
Introduction
Squamous cell cancer of the head and neck (SCCHN) is the sixth leading cause for cancer deaths worldwide [1]. Despite recent progress in understanding SCCHN biology and improved treatment, the 5 year survival has remained 50 percent for the past two decades. There is a pressing need to improve therapy in particular for patients with metastatic disease or local recurrence, where the median progression-free and overall survival is only ~ 6 months and ~11 months, respectively [2-4].
Several genetic alterations have been described in SCCHN, including mutations in the p53 tumor suppressor gene and mutations in genes that encode cell cycle proteins such as p16 and cyclin D1. In addition, several oncogenic pathways including Ras, PI3K/PTEN/Akt, TGF-β/BMP and EGFR/STAT3 are up-regulated in SCCHN [4-11]. Epidermal growth factor receptor (EGFR) overexpression in SCCHN is often caused by gene amplification [12], and elevated expression correlates with poor disease control and metastasis [13-14]. Furthermore, overexpression of two of its ligands, EGF and transforming growth factor-alpha (TGF-alpha), has been linked to a poor prognosis [15]. The major signaling pathways activated by EGFR are the RAS-RAF-MAP kinase pathway, which is mainly involved in proliferation, and the PI3K-PTEN-AKT pathway, which is mainly involved in survival [16]. The addition of the monoclonal antibody C225 (cetuximab) to the standard first-line regimen cisplatin/5-fluorouracil [17] not only increased the rate of objective responses but also improved progression-free and overall survival in patients with recurrent or metastatic SCCHN [2].
The Aurora kinases A and B (Aurora-A and Aurora-B) are highly conserved serine/threonine kinases that play essential and distinct roles in mitosis [18]. Specifically, Aurora-A is required for the assembly of the mitotic spindle, where it accumulates on centrosomes at the spindle poles during prophase until metaphase. Recently a kinase-independent role in mitotic spindle assembly has been reported for Aurora-A [19]. Aurora-B is required for mitotic progression and cytokinesis, and is localized, along with inner centromeric protein (INCENP) and survivin, at centromeres and the spindle midzone during the metaphase to anaphase transition [18, 20]. AURORA-A (AURKA) mRNA is amplified in a variety of human cancers including SCCHN, where it is associated with poor prognosis [21]. Increased levels of Aurora-B have been reported in various aggressive malignancies [20].
Both Aurora-A and EGFR overexpression have been implicated in SCCHN tumorigenesis and are established adverse prognostic factors. Aurora-A and EGFR share downstream signaling pathways, and each by itself represents an attractive therapeutic target. Here we report that joint protein overexpression of EGFR and Aurora-A defines a poor risk group among SCCHN patients. Combining drugs that target Aurora kinases and EGFR may overcome resistance to single agent treatment in SCCHN cells.
Results
High levels of EGFR and Aurora-A assessed by IHC identify adverse prognosis in SCCHN
Publicly available gene expression data [22] (www.oncomine.org) were analyzed for the expression and prognostic relevance of EGFR and AURORA-A expression. AURORA-A transcripts were expressed at significantly higher levels in SCCHN tumor samples as compared to normal control tissue (p=0.002, Figure 1), and the median relative expression in surviving patients was lower as compared to patients dying from SCCHN (n.s.). In a previous report the level of AURORA-A transcript was associated with survival [21]. We therefore next addressed the prognostic relevance of Aurora-A and EGFR protein levels in the SCCHN patient cohort (n=180) described in Table 1. There was a highly significant difference between patients’ protein levels when comparing normal adjacent mucosa with the levels expressed in tumor cells for both Aurora-A and EGFR (Figure 2A), with independent expression of EGFR and Aurora-A for each patient (r=0.03 / p=0.74). Furthermore, there were clear differences in expression levels for Aurora-A and EGFR within the patient tumor tissue assessed (Figure 2). While protein levels of EGFR (Figure 3A) or Aurora-A (Figure 3B) above median assessed by IHC in a Kaplan Meier analysis did not identify a population with a significantly reduced disease-free survival (EGFR: p=0.10; Aurora-A; p=0.21), our analysis identifies a poor risk population with regard to overall and disease-free survival that is characterized by above median levels of EGFR (EGFRhigh) and Aurora-A (Aurora-Ahigh) (p=0.024, compared to EGFRlow and Aurora-Alow, Figure 2C). Thus, the coexpression of elevated levels of Aurora-A and EGFR is an adverse prognostic factor in SCCHN.
Table 1: Patient characteristics (n=180). |
||
characteristic |
number (%) |
|
| sex | female | 17 (9) |
| male | 163 (91) | |
| localisation | oral cavity | 33 (18) |
| oropharynx | 58 (32) | |
| hypopharynx | 33 (18) | |
| larynx | 56 (31) | |
| primary tumor category (pT) | pT1 | 25 (14) |
| pT2 | 66 (37) | |
| pT3 | 48 (27) | |
| pT4 | 41 (23) | |
| lymph node category (c/pN) | c/pN0 | 94 (52) |
| pN1 | 23 (13) | |
| pN2a | 2 (1) | |
| pN2b | 39 (22) | |
| pN2c | 20 (11) | |
| pN3 | 2 (1) | |
| tumor grade | G1 | 10 (6) |
| G2 | 110 (61) | |
| G3 | 60 (33) | |
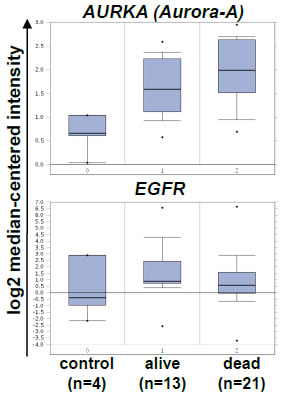
Figure 1: EGFR and Aurora-A transcript levels in SCCHN and clinical outcome. A public database (www.oncomine.com) was searched for gene expression analyses studies that compare AURORA-A transcript levels in control tissue and SCCHN samples from patients who were alive or dead [22]. Shown is the log2 median-centered relative intensity of expression for AURORA-A (AURKA, upper panel, tumor versus control tissue: p= 0.002, [reporter: 34851_at]) and EGFR (lower panel, tumor versus control tissue: n.s., [reporter: 1537_at]).
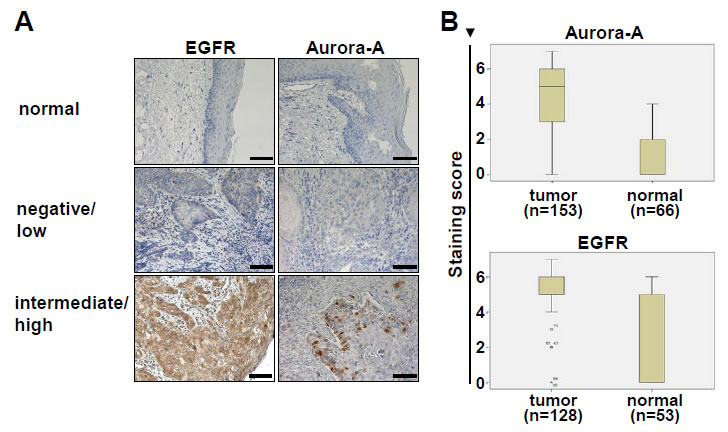
Figure 2: EGFR and Aurora-A expression in tumor tissue and adjacent normal mucosa. (A) Histological assessment of EGFR and Aurora-A protein expression by immunohistochemistry. Shown are representative tumor samples that were graded as negative/low expression (middle panel), high expression (lower panel) and normal mucosa control tissue (upper panel). Bar equals 100µm. (B) Within each patient sample the expression of Aurora-A and EGFR was assessed in normal adjacent tissue and tumor tissue. The differences are highly significant. Aurora-A: p<0.001; EGFR: p<0.001. The staining score is defined in the material and method section.
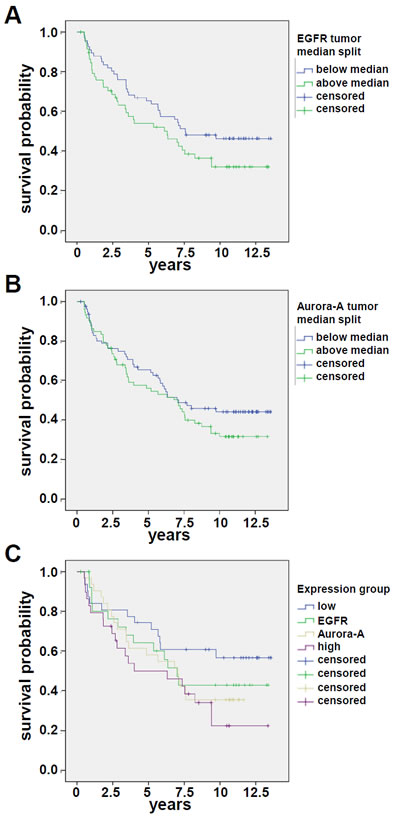
Figure 3: EGFR and Aurora-A expression assessed by IHC is an adverse prognostic factor in SCCHN. (A) EGFR: the difference in disease-free survival for patients with expression above median (green curve; n=90) is not statistically different from the survival of patients with expression below median (blue curve; n=90). p=0.10. (B) Aurora-A: the difference in disease-free survival for patients with expression below median (blue curve; n=90) is not statistically different from the survival of patients with expression above median (green curve; n=90). p=0.21. (C) The difference in disease-free survival of patients with EGFRhigh and Aurora-Ahigh is statistically different from the survival of patients who are characterized by EGFRlow and Aurora-Alow. p=0.024. The staining score is defined in the material and method section.
Aurora kinase inhibition results in defective cytokinesis and polyploidy irrespective of the EGFR status
Given our results (Figure 3) and mRNA data showing that Aurora-A expression is an adverse prognostic factor [21], molecular targeted therapy towards Aurora kinases could be an attractive approach. We first characterized six SCCHN cell lines for the expression of EGFR, Aurora-A and Aurora-B. As expected all cell lines showed detectable levels of Aurora kinases as well as phosphorylation of the Aurora kinase substrate Serin10-phosphorylated Histone H3 (S10-HH3) [23](Figure 4A). Real-time PCR analysis revealed no clear correlation between transcript and protein level for Aurora-A or Aurora-B (Figure 4B, upper panel). We next assessed the presence of the EGFR variant III (EGFRvIII), which has been reported to contribute to tumor growth and resistance to EGFR targeting [24]. EGFRvIII was not present in any of the cell lines analyzed by RT-PCR, where NIH-3T3 cells that were engineered to ectopically express EGFRvIII were included as a control (Figure 4B, lower panel). We next analyzed the effects of the EGFR antibody cetuximab and the small molecule pan-Aurora kinase inhibitor R763 [25] on SCCHN cells. Treatment with 200 nM cetuximab resulted in reduced autophosphorylation of EGFR after 5 minutes, which subsequently resumed to normal and above normal levels consistent with a previous report [26]. In accord, the abundance of phosphorylated Akt and Erk upon cetuximab treatment was reduced [27] (Figure 4C, upper panel). We then assessed the abundance of S10-HH3 as a measure of Aurora kinase activity. The exposure to 5 nM R763 lead to a rapid and efficient decrease in S10-HH3 levels (Figure 4C, lower panel). In order to assess the Aurora kinase inhibition effects on ploidy and cell death we next treated SCCHN cell lines for a 24 hour period with R763 at various concentrations. There was a strong effect with regard to G2-M arrest and/or ploidy (p<0.05 in all cell lines, Figure 4D) and to a lesser extent to the subG1 fraction of SCCHN cells, indicating that mitosis and cytokinesis were effectively blocked. R763 treatment did however result in low apoptosis rates. In conclusion, a low nanomolar concentration of the Aurora kinase inhibitor R763 resulted in effective inhibition of Aurora kinase activity, of cytokinesis and caused polyploidy.
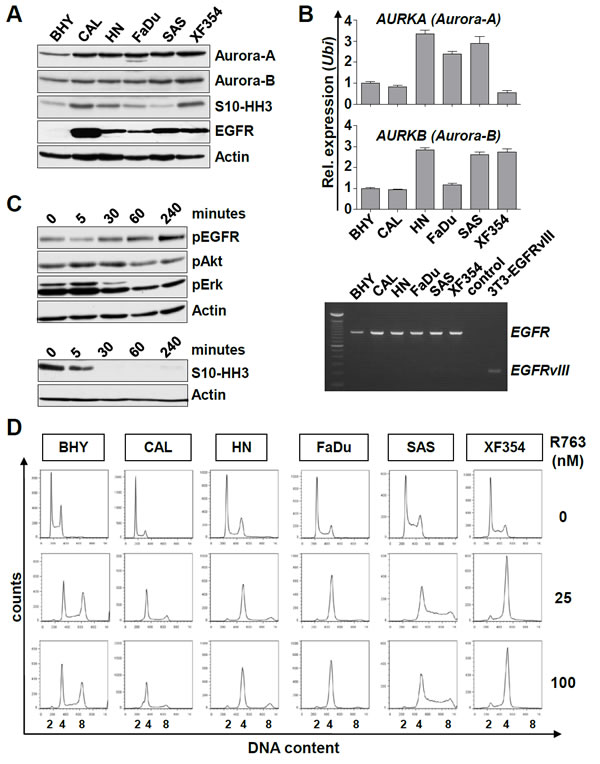
Figure 4: Expression and activity of Aurora kinases and EGFR in SCCHN cell lines. (A) Six SCCHN cell lines were assessed by immunoblotting for the expression of Aurora-A and Aurora-B, for Aurora kinase activity measured by Histone H3 phosphorylation at serine10 (S10-HH3), and for EGFR protein levels. (B) Upper panel: AURORA-A and AURORA-B transcript levels were assessed by real-time qRT-PCR. Shown is the relative expression normalized to the expression of Ubiquitin. Lower panel: Expression of EGFR analyzed by RT-PCR. None of the SCCHN cell lines express the EGFRvIII mutant. Transiently transfected NIH-3T3 cells expressing EGFRvIII (3T3-EGFRvIII) were included as a control. (C) Upper panel: CAL cells were treated with 200 nM Cetuximab for the indicated time and assessed by immunoblotting for suppression of EGFR downstream target phosphorylation. Lower panel: Treatment of FADU cells with 5 nM Pan-Aurora kinase inhibitor R763 for the indicated time. The activity of Aurora kinases was assessed by immunoblotting for S10-HH3. (D) SCCHN cell lines were treated for 24 hr with R763 at the indicated concentrations or carrier alone (0 nM). The representative histograms show the DNA content assessed by propidium iodide (PI) staining.
Additive effects of combined Aurora kinase and EGFR targeting
Given that we found Aurora-A and EGFR protein expression as adverse prognostic factor in SCCHN, targeting both is an attractive therapeutic approach. We therefore assessed whether combined targeting using R763 and cetuximab would result in increased cell cycle effects and/or apoptosis. To mimic the in vivo drug action we estimated the long term effects of EGFR and/or Aurora kinase targeting in asynchronously growing SCCHN cultures. SCCHN growth curves revealed that the addition of 200 nM cetuximab or 5 nM R763 results in a delayed growth inhibition starting at around 7 days after treatment initiation (data not shown). The effects of a combination treatment in longer term cell culture were significantly pronounced (Figure 5A). Quite surprisingly, in cell lines that showed no or very moderate growth inhibition upon cetuximab only treatment (BHY and FaDu), addition of the Aurora kinase inhibitor led to an additive growth inhibition (Figure 4A), even in cells that are characterized by very low EGFR expression (BHY, Figure 4A). Thus, the combination of Aurora kinase inhibition and EGFR targeting is highly efficient in vitro and may overcome cetuximab resistance.
To mechanistically address the additive effect SCCHN cells were incubated with 5 nM R763, which blocked kinase activity effectively (Figure 4C), 200 nM cetuximab or the combination of both drugs, and compared to untreated controls. 48 hour treatment with cetuximab showed minor efficacy with regard to cell cycle arrest and polyploidy or apoptosis induction assessed by PI staining or AnnexinV positivity. 48 hour treatment with R763 resulted in a significant increase in polyploid (Figure 5A) and apoptotic cells (Figure 5B). The combination of cetuximab and R763 did not lead to a significantly increased fraction of cells with a polyploid phenotype representing defective mitosis and cytokinesis as compared to R763 monotherapy (Figure 5A), but, importantly, in several cell lines to a significantly elevated percentage of cell death (subG1 DNA content, data not shown), and AnnexinV positive apoptotic cells (Figure 5B). Thus, combined EGFR and Aurora kinase targeting results in additive effects, potentially by sensitizing mitotic checkpoints.
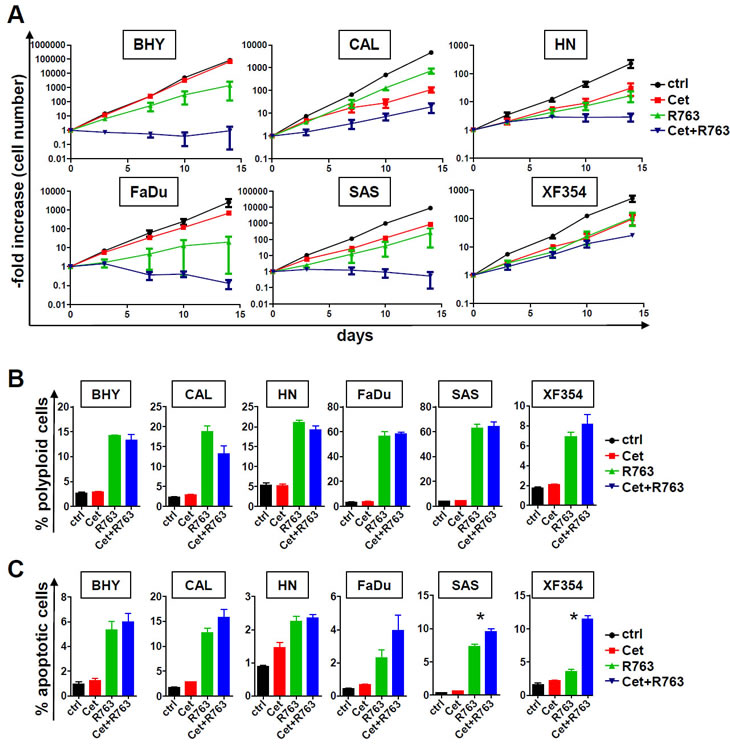
Figure 5: Combined exposure to EGFR antibody and Aurora kinase inhibitor results in fortified growth inhibition and apoptosis. (A) SCCHN cells were treated for a total of 14 days with cetuximab (200 nM), the Aurora kinase inhibitor R763, the combination of both (Cet+ R763), or carrier only (control). The cell number was counted at the indicated times and the -fold increase in cell number calculated. Note that the increase in cell number is given in a logarythmic scale. The combination of cetuximab and Aurora kinase inhibitor resulted in a significantly reduced -fold increase after 14 day treatment period in all cell lines investigated in comparison to all other conditions (Cet alone, R763 alone, control; p<0.05). (B) The indicated SCCHN cells were cultured for a 48 hr period with the indicated conditions (cetuximab 200 nM, R7635 nM) and assessed for DNA content by PI staining. The percentage of polyploid cells with a DNA content >4n is given. (C) Analysis of the cells shown in (B) for apoptosis (Annexin V-positive, PI-negative cell fraction) by flow cytometry. The bars represent the mean ± SD of 3 independently performed experiments. Statistically significant differences are marked (* indicates p<0.05).
Selective Aurora-A inhibition is less effective than combined Aurora kinase inhibition
R763 is a pan-Aurora kinase inhibitor that inhibits Aurora-A and Aurora-B [28]. To further analyze whether Aurora-A, a prognostic factor in SCCHN [21](and this report Figure 3), or Aurora-B is the major target of R763 in SCCHN, we next directly compared R763 with the Aurora-A specific kinase inhibitor MLN8237 (Mln). Mln effectively blocked S10-HH3 phosphorylation at 10nM (Figure 6A). Mln treatment furthermore resulted in an increase of the fraction of polyploid cells (Figure 6B), and combined EGFR and Aurora-A targeting using Mln decreased the growth of SCCHN cells significantly (Figure 6C). A direct comparison of the Pan-Aurora kinase inhibitor R763 (5nM) and the Aurora-A specific kinase inhibitor Mln (10nM) at concentrations that each block S10-HH3 phosphorylation effectively revealed that the R763/cetuximab combination was much more potent in inducing polyploidy (Figure 6D) as well as apoptosis (Figure 6E) compared to cetuximab in combination with the specific Aurora-A inhibitor Mln. Thus, the superior effects of R763 are most likely mediated by its blockage of Aurora-B activity or its dual Aurora kinase inhibition.
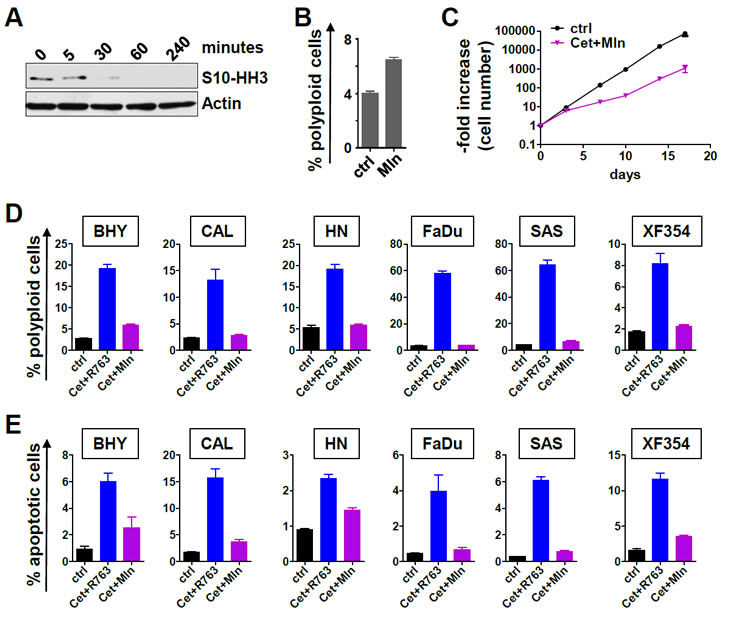
Figure 6: Selective Aurora-A inhibition versus pan-Aurora kinase inhibition in combination with Cetuximab. (A) FADU cell were treated with 10 nM Mln for the indicated time. The effect of Aurora-A inhibition was assessed by immunoblotting for serine10-phosphorylated Histone H3 (S10-HH3). (B) Mln treatment (10 nM) for 48 hr resulted in a significant (p<0.05) but moderate increase of polyploid cells (>4n DNA content) as evaluated by PI flow cytometry. (C) Combined Aurora-A inhibiton with 10 nM Mln and EGFR inhibition with 200 nM cetuximab treatment results a significantly reduced cell number increase. (D) The indicated SCCHN cell lines were treated for 48 hr with carrier only or cetuximab plus R763 (Cet+ R763) or cetuximab plus Mln (Cet+Mln). The percentage of polyploid cells as defined by a DNA content >4n was measured by flow cytometry of PI stained cells. (E) Cells were treated as in (C). The percentage of apoptotic cells was assessed by Annexin V flow cytometry. The bars represent the mean ± SD of 3 independent experiments. The differences between Cet+ R763 versus Cet+Mln treatment are significant (p<0.05) for all cell lines tested with regard to polyploidy and with regard to apoptosis.
Discussion
Other than EGFR blockage through cetuximab, none of the targeted approaches have yet shown clinically convincing results or changed the standard of care in relapsed or metastatic SCCHN. We identify the Aurora kinases as potential targets in this disease. Aurora kinases are upregulated in multiple human cancers, correlating in some cases with poor prognosis [18, 20-21, 29]. By investigating 180 patient samples of SCCHN tumors we show that both Aurora-A and EGFR are significantly overexpressed in tumor tissue. The spearman correlation coefficient showed that the expression of Aurora-A and EGFR was independent. Our findings thus establish that the joint overexpression of EGFR and Aurora-A defines a subgroup of SCCHN patients with inferior prognosis regarding disease-free and overall survival. These results prompt the analysis of combined targeted treatment strategies in this disease. We used a dual Aurora-A/Aurora-B inhibitor in combination with EGFR blockage through cetuximab and established an additive or possibly even synergistic effect on SCCHN cells in vitro. At this time it is however not clear whether Aurora-B was the main therapeutic target in our SCCHN studies or whether combined inhibition of Aurora-A and Aurora-B is beneficial. In a targeted small interfering RNA screen others identified Aurora-A as a component of an EGFR-centered network. When the Aurora kinase inhibitor PHA-680632 (PHA) was combined with EGFR inhibition, therapeutic synergism was observed in EGFR-dependent cell lines [30]. It has however to be noted that the applied concentrations of PHA most likely also inhibit Aurora-B [31]. There is further linkage between EGFR activation and Aurora-A. A study demonstrated that the nuclear EGFR can cooperate with STAT5A to target the promoter region of AURORA-A and enhance its expression in cancer cells [32].
A consistent finding in our in vitro study is that there is a uniform additive inhibition of cell growth when cetuximab and Aurora kinase inhibition was combined, even in cell lines that were resistant towards EGFR-directed treatment or that showed moderate growth inhibition upon single Aurora kinase targeting. Our immunohistochemical studies did not address the frequency of the EGFRvIII mutant that might be associated with resistance towards cetuximab [24]. The cell lines we used did not express EGFRvIII. At this time we cannot conclude whether EGFRvIII bearing SCCHN patients have an inferior prognosis (our retrospective cohort) or whether EGFRvIII mutant cell lines are different with regard to sensitivity towards Aurora kinase inhibition. A recent clinical trial indicated that high EGFRvIII expression levels identify SCCHN patients who are less likely to benefit from combination treatment with cetuximab and docetaxel [33]. However, our studies suggest that even inhibiting a very low level of EGFR expression might be sufficient to sensitize for Aurora kinase inhibition. This could occur by either concertedly targeting the same growth and/or survival pathways or by blocking resistance-mediating mechanisms.
The G2-M targeting approach is of particular interest since conventional chemotherapy usually targets cancer cells at the G1-S transition of the cell cycle. The cell cycle is driven by Cyclin-dependent kinases (Cdk). Of particular importance is the negative regulation of Cdk by checkpoints when defects such as DNA damage occur. Following DNA damage the transcription factor p53 is activated, which results in transcription of the Cdk inhibitor p21 and cell cycle arrest in G1, or induction of apoptosis [34]. Loss of p53 function, a frequent event in SCCHN [4, 35], therefore has the dual effect of loss of the G1-S checkpoint and loss of an important pathway leading to death [36]. On the other hand G2-M checkpoint genes are rarely if ever mutated in cancer. Therefore therapeutics targeting cancer cells at G2-M and during cytokinesis are highly interesting. Current therapeutic strategies in SCCHN use mitotic poisons such as taxanes, which act directly on spindle microtubules inducing spindle assembly checkpoint (SAC) activation, and prolonged mitotic arrest that frequently ends in cell death [37]. A second approach is to directly target mitotic checkpoint kinases such as Aurora kinases. Several of the currently available Aurora kinase inhibitors target both Aurora-A and Aurora-B. Comparing the pan Aurora kinase inhibitor R763 [25] with the Aurora-A specific inhibitor MLN our results establish Aurora-B as the potentially more powerful target in SCCHN, but cannot rule out that a combined Aurora-A and Aurora-B inhibition might be beneficial to induce mitotic failure and cell death. Importantly, there are interactions between Aurora-A and p53, where Aurora-A directly phosphorylates p53 to augment p53 protein turnover and transcriptional activity [38]. In addition, a differential effect of Aurora kinase inhibition related to function p53 has been suggested [39].
The G2-M checkpoint is a particularly interesting therapeutic target in SCCHN, where due to the high frequency of mutations in the p53 apoptotic pathway the G1-S checkpoint is often dysfunctional. Our results define a new risk group in SCCHN and provide a rationale for testing combined EGFR and Aurora kinase targeting in clinical studies.
Materials and Methods
Patient selection and tissue samples
Paraffin wax-embedded tumor samples from 180 patients (mean age 54 years, range 30-70 years) with a squamous cell carcinoma of the oral cavity, oropharynx, hypopharynx and larynx were investigated. Patients had been treated by radical surgical resection between 1993 and 1997 in the Department of Head and Neck Surgery, Klinikum rechts der Isar, Technische Universität München (TUM), Munich, Germany or in the Department of Head and Neck Surgery, University of Regensburg, Regensburg, Germany.
The pT and pN categories of the tumor were determined according to the tumor-node-metastasis classification [40] and tumor grading according to the World Health Organization (WHO) classification [41]. For all tumors and patients, histopathological and clinical follow-up data were available (mean follow-up period 6.6 years, follow-up period of 0.02 to 13.6 years). Clinical and histopathological data were correlated with expression patterns of Aurora-A and EGFR. The study was approved by the Ethics Committee of the Medical Faculty of the TUM. Detailed patient characteristics and histomorphological features are shown in Table 1.
Preparation of Tissue MicroArrays (TMA), Immunohistochemistry (IHC), and Scoring
For each of the 180 SCCHN, one paraffin block was selected. An experienced pathologist marked the viable, representative areas of tumor specimens. Core needle biopsy specimens were retrieved from the original tumor blocks by using a manual arrayer (Beecher Instruments, Sun Prairie, WI, USA) and positioned in a recipient paraffin wax array block. We aimed to obtain at least three tissue cylinders per tumor with a diameter of 0.6 mm from each biopsy specimen.
IHC was performed on deparaffinized tissue sections (2µm), stained with antibodies against Aurora kinase A (Novocastra, Leica-Microsystems, Wetzlar, Germany) and EGFR (Santa Cruz Biotechnology, Santa Cruz, CA), visualized with peroxidase-conjugated secondary antibody (LSAB Kit, DAKO, Hamburg, Germany). The tissue sections were counterstained with Mayer hematoxylin solution. For positive controls, we used tissues with known expression of the respective antigens. For negative controls, we used irrelevant antibodies with the same immunoglobulin isotype.
According to previously published criteria cytoplasmatic and/or nuclear immunoreactivity of Aurora-A [21] and the membrane and/or cytoplasmatic staining of EGFR [42] was evaluated in three tumor areas of each case. Immunoreactivity was scored into seven groups according to the percentage and intensity of cytoplasmic, nuclear and membrane staining of the positively stained tumor cells. Specimens with > 60% of cells stained were scored as strongly positive (4+), those with 30-60% of cells stained were scored as moderately positive (3+), those with 10-20% of cells stained were scored as weakly positive (2+), those with < 10% cells stained were scored as less weakly positive (1+). Specimens with no staining were scored as negative. The intensity of staining was grouped in strong (3+), moderate (2+) and weak (1+). Intensity and percentage of staining cells were added up identifying the seven groups. All scoring analysis was done by two independent investigators. To compare high with low expression levels, a median split analysis was applied. EGFR≥5 and Aurora-A≥5 were specified as high expression.
Cell culture, transfection and plasmids
All cell lines were obtained from ATCC-LGC (Wesel, Germany) or DSMZ (Braunschweig, Germany). SCCHN cells were cultured in DMEM (Invitrogen, Darmstadt, Germany) supplemented with 10% heat inactivated fetal bovine serum (FBS, PAA, Cölbe, Germany), 1% glutamine, 1% penicillin-streptomycin and 1% non-essential amino acids (all from Invitrogen, Darmstadt, Germany) . NIH-3T3 cells were cultured in DMEM supplemented with 10% heat inactivated bovine serum and 1% penicillin-streptomycin. NIH-3T3 cells were transfected with pLERN-EGFRvIII (kind gift of Frank Furnari, La Jolla, CA) with Lipofectamine 2000 according to the manufacturer’s instructions (Invitrogen, Darmstadt, Germany) and selected with G418 (1000µg/ml, Sigma, Munich, Germany). To measure proliferation, SCCHN cells were split, reseeded (5x105 in 25cm2 flasks), and counted at the indicated time points. Cells were then replated at the initial density. The -fold increase in cell number was calculated, all given results are based on triplicate experiments. R763 [25] was kindly provided by EMD-Serono (Rockland, MA). MLN8237 was purchased from Selleck (Houston, TX).
Flow cytometry and apoptosis assays
To assess apoptosis, 5x105 cells were stained with FITC-labeled Annexin V (BD Pharmingen, Heidelberg, Germany) and counterstained with propidium iodide (PI, Sigma, Munich, Germany). Following incubation cells were washed, resuspended in PBS, and analyzed by flow cytometry. The fraction of Annexin V–positive (FL2 channel) PI-negative (FL3 channel) cells was reported as apoptotic. For analysis of cell cycle distribution, cells were fixed with 70% ethanol and stained with PI. Flow cytometric analysis of DNA content was performed using PI in the FL2 channel in linear mode. Cells with less than diploid DNA content were considered dead (sub-G1), cells with more than 2N DNA content were considered polyploid.
RNA preparation and analyses
For reverse transcriptase quantitative PCR (qRT-PCR), RNA was prepared from cultured cells using the RNeasy kit (Qiagen, Hilden, Germany). cDNA was prepared from 2µg RNA using the SuperScript II reverse transcriptase cDNA synthesis kit (Invitrogen, Darmstadt, Germany). qRT-PCR was performed on an ABI Prism 7900 cycler (Applied Biosystems, Darmstadt, Germany) with the Platinum SYBR Green qPCR SuperMIX-UDG kit (Invitrogen, Darmstadt, Germany). Data were analyzed by using the ∆Ct method, where Ubiquitin served as an internal control, with one sample set as 1. RT-PCR was performed to validate the expression of the EGFRvIII mutant in NIH-3T3 cells. Primer sequences can be obtained from the authors upon request.
Immunoblotting
Protein extracts (50µg per lane) were electrophoretically separated on SDS-PAGE gels, transferred to membranes (Protran, Schleicher&Schuell, Dassel, Germany) and blotted with specific antibodies as described earlier [43] (Actin, Aurora-A, Aurora-B: all from Sigma, Munich, Germany; S10-HH3: Millipore, Schwalbach, Germany; EGFR: Santa Cruz, Heidelberg, Germany; pEGFR: Invitrogen, Darmstadt, Germany, pAKT, pERK: both from New England Biolabs, Frankfurt, Germany).
Statistical analysis
Statistical analyses were performed using the statistical functions (t-test if not otherwise indicated) of GraphPad Prism (GraphPad Software, La Jolla, CA). For quantitative variables, means and standard deviations are given, for categorical data absolute and relative frequencies. The bars shown represent the mean ± standard deviation (SD) or standard error of the mean (SEM). Spearman rank correlation coefficient (r) was correlated to assess the relationship between Aurora-A and EGFR expression. Also for box plots showing medians, quantiles and ranges as well as Kaplan-Meier survival analyses the Statistical Package for Social Sciences (SPSS 17.0 Package Facility, SPSS Inc, IL, USA) was used. Survival curves were compared with the log-Rank test. Any p values given are two-sided and subject to a local significance level of 0.05.
Conflict of Interests
The authors declare no potential conflicts of interests.
Acknowledgements
We thank Frank Furnari (La Jolla, CA) for kindly providing the pLERN-EGFRvIII expression plasmid. EMD-Serono (Rockland, MA) kindly provided R763.
Grant Support
This work was supported by the Medical Faculty of the TU München.
References
1. Hunter KD, Parkinson EK, Harrison PR: Profiling early head and neck cancer. Nat Rev Cancer 2005, 5:127-135.
2. Vermorken JB, Mesia R, Rivera F, Remenar E, Kawecki A, Rottey S, Erfan J, Zabolotnyy D, Kienzer HR, Cupissol D, et al: Platinum-based chemotherapy plus cetuximab in head and neck cancer. N Engl J Med 2008, 359:1116-1127.
3. Forastiere A, Koch W, Trotti A, Sidransky D: Head and neck cancer. N Engl J Med 2001, 345:1890-1900.
4. Haddad RI, Shin DM: Recent advances in head and neck cancer. N Engl J Med 2008, 359:1143-1154.
5. Jin C, Jin Y, Wennerberg J, Akervall J, Dictor M, Mertens F: Karyotypic heterogeneity and clonal evolution in squamous cell carcinomas of the head and neck. Cancer Genet Cytogenet 2002, 132:85-96.
6. Jang SJ, Chiba I, Hirai A, Hong WK, Mao L: Multiple oral squamous epithelial lesions: are they genetically related? Oncogene 2001, 20:2235-2242.
7. Braakhuis BJ, Tabor MP, Kummer JA, Leemans CR, Brakenhoff RH: A genetic explanation of Slaughter’s concept of field cancerization: evidence and clinical implications. Cancer Res 2003, 63:1727-1730.
8. Copper MP, Jovanovic A, Nauta JJ, Braakhuis BJ, de Vries N, van der Waal I, Snow GB: Role of genetic factors in the etiology of squamous cell carcinoma of the head and neck. Arch Otolaryngol Head Neck Surg 1995, 121:157-160.
9. Sparano A, Quesnelle KM, Kumar MS, Wang Y, Sylvester AJ, Feldman M, Sewell DA, Weinstein GS, Brose MS: Genome-wide profiling of oral squamous cell carcinoma by array-based comparative genomic hybridization. Laryngoscope 2006, 116:735-741.
10. Zhang Z, Stiegler AL, Boggon TJ, Kobayashi S, Halmos B: EGFR-mutated lung cancer: a paradigm of molecular oncology. Oncotarget 2010, 1:497-514.
11. D’Agostino L, Giordano A: NSP 5a3a: a potential novel cancer target in head and neck carcinoma. Oncotarget 2010, 1:423-435.
12. Chung CH, Ely K, McGavran L, Varella-Garcia M, Parker J, Parker N, Jarrett C, Carter J, Murphy BA, Netterville J, et al: Increased epidermal growth factor receptor gene copy number is associated with poor prognosis in head and neck squamous cell carcinomas. J Clin Oncol 2006, 24:4170-4176.
13. Ang KK, Berkey BA, Tu X, Zhang HZ, Katz R, Hammond EH, Fu KK, Milas L: Impact of epidermal growth factor receptor expression on survival and pattern of relapse in patients with advanced head and neck carcinoma. Cancer Res 2002, 62:7350-7356.
14. Hitt R, Ciruelos E, Amador ML, Benito A, Sanchez JJ, Ballestin C, Cortes-Funes H: Prognostic value of the epidermal growth factor receptor (EGRF) and p53 in advanced head and neck squamous cell carcinoma patients treated with induction chemotherapy. Eur J Cancer 2005, 41:453-460.
15. Rubin Grandis J, Melhem MF, Gooding WE, Day R, Holst VA, Wagener MM, Drenning SD, Tweardy DJ: Levels of TGF-alpha and EGFR protein in head and neck squamous cell carcinoma and patient survival. J Natl Cancer Inst 1998, 90:824-832.
16. Baselga J: The EGFR as a target for anticancer therapy--focus on cetuximab. Eur J Cancer 2001, 37 Suppl 4:S16-22.
17. Forastiere AA, Metch B, Schuller DE, Ensley JF, Hutchins LF, Triozzi P, Kish JA, McClure S, VonFeldt E, Williamson SK, et al.: Randomized comparison of cisplatin plus fluorouracil and carboplatin plus fluorouracil versus methotrexate in advanced squamous-cell carcinoma of the head and neck: a Southwest Oncology Group study. J Clin Oncol 1992, 10:1245-1251.
18. Andrews PD, Knatko E, Moore WJ, Swedlow JR: Mitotic mechanics: the auroras come into view. Curr Opin Cell Biol 2003, 15:672-683.
19. Toya M, Terasawa M, Nagata K, Iida Y, Sugimoto A: A kinase-independent role for Aurora A in the assembly of mitotic spindle microtubules in Caenorhabditis elegans embryos. Nat Cell Biol 2011, 13:710-716.
20. Vader G, Lens SM: The Aurora kinase family in cell division and cancer. Biochim Biophys Acta 2008, 1786:60-72.
21. Reiter R, Gais P, Jutting U, Steuer-Vogt MK, Pickhard A, Bink K, Rauser S, Lassmann S, Hofler H, Werner M, Walch A: Aurora kinase A messenger RNA overexpression is correlated with tumor progression and shortened survival in head and neck squamous cell carcinoma. Clin Cancer Res 2006, 12:5136-5141.
22. Cromer A, Carles A, Millon R, Ganguli G, Chalmel F, Lemaire F, Young J, Dembele D, Thibault C, Muller D, et al: Identification of genes associated with tumorigenesis and metastatic potential of hypopharyngeal cancer by microarray analysis. Oncogene 2004, 23:2484-2498.
23. Hirota T, Lipp JJ, Toh BH, Peters JM: Histone H3 serine 10 phosphorylation by Aurora B causes HP1 dissociation from heterochromatin. Nature 2005, 438:1176-1180.
24. Sok JC, Coppelli FM, Thomas SM, Lango MN, Xi S, Hunt JL, Freilino ML, Graner MW, Wikstrand CJ, Bigner DD, et al: Mutant epidermal growth factor receptor (EGFRvIII) contributes to head and neck cancer growth and resistance to EGFR targeting. Clin Cancer Res 2006, 12:5064-5073.
25. McLaughlin J, Markovtsov V, Li H, Wong S, Gelman M, Zhu Y, Franci C, Lang DW, Pali E, Lasaga J, et al: Preclinical characterization of Aurora kinase inhibitor R763/AS703569 identified through an image-based phenotypic screen. J Cancer Res Clin Oncol 2010, 136:99-113.
26. Raben D, Helfrich B, Chan DC, Ciardiello F, Zhao L, Franklin W, Baron AE, Zeng C, Johnson TK, Bunn PA, Jr.: The effects of cetuximab alone and in combination with radiation and/or chemotherapy in lung cancer. Clin Cancer Res 2005, 11:795-805.
27. Yoshida T, Okamoto I, Okabe T, Iwasa T, Satoh T, Nishio K, Fukuoka M, Nakagawa K: Matuzumab and cetuximab activate the epidermal growth factor receptor but fail to trigger downstream signaling by Akt or Erk. Int J Cancer 2008, 122:1530-1538.
28. McLaughlin J, Markovtsov V, Li H, Wong S, Gelman M, Zhu YH, Franci C, Lang DW, Pali E, Lasaga J, et al: Preclinical characterization of Aurora kinase inhibitor R763/AS703569 identified through an image-based phenotypic screen. Journal of Cancer Research and Clinical Oncology 2010, 136:99-113.
29. Perez de Castro I, de Carcer G, Malumbres M: A census of mitotic cancer genes: new insights into tumor cell biology and cancer therapy. Carcinogenesis 2007, 28:899-912.
30. Astsaturov I, Ratushny V, Sukhanova A, Einarson MB, Bagnyukova T, Zhou Y, Devarajan K, Silverman JS, Tikhmyanova N, Skobeleva N, et al: Synthetic lethal screen of an EGFR-centered network to improve targeted therapies. Sci Signal 2010, 3:ra67.
31. Soncini C, Carpinelli P, Gianellini L, Fancelli D, Vianello P, Rusconi L, Storici P, Zugnoni P, Pesenti E, Croci V, et al: PHA-680632, a novel Aurora kinase inhibitor with potent antitumoral activity. Clin Cancer Res 2006, 12:4080-4089.
32. Hung LY, Tseng JT, Lee YC, Xia W, Wang YN, Wu ML, Chuang YH, Lai CH, Chang WC: Nuclear epidermal growth factor receptor (EGFR) interacts with signal transducer and activator of transcription 5 (STAT5) in activating Aurora-A gene expression. Nucleic Acids Res 2008, 36:4337-4351.
33. Tinhofer I, Klinghammer K, Weichert W, Knodler M, Stenzinger A, Gauler T, Budach V, Keilholz U: Expression of Amphiregulin and EGFRvIII Affect Outcome of Patients with Squamous Cell Carcinoma of the Head and Neck (SCCHN) Receiving Cetuximab-Docetaxel Treatment. Clin Cancer Res 2011.
34. Giono LE, Manfredi JJ: The p53 tumor suppressor participates in multiple cell cycle checkpoints. J Cell Physiol 2006, 209:13-20.
35. Poeta ML, Manola J, Goldwasser MA, Forastiere A, Benoit N, Califano JA, Ridge JA, Goodwin J, Kenady D, Saunders J, et al: TP53 mutations and survival in squamous-cell carcinoma of the head and neck. N Engl J Med 2007, 357:2552-2561.
36. Kuntz K, O’Connell MJ: The G(2) DNA damage checkpoint: could this ancient regulator be the Achilles heel of cancer? Cancer Biol Ther 2009, 8:1433-1439.
37. Jordan MA, Wilson L: Microtubules as a target for anticancer drugs. Nat Rev Cancer 2004, 4:253-265.
38. Katayama H, Sasai K, Kawai H, Yuan ZM, Bondaruk J, Suzuki F, Fujii S, Arlinghaus RB, Czerniak BA, Sen S: Phosphorylation by aurora kinase A induces Mdm2-mediated destabilization and inhibition of p53. Nat Genet 2004, 36:55-62.
39. Rao B, van Leeuwen IM, Higgins M, Campbel J, Thompson AM, Lane DP, Lain S: Evaluation of an Actinomycin D/VX-680 aurora kinase inhibitor combination in p53-based cyclotherapy. Oncotarget 2010, 1:639-650.
40. Sobin LW, C. (Ed.). TNM classification of malignant tumours (UICC). , Sixth Edition edition: Wiley; 2002.
41. Hamilton S.R. ALA (Ed.). Classification of tumours. Pathology and genetics of tumours of the digestive system. Lyon: IARC Press; 2000.
42. Gamboa-Dominguez A, Dominguez-Fonseca C, Quintanilla-Martinez L, Reyes-Gutierrez E, Green D, Angeles-Angeles A, Busch R, Hermannstadter C, Nahrig J, Becker KF, et al: Epidermal growth factor receptor expression correlates with poor survival in gastric adenocarcinoma from Mexican patients: a multivariate analysis using a standardized immunohistochemical detection system. Mod Pathol 2004, 17:579-587.
43. den Hollander J, Rimpi S, Doherty JR, Rudelius M, Buck A, Hoellein A, Kremer M, Graf N, Scheerer M, Hall MA, et al: Aurora kinases A and B are up-regulated by Myc and are essential for maintenance of the malignant state. Blood 2010, 116:1498-1505.