikowski JS, McBryan T, Doyle B, McKay C, Oien KA, Enders GH, Zhang R, Sansom OJ, Adams PD. Activation of the PIK3CA/AKT pathway suppresses senescence induced by an activated RAS oncogene to promote tumorigenesis. Mol Cell. 2011;42: 36-49.
208. Hafner C, Toll A, Fernández-Casado A, Earl J, Marqués M, Acquadro F, Méndez-Pertuz M, Urioste M, Malats N, Burns JE, Knowles MA, Cigudosa JC, Hartmann A, Vogt T, Landthaler M, Pujol RM, Real FX. Multiple oncogenic mutations and clonal relationship in spatially distinct benign human epidermal tumors. Proc Natl Acad Sci U S A. 2010;107:20780-20785.
209. Axanova LS, Chen YQ, McCoy T, Sui G, Cramer SD. 1,25-dihydroxyvitamin D(3) and PI3K/AKT inhibitors synergistically inhibit growth and induce senescence in prostate cancer cells. Prostate. 2010;70:1658-1671
210. Hirose Y, Katayama M, Mirzoeva OK, Berger MS, Pieper RO. Akt activation suppresses Chk2-mediated, methylating agent-induced G2 arrest and protects from temozolomide-induced mitotic catastrophe and cellular senescence. Cancer Res. 2005;65:4861-4869.
211. Wozniak K, Kolacinska A, Blasinska-Morawiec M, Morawiec-Bajda A, Morawiec Z, Zadrozny M, Blasiak J. The DNA-damaging potential of tamoxifen in breast cancer and normal cells. Arch Toxicol. 2007;81:519-527.
212. Siedlakowski P, McLachlan-Burgess A, Griffin C, Tirumalai SS, McNulty J, Pandey S. Synergy of Pancratistatin and Tamoxifen on breast cancer cells in inducing apoptosis by targeting mitochondria. Cancer Biol Ther. 2008;7:376-384.
213. Vessières A, Corbet C, Heldt JM, Lories N, Jouy N, Laïos I, Leclercq G, Jaouen G, Toillon RA. A ferrocenyl derivative of hydroxytamoxifen elicits an estrogen receptor-independent mechanism of action in breast cancer cell lines. J Inorg Biochem. 2010;104:503-511.
214. Bansal R, Nikiforov MA. Pathways of oncogene-induced senescence in human melanocytic cells. Cell Cycle. 2010;9:2782-2788.
215. Miller KR, Kelley K, Tuttle R, Berberich SJ. HdmX overexpression inhibits oncogene induced cellular senescence. Cell Cycle. 2010;9:3376-3382.
216. Hill R, Lee PW. The DNA-dependent protein kinase (DNA-PK): More than just a case of making ends meet? Cell Cycle. 2010;9:3460-3469.
217. Gomes NP, Espinosa JM. Disparate chromatin landscapes and kinetics of inactivation impact differential regulation of p53 target genes. Cell Cycle. 2010;9:3428-3437.
218. Neise D, Sohn TD, Budach W, Janicke RU. Evidence for a differential modulation of p53-phosphorylating kinases by the cyclin-dependent kinase inhibitor p21WAF1/CIP1. Cell Cycle. 2010;9:3575-3583.
219. Bajaj A, Zheng Q, Adam A, Vincent P, Pumiglia K. Activation of endothelial ras signaling bypasses senescence and causes abnormal vascular morphogenesis. Cancer Res. 2010;70:3803-3812.
220. Adams JR, Schachter NF, Liu JC, Zacksenhaus E, Egan SE. Elevated PI3K signaling drives multiple breast Cancer subtypes. Oncotarget. 2011;2:435-447.
221. Eckert MA, Yang J. Targeting invadopodia to block breast cancer metastasis. Oncotarget. 2011;2:562-568.
222. Eckert MA, Lwin TM, Chang AT, Kim J, Danis E, Ohno-Machado L, Yang J. Twist1-induced invadopodia formation promotes tumor metastasis. Cancer Cell. 2011;19:372-386.
223. Hydbring P, Larsson LG. Cdk2: a key regulator of the senescence control function of Myc. Aging. 2010;2:244-250.
224. Romanov VS, Abramova MV, Svetlikova SB, Bykova TV, Zubova SG, Aksenov ND, Fornace AJ Jr, Pospelova TV, Pospelov VA. p21(Waf1) is required for cellular senescence but not for cell cycle arrest induced by the HDAC inhibitor sodium butyrate. Cell Cycle. 2010;9:3945-3955.