
Introduction
MYC is a basic helix-loop-helix leucine zipper (bHLHLZ) protein that controls cell proliferation, differentiation, metabolism, apoptosis, and the maintenance of pluripotency. It is a key component of a broad transcription factor network, forming heterodimers with the bHLHLZ protein MAX [1-4]. MYC-MAX dimers bind to DNA Enhancer (E)-box elements with the consensus sequence CACGTG at target gene promoters and positively or negatively regulate gene expression [5].
MYC plays a critical role in human cancer [3, 6]. In numerous tumor types, it shows gain of function, primarily through overexpression or amplification [7, 8]. The classic example is Burkitt’s lymphoma in which a t(8;14) translocation brings the transcription of MYC under the control of the immunoglobulin heavy chain locus [9], leading to ectopically high levels of MYC expression. In other tumor types, the incidence of MYC gain of function extends from 5 to 45% [7, 8]. The traditional view of MYC-driven tumorigenesis is that the MYC protein behaves as a classical transcription factor, which regulates the expression of a specific set of downstream genes that contribute to cancer progression. However, it has been shown recently that MYC-mediated transcriptional regulation is so widespread that MYC can be considered an ‘amplifier’ of transcription on a global scale [10-13].
Studies on MYC-mediated transcriptional regulation have focused mainly on coding transcripts; specific links between MYC and non-coding RNAs (ncRNAs) have been investigated only recently [14-19]. The importance of ncRNAs, such as transfer RNAs, ribosomal RNAs and spliceosome-associated RNAs, has been recognized since the formative years of molecular biology. However, the recent development of high-throughput sequencing and RNA tiling array technologies has revealed the full extent to which mammalian genomes are pervasively transcribed [20, 21]. It is now known that the majority of gene loci produce multiple interlaced [22] and overlapping transcripts [23] in both sense and antisense orientations [24, 25]. Of particular interest are the long non-coding RNAs (lncRNAs). lncRNAs are mRNA-like transcripts (>200 nucleotides) with low protein coding potential. A variety of diverse functions have been ascribed to lncRNAs including post-transcriptional gene silencing [26-29], epigenetic regulation [30-34], modulation of transcription factor function [35], modulation of alternative splicing [36, 37], nuclear organization [38-40], sub-cellular trafficking [41], generation of small RNAs [42-44], sequestration of signaling proteins [45], telomere function [46], and regulation of 3-dimensional chromatin structure [47]. lncRNAs are therefore important components of the gene regulatory apparatus of the cell. Yet the control and functional mechanisms of most lncRNAs are only just beginning to be explored [48]. Here we show that the broad transcriptional control that MYC exerts on the coding transcriptome also extends to the lncRNA transcriptome.
Results
Numerous lncRNAs are regulated by MYC in P493-6 cells
To study the effect of MYC on the non-coding transcriptome, we used the human B-cell line P493-6. P493-6 cells express high levels of MYC from an integrated transgene under the control of a tet-off promoter [49]. These cells are therefore a useful system for studying MYC target genes, as the addition of doxycycline reduces MYC expression by >40 fold. To investigate the possible role of MYC in regulating the non-coding RNA transcriptome, triplicate cultures of P493-6 cells were prepared in the presence (low, endogenous MYC) or absence (high MYC) of doxycycline. Total RNA was harvested after 24 hours and transcript expression levels determined by RNA sequencing (RNA-seq). Total RNA samples were depleted of ribosomal RNAs and strand-specific cDNA libraries were prepared in order to maximize sequencing depth, distinguish between sense and antisense RNAs, and to enable the measurement of transcripts irrespective of their 3′ terminal polyadenylation status. Raw reads were aligned to the hg19 build of the human genome, and reads uniquely aligning with non-coding transcripts (as defined in the Gencode v19 non-coding transcripts [50, 51]) were counted [52]. A total of 1,273 non-coding transcripts were detected (Supplemental Table 1). MYC significantly upregulated 296 and downregulated 238 non-coding transcripts by more than 2-fold (P<10-3). A visualization of expression ratios by heat map (Fig. 1a) or volcano plot (Fig. 1b) reveals the widespread extent of MYC-dependent regulation of the non-coding transcriptome. We also observed increased levels of total RNA and nuclear RNA per cell at high levels of MYC (Fig. 2). This is in accord with the MYC-driven transcriptional amplification effect reported in previous studies [10, 11].
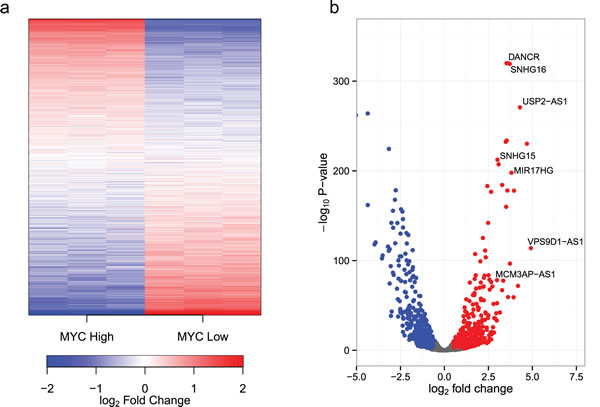
Figure 1: MYC regulates the expression of ncRNAs. (a) A heatmap of ncRNAs detected in P493-6 cells. P493-6 cells were analyzed by RNA-seq in triplicate under conditions of high and low MYC expression. Transcripts are ordered by the change in expression upon MYC upregulation with those most upregulated at the top and those most downregulated at the bottom. ![]() where i refers to the sample (column), gene refers to the gene (row) and N refers to the total number of samples (6). (b) A volcano plot of observed fold changes in ncRNA expression vs significance.
where i refers to the sample (column), gene refers to the gene (row) and N refers to the total number of samples (6). (b) A volcano plot of observed fold changes in ncRNA expression vs significance.
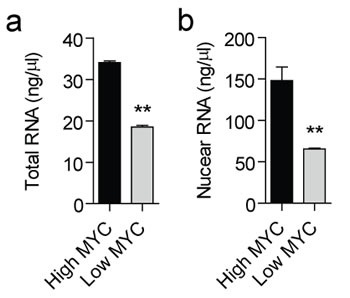
Figure 2: Total and nuclear RNA content at high and low MYC levels. Total RNA was prepared using an RNA extraction robot from (a) ~2x105 P493-6 cells with high or low expression of MYC, or (b) ~4x106 purified nuclei in triplicate. Samples were eluted in a fixed volume of water and RNA concentration determined by Nanodrop. Values are mean + SEM, **P<0.01.
MYC is present at the promoters of ncRNA transcripts
MYC could influence the expression of lncRNA genes either by directly binding to their promoters or by regulating the expression of other transcription factors which subsequently target the non-coding genes. To test for direct regulation by MYC, we have interrogated publicly available P493-6 ChIP-seq (chromatin immunoprecipitation sequencing) data for MYC enrichment proximal to the transcription start sites (TSS) of the 1,273 detected lncRNA genes (Fig. 3). Some MYC-responsive lncRNAs show an increase in MYC occupancy near their TSS similar to that seen for coding transcripts, and MYC typically binds within 1 kb of the TSS. MYC directly binds the TSS in 616 out of 1,273 lncRNAs (48%); this is similar to the situation for coding genes where 6,911 of 10,382 (67%) have MYC present at the TSS. Notably, MYC binding to near TSS was not always associated with a detectable change of expression. This fact may reflect the global effect of MYC on RNA synthesis. These data suggest that lncRNA genes are direct targets of MYC.
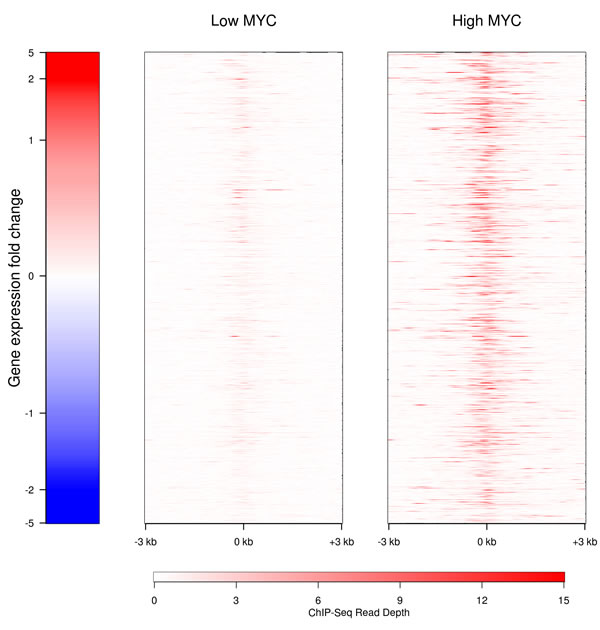
Figure 3: Binding of MYC to the promoters of lncRNAs in the presence of high (right) and low (left) levels of MYC. The MYC binding to the proximal promoters of the lncRNAs with detectable transcription is shown in red. Genes are ranked according to their level of expression.
MYC regulates lncRNA transcripts in other cell lines
Public ChIP-seq and RNA-seq datasets are also available for two additional cell lines that show varying levels of MYC expression. The multiple myeloma cell line MM.1S overexpresses MYC as a consequence of a translocation [53], whereas the U87-MG glioblastoma cell line exhibits a more modest increase in MYC expression [54]. The number of promoters bound by MYC is correlated with the amount of MYC present in the cell line. MYC binding is observed at a very similar subset of promoters across all three cell lines. However, at high cellular concentrations of MYC more promoters are targeted than at low levels of MYC (Fig. S1). Yet despite this concordant pattern of binding, each cell line expresses a distinct set of lncRNAs (Fig. 4). Only a minority of lncRNAs are expressed in all three cell lines (14%, 390/2,755). In contrast, a majority of protein-coding genes are expressed in the three cell lines (65%, 8,830/13,650), with fewer transcripts specific to an individual cell line. This observation suggests the existence of additional, possibly differentiation-specific factors that control the transcriptional regulator activity of promoter-bound MYC.
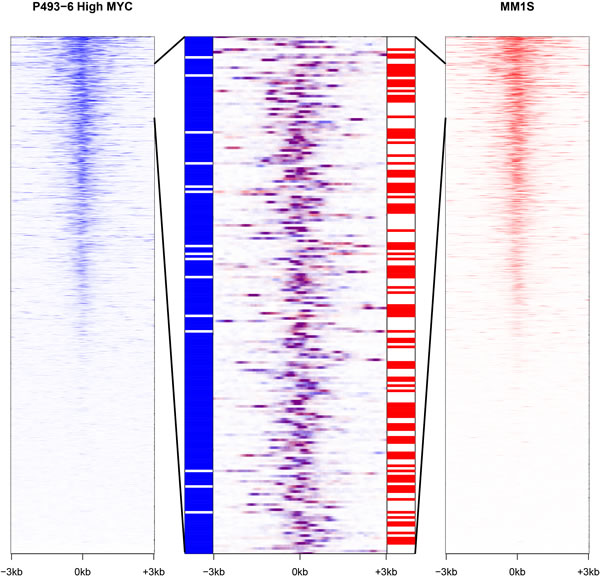
Figure 4: MYC stimulates the transcription of distinct sets of lncRNAs in different cell lines. In the extreme right and left panels, lncRNA promoters are ranked according to MYC occupancy for P493-6 cells (high MYC levels) and MM.1S cells respectively. The central panel depicts an overlap of an enlarged segment from the side panels and shows that MYC promoter occupancy is virtually the same in both cell lines (purple color). However, the two cell lines differ starkly in the expression of the corresponding lncRNAs represented in the bar strips flanking the central panel (with blue signifying expression in P493-6 cells and red indicating expression in MM.1S cells).
Classification of MYC-regulated lncRNA transcripts
lncRNAs are frequently produced by bidirectional transcription [24, 55]. There are 2,747 bidirectional promoters which are bound by MYC, and 444 of these involve a lncRNA transcript. The MYC-bound bidirectional promoters were divided into functional categories: bidirectional (both genes expressed) and unidirectional (one of the two genes expressed) and classified according to the impact of high MYC levels upon expression of their respective transcripts (Fig. 5). Many of the bidirectional promoters behave in a discordant manner (19%, 516/2,747) showing a significant preference for one direction over the other. Concordant regulation of both transcripts is seen in the minority of the bidirectional promoters (5%, 141/2,747).
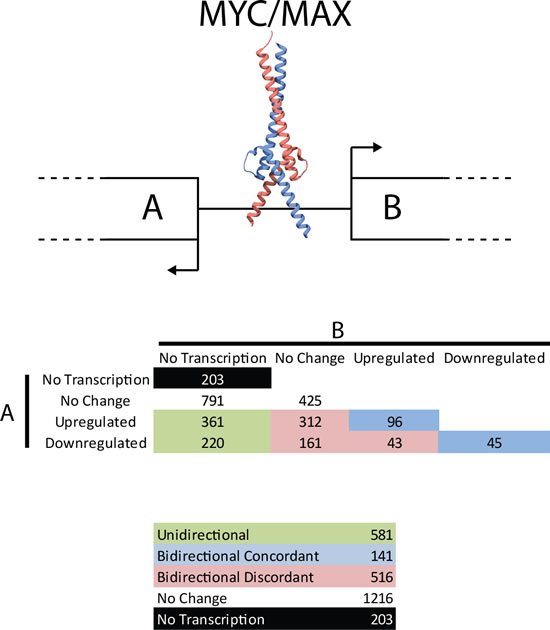
Figure 5: Transcriptional activity from bidirectional promoters. MYC binds to bidirectional promoters and drives transcription. ncRNAs are frequently produced from bidirectional promoters. Shown here are the number of MYC-bound bidirectional promoters producing 0 (no transcription), 1 (unidirectional) or 2 (bidirectional) transcripts and whether MYC regulates the genes involved (red, blue and green colored are MYC-regulated, black and white are not MYC-regulated). Despite the symmetric nature of the E-box motif, MYC frequently acts asymmetrically at bidirectional promoters. Numbers indicate the number of bidirectional promoters acting in a specific way.
Polyadenylation of lncRNAs
Whereas the vast majority of coding genes are polyadenylated, many lncRNAs are not [56-59]. To investigate the polyadenylation status of the lncRNA transcriptome, we compared our ribosomal RNA-depleted RNA-seq data with published poly-A-enriched sequencing data prepared from the same cells under identical conditions [13]. A linear relationship was observed between read counts for ribosomal RNA-depleted RNA and poly-A experiments for coding genes (R2=0.76, P<10-16 for low MYC and R2=0.58, P<10-16 for high MYC). Using a linear discriminator classifier, we found only a small number of coding transcripts that lack polyadenylation (0.6%, 69/11,154). Performing a similar analysis for the lncRNA transcripts, we found a weaker, but significant, linear relationship between ribosome-depleted and poly-A methods (low MYC R2=0.44, P<10-16; high MYC R2=0.36, P<10-16). Applying the discriminator established with coding transcripts, a much larger number of non-coding transcripts (18%, 241/1,371) were found to lack polyadenylation.
Validation of RNA-seq data
To validate the RNA-seq data, we selected eight representative lncRNAs (Table 1) based on the following criteria: (1) significantly upregulated by MYC by >8 fold in the RNA-seq dataset, (2) well annotated in UCSC (University of California Santa Cruz) genome browser, (3) detected with >5 reads per sample, and (4) presence of a MYC binding ChIP-seq peak proximal (+/- 0.5kb) to the lncRNA TSS. These eight lncRNAs could be grouped into three general classes: (1) host genes for small RNAs (DANCR, MIR17HG, SNHG15, SNHG16), (2) concordantly regulated sense-antisense pairs (MCM3AP-AS1, USP2-AS1), and (3) discordantly regulated sense-antisense pairs (KTN1-AS1, VPS9D1-AS1). Analysis of exon-exon junction spanning split reads supported the intron-exon gene models annotated in the UCSC genome browser hg19 for all eight lncRNA genes. Parallel nuclear run-on (NRO) and steady-state gene expression analysis by RT-qPCR (quantitative reverse transcription PCR) were performed on P493-6 cultures in the presence and absence of doxycycline. Given the precedent for sense gene regulation by proximal antisense transcripts, we also investigated expression of neighboring or overlapping transcripts where applicable. Using this strategy, RNA-seq expression data were validated at both the transcriptional and steady-state level for all eight lncRNAs. A detailed example of two of these validated genes is presented in Fig. 6; the data on the remaining 6 validated lncRNAs are shown in Figs. S2 through S7. A positive correlation (Pearson r=0.927, P=0.0027) was observed between NRO and RT-qPCR expression ratios indicating that these lncRNAs are predominantly regulated at the level of transcription.
The properties of lncRNAs can provide clues as to their function. For example, lncRNAs which execute their functions at the transcriptional level are expected to be enriched in the nucleus. To investigate the sub-cellular localization of the eight representative lncRNAs, P493-6 cells were separated into nuclear and cytoplasmic fractions and lncRNA levels assessed by RT-qPCR. Two transcripts were predominantly cytoplasmic (DANCR and SNHG16) whereas three transcripts were enriched in the nucleus (MCM3AP-AS1, MIR17HG and USP2-AS1). The remaining transcripts were evenly distributed between nucleus and cytoplasm (Fig. S8). Interestingly, a positive correlation (Pearson r=0.675, P=0.023) was observed between transcript abundance and the cytoplasmic:nuclear distribution ratio such that the more abundant lncRNAs tended to be enriched in the cytoplasm. All eight of the validated lncRNAs could be amplified from oligo dT primed cDNA libraries suggesting that these transcripts are polyadenylated, and that polyadenylation is unlikely to be a determinant of sub-cellular localization for this subset of transcripts.
Determination of transcript stability can be informative with respect to function as one class of lncRNAs (the PROMPTs or PROMoter uPstream Transcripts) is known to be highly unstable [60]. Equally, non-functional transcripts which arise as a result of ‘noisy transcription’ might also be expected to be short-lived. To investigate the transcript half-lives of the eight representative lncRNAs, P493-6 cultures were treated with Actinomycin D in order to inhibit transcription [61], RNA was collected over a series of time points, transcript levels were measured by PCR, and transcript half-lives (t1/2) calculated (Fig. S9, Table 1). The half-lives of MYC (fast turnover: t1/2=30 minutes) and GAPDH (slow turnover: t1/2=215 minutes) were also measured for comparison. The eight lncRNAs showed transcript stabilities that extended from half-lives of 132 minutes (DANCR) to 25 minutes (MIR17HG). These results demonstrate that the eight validated lncRNAs have mRNA-like stability properties and are unlikely to be PROMPTs (despite some being promoter-overlapping) or a consequence of transcriptional noise.
Table 1: Representative MYC-regulated lncRNAs. Eight putative MYC-regulated lncRNAs were selected for validation studies. CPM, counts per million.
lncRNA (RefSeq ID) |
Fold up-regulated RNA-seq (RT-qPCR) |
Mean CPM |
Nuclear/ Cytoplasmic |
Poly-A |
t1/2 (minutes) |
Predicted function |
DANCR (NR_024031.1) |
+13.3 (+9.4) |
48 |
Cytoplasmic |
+ |
132 |
Small RNA host gene/ Unknown cytoplasmic function |
KTN1-AS1 (NR_027123.1) |
+21.9 (+12.5) |
6.6 |
Both |
+ |
66 |
Discordant bidirectional promoter (KTN1) |
MCM3AP-AS1 (NR_002776.4) |
+8.5 (+1.7) |
8.9 |
Nuclear |
+ |
57 |
Concordant bidirectional promoter (LSS) |
MIR17HG (NR_027349.1) |
+15.5 (+17.0) |
24 |
Nuclear |
+ |
25 |
Small RNA host gene |
SNHG15 (NR_003697.1) |
+9.0 (+3.0) |
23 |
Both |
+ |
31 |
Small RNA host gene |
SNHG16 (NR_038111.1) |
+14.5 (+10.4) |
69 |
Cytoplasmic |
+ |
125 |
Small RNA host gene |
VPS9D1-AS1 (NR_036480.1) |
+33.3 (+10.5) |
8.0 |
Both |
+ |
107 |
Post-transcriptional regulator of VPS9D1 |
USP2-AS1 (NR_034160.1) |
+21.9 (+21.2) |
22 |
Both |
+ |
127 |
Discordant bidirectional promoter (USP2) |
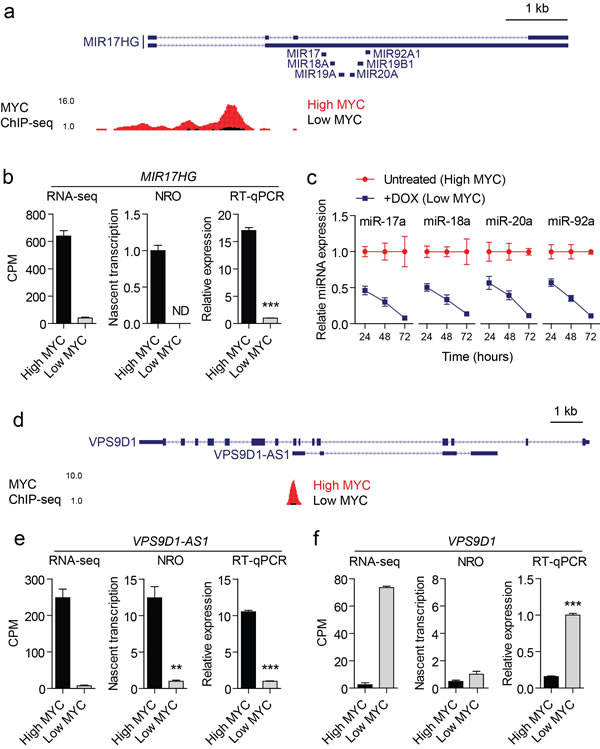
Figure 6: Validation data for two representative MYC-regulated lncRNAs. (a) MYC ChIP-seq data were visualized in the UCSC genome browser for the MIR17HG locus. The locations of miRNA precursors are indicated. (b) Expression of MIR17HG in high MYC and low MYC conditions was determined by RNA-seq, NRO and steady-state RT-qPCR. (c) Mature miRNAs from the miR-17-92 cluster were also affected by MYC levels. Addition of doxycycline (DOX) reduced miRNA levels. This effect of the repression increased over 72 hours. (d) MYC ChIP-seq data were visualized at the VPS9D1-AS1 locus. The VPS9D1-AS1 gene is antisense to, and resides completely within, its corresponding sense coding gene VPS9D1. Expression of (e) VPS9D1-AS1 and (f) VPS9D1 were determined by RNA-seq, NRO and RT-qPCR. This sense-antisense pair was discordantly regulated such that VPS9D1-AS1 was transcriptionally upregulated under high MYC expression, whereas VPS9D1 was correspondingly downregulated suggesting possible regulatory relationship. The change in VPS9D1 expression observed by RNA-seq and steady-state RT-qPCR was not recapitulated in the NRO data indicating that VPS9D1 is regulated at the post-transcriptional level. Values are mean +/- SEM, n=3, **P<0.01, ***P<0.001, ND not detected.
Discussion
Recent work has suggested that MYC acts as an amplifier of global transcriptional output [10, 11]. The present study shows that in addition to affecting the coding transcriptome, overexpression of MYC also induces widespread changes in non-coding transcription. MYC binds to the promoters of lncRNA transcripts, but that interaction is not always sufficient to modulate transcription of the target promoter. The general picture that emerges from the data is that the extent and degree of MYC-mediated transcriptional activity is similar for non-coding and coding transcripts. However, there is one significant difference. Expression from MYC-binding lncRNA promoters is, in contrast to the observations on coding genes, largely distinct in different cell types. There are cell type-specific subsets of lncRNAs. This phenomenon has been documented previously. An investigation of the transcriptional landscape of multiple human cell lines found that 29% of lncRNAs are expressed specifically in a single cell type, whereas only 10% are expressed in all cell types [62]. This specificity could reflect the regulatory functions of many lncRNAs and suggests the existence of additional, possibly differentiation-specific factors that control the effectiveness of promoter-bound MYC.
The functions of most lncRNAs are currently unknown. There are, however, broad criteria that define various categories of lncRNAs and that are relevant to function. These include antisense RNAs likely to control their sense counterparts and nuclear versus cytoplasmic localization that could serve as broad indicator of function. Transcriptional activities initiated from bidirectional promoters can be divided into concordant and discordant with implied consequences on function.
Eight lncRNAs that were upregulated in high MYC expression conditions were validated by RT-qPCR. MYC binding was enriched at the promoters of these genes when MYC expression was high, and regulation at the transcriptional level confirmed by NRO. This set of representative MYC-regulated lncRNAs exhibited diverse properties in terms of their genomic organization, sub-cellular localization, abundance, and transcript stability (Table 1) although all eight lncRNAs were found to be polyadenylated. Some functions of the eight validated lncRNAs are apparent. DANCR, MIR17HG, SNHG15 and SNHG16 act as precursors of small RNA species including miRNAs and snoRNAs. These function in post-transcriptional gene regulation and rRNA editing/maturation respectively.
The oncogenic activities of MYC are likely to include regulation of lncRNAs. The transcriptional activation of negative regulators could be a major mechanism of MYC-mediated gene silencing. Among the eight validated lncRNAs, there are three with relevance to cancer, MIR17HG, USP2-AS1, and DANCR; the roles of the remaining five are currently not known. MIR17HG encodes the miR-17-92 cluster. This miRNA cluster is upregulated by MYC (Fig. 6), and we have confirmed this upregulation for the processed, mature miRNAs. Their roles in cancer have been widely documented [16, 63-66]. The transcription of USP2-AS1 is also activated by MYC. The corresponding sense gene USP2 is a ubiquitin-specific cysteine protease that shows diverse pro-oncogenic activities [67] and could be controlled by a regulatory circuit that includes USP2-AS1. DANCR is also upregulated by MYC. It generates miR-4449 and SNORA26 and is downregulated during differentiation [68, 69], hence upregulated DANCR might contribute to cancer by maintaining a pro-proliferative state.
The regulatory effect of MYC on a broad segment of the non-coding transcriptome opens up a new area of MYC activity. The challenge is now to identify the cancer-relevant lncRNA targets of MYC and determine their functions.
Materials and Methods.
Cell Culture
P493-6 cells suspensions were maintained in RPMI 1640 (Life Technologies) containing 10% tetracycline-free FBS (Gemini) and supplemented with 2 mM L-glutamine, 100 U penicillin, 0.1 mg/mL streptomycin (Sigma-Aldrich) at 37°C and 5% CO2 at cell densities between 105 and 106 cells/mL. To inhibit MYC expression, doxycycline (Sigma-Aldrich) was added to the media to a final concentration of 0.1 µg/mL.
Bioinformatics
Both ribosome-depleted and poly-A-enriched RNA-seq +/- doxycycline P493-6 were previously published and were obtained from NCBI SRA accession numbers: SRR1313741, SRR1313742, SRR1313743, SRR1313735, SRR1313736, SRR1313737, SRR567561, SRR567562 [13, 70]. U87MG (SRR492058, SRR492059) [23] and MM.1S (SRR931814) [71] were also obtained through NCBI SRA. Reads were mapped using the RNA STAR aligner (2 pass method) [72] to hg19. Counts aligning to Gencode v19 coding and non-coding genes [50, 51]were obtained using HTSeq-count [50, 52]. Differential expression was analyzed using EdgeR. Coverage data was obtained using Bedtools genomecov [73] and UCSC Kent utilities [74].
ChIP-seq data for P493-6, MM.1S and U87 were previously published [11] and were obtained through NCBI SRA accession numbers: SRR444481, SRR444479, SRR444467, SRR444466, SRR444464, SRR444432, SRR444430, SRR444429. The raw reads were mapped to hg19 using BWA [75, 76]. Binding peaks for MYC were determined using MACS [77] and annotated using Homer [78]. Coverage plots were generated from Homer annotation data and plotted in R.
Poly-A and rRNA-depleted RNA-seq data were compared. Raw counts were normalized to counts per million and log2 transformed. Genes with less than 5 counts in the lowest sample were filtered out. Known polyadenylated transcripts and those lacking polyadenylation [57] were used to construct a training set for linear discriminant analysis.
Nuclear Run-On
Treated P493-6 cultures were counted using a Countess Automated Cell Counter (Life Technologies) and 3.6x106 cells were aliquoted per NRO sample. 5% of the cells were removed and stored separately for parallel steady-state RT-qPCR analysis. Nuclei were harvested in NP-40 Lysis Buffer (10 mM Tris-HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.5% NP-40) and resuspended in Nuclei Storage Buffer (50 mM Tris-HCl pH 8.3, 0.1 mM EDTA, 5 mM MgCl2, 40% glycerol). NRO transcription was performed by adding 2x transcription buffer (20 mM Tris-HCl pH 8.3, 5 mM MgCl2, 300 mM KCl, 4 mM DTT) supplemented with 1 mM ATP, 1 mM CTP, 1 mM GTP, 0.5 mM UTP, 0.5 mM BrUTP and 100 U RNaseOUT and incubating for 30 minutes at 30°C. A spike-in control RNA (Renilla luciferase mRNA prepared by T7 in vitro transcription in the presence of BrUTP) was added to act as an external reference for data normalization that was independent of global changes in nuclear RNA content. NRO reactions were terminated by immediately lysing the nuclei and extracting RNA using the Maxwell-16 System RNA LEV kit (Promega). Genomic DNA was removed by a DNase I treatment step included in the extraction protocol. ~500 ng of NRO-RNA per sample were immunoprecipitated using mouse monoclonal anti-BrdU antibodies (Santa Cruz Biotechnology) and Protein G Dynabeads (Life Technologies). Beads were blocked with blocking buffer (PBST, 0.1% Polyvinylpyrrolidone, 0.1% UltraPure BSA) and NRO-RNA samples incubated with the beads for 30 minutes at room temperature on a rotating platform. Subsequently, the beads were washed three times with PBST (supplemented with 8 U/ml RNaseOUT). Immunoprecipitated NRO-RNAs were extracted using TRIzol Reagent (Life Technologies) according to manufacturer’s instructions and RT-qPCR performed.
Cellular Fractionation
P493-6 nuclear and cytoplasmic RNA samples were prepared using the Protein and RNA Isolation System (PARIS) kit (Life Technologies) and genomic DNA removed using the TURBO DNA-free kit (Life Technologies) according to manufacturer’s instructions.
Determination of Transcript Half-life
P493-6 cultures were treated with 2 µg/mL Actinomycin D and then harvested at 30, 60, 120 and 240 minutes later. RNA was extracted and transcript levels assessed by RT-qPCR. Transcript half-lives were calculated using the formula:
![]()
RT-qPCR
RNA samples were reverse-transcribed using the High Capacity cDNA Synthesis kit (Life Technologies) according to manufacturer’s instructions (using a random priming or oligo dT-priming strategy as appropriate). qPCR was performed on either a Mastercycler ep RealPlex 2 (Eppendorf) or LightCycler 96 (Roche) real-time PCR instrument. Reactions were prepared using KAPA SYBR FAST qPCR Master Mix (KAPA Biosystems) and universal cycling conditions (95°C for 3 minutes followed by 40 cycles of 95°C for 3 seconds and 60°C for 30 seconds). Reaction specificity was confirmed by melt curve analysis. All primer sequences are listed in Table S2. For miRNA quantification the TaqMan MicroRNA Reverse Transcription Kit (Life Technologies) was used with the following Small RNA TaqMan Assays: miR-17a, miR-18a, miR-20a and miR-92a (#4427975, #000394, #000580 and #000431). Steady-state gene expression measurements were normalized to the geometric mean [79] of RPLP0 and RPL10 (two transcripts that were found to be largely unaffected by MYC-driven transcriptional amplification). For NRO-RT-qPCR, data were normalized to a bromouridylated spike-in control transcript (RLuc) added at the NRO phase in order to provide an external reference transcript independent of MYC-induced changes in global transcriptional activity. miRNA data were normalized to RNU6 (#001973) expression.
Acknowledgments
The authors thank Chi Van Dang for generously providing the P493-6 cells. The authors are grateful to Lynn Ueno for superb technical support and to Anja Zembrzycki for assistance with organizing and producing this manuscript. This work was supported by the National Institutes of Health under award numbers R01 CA153124, R01 CA151574 and R01 CA078230. This is manuscript 29023 of The Scripps Research Institute.
References
1. Blackwood EM, Eisenman RN. Max: a helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science 1991; 251:1211-1217.
2. Conacci-Sorrell M, McFerrin L, Eisenman RN. An overview of MYC and its interactome. Cold Spring Harb Perspect Med 2014; 4:a014357.
3. Dang CV. MYC on the path to cancer. Cell 2012; 149:22-35.
4. Eisenman RN. Deconstructing myc. Genes & development 2001; 15:2023-2030.
5. Eilers M, Eisenman RN. Myc’s broad reach. Genes & development 2008; 22:2755-2766.
6. Soucek L, Whitfield J, Martins CP, Finch AJ, Murphy DJ, Sodir NM, Karnezis AN, Swigart LB, Nasi S, Evan GI. Modelling Myc inhibition as a cancer therapy. Nature 2008; 455:679-683.
7. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012; 2:401-404.
8. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013; 6:pl1.
9. Dalla-Favera R, Bregni M, Erikson J, Patterson D, Gallo RC, Croce CM. Human c-myc onc gene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proceedings of the National Academy of Sciences of the United States of America 1982; 79:7824-7827.
10. Nie Z, Hu G, Wei G, Cui K, Yamane A, Resch W, Wang R, Green DR, Tessarollo L, Casellas R, Zhao K, Levens D. c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell 2012; 151:68-79.
11. Lin CY, Loven J, Rahl PB, Paranal RM, Burge CB, Bradner JE, Lee TI, Young RA. Transcriptional amplification in tumor cells with elevated c-Myc. Cell 2012; 151:56-67.
12. Sabo A, Kress TR, Pelizzola M, de Pretis S, Gorski MM, Tesi A, Morelli MJ, Bora P, Doni M, Verrecchia A, Tonelli C, Faga G, Bianchi V, et al. Selective transcriptional regulation by Myc in cellular growth control and lymphomagenesis. Nature 2014; 511:488-492.
13. Loven J, Orlando DA, Sigova AA, Lin CY, Rahl PB, Burge CB, Levens DL, Lee TI, Young RA. Revisiting global gene expression analysis. Cell 2012; 151:476-482.
14. Prensner JR, Chen W, Han S, Iyer MK, Cao Q, Kothari V, Evans JR, Knudsen KE, Paulsen MT, Ljungman M, Lawrence TS, Chinnaiyan AM, Feng FY. The Long Non-Coding RNA PCAT-1 Promotes Prostate Cancer Cell Proliferation through cMyc. Neoplasia 2014; 16:900-908.
15. Zhou X, Chen J, Tang W. The molecular mechanism of HOTAIR in tumorigenesis, metastasis, and drug resistance. Acta Biochim Biophys Sin (Shanghai) 2014; 46:1011-1015.
16. Li Y, Choi PS, Casey SC, Dill DL, Felsher DW. MYC through miR-17-92 suppresses specific target genes to maintain survival, autonomous proliferation, and a neoplastic state. Cancer Cell 2014; 26:262-272.
17. Ma MZ, Li CX, Zhang Y, Weng MZ, Zhang MD, Qin YY, Gong W, Quan ZW. Long non-coding RNA HOTAIR, a c-Myc activated driver of malignancy, negatively regulates miRNA-130a in gallbladder cancer. Mol Cancer 2014; 13:156.
18. Zheng GX, Do BT, Webster DE, Khavari PA, Chang HY. Dicer-microRNA-Myc circuit promotes transcription of hundreds of long noncoding RNAs. Nat Struct Mol Biol 2014; 21:585-590.
19. Hung CL, Wang LY, Yu YL, Chen HW, Srivastava S, Petrovics G, Kung HJ. A long noncoding RNA connects c-Myc to tumor metabolism. Proceedings of the National Academy of Sciences of the United States of America 2014.
20. Clark MB, Amaral PP, Schlesinger FJ, Dinger ME, Taft RJ, Rinn JL, Ponting CP, Stadler PF, Morris KV, Morillon A, Rozowsky JS, Gerstein MB, Wahlestedt C, et al. The reality of pervasive transcription. PLoS Biol 2011; 9:e1000625; discussion e1001102.
21. Mattick JS, Makunin IV. Non-coding RNA. Hum Mol Genet 2006; 15 Spec No 1:R17-29.
22. Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, Maeda N, Oyama R, Ravasi T, Lenhard B, Wells C, Kodzius R, Shimokawa K, Bajic VB, et al. The transcriptional landscape of the mammalian genome. Science 2005; 309:1559-1563.
23. Encode Project Consortium, Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, Thurman RE, Kuehn MS, Taylor CM, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 2007; 447:799-816.
24. Katayama S, Tomaru Y, Kasukawa T, Waki K, Nakanishi M, Nakamura M, Nishida H, Yap CC, Suzuki M, Kawai J, Suzuki H, Carninci P, Hayashizaki Y, et al. Antisense transcription in the mammalian transcriptome. Science 2005; 309:1564-1566.
25. Engstrom PG, Suzuki H, Ninomiya N, Akalin A, Sessa L, Lavorgna G, Brozzi A, Luzi L, Tan SL, Yang L, Kunarso G, Ng EL, Batalov S, et al. Complex Loci in human and mouse genomes. PLoS Genet 2006; 2:e47.
26. Kretz M, Siprashvili Z, Chu C, Webster DE, Zehnder A, Qu K, Lee CS, Flockhart RJ, Groff AF, Chow J, Johnston D, Kim GE, Spitale RC, et al. Control of somatic tissue differentiation by the long non-coding RNA TINCR. Nature 2013; 493:231-235.
27. Faghihi MA, Zhang M, Huang J, Modarresi F, Van der Brug MP, Nalls MA, Cookson MR, St-Laurent G, 3rd, Wahlestedt C. Evidence for natural antisense transcript-mediated inhibition of microRNA function. Genome biology 2010; 11:R56.
28. Annilo T, Kepp K, Laan M. Natural antisense transcript of natriuretic peptide precursor A (NPPA): structural organization and modulation of NPPA expression. BMC Mol Biol 2009; 10:81.
29. Faghihi MA, Modarresi F, Khalil AM, Wood DE, Sahagan BG, Morgan TE, Finch CE, St Laurent G, 3rd, Kenny PJ, Wahlestedt C. Expression of a noncoding RNA is elevated in Alzheimer’s disease and drives rapid feed-forward regulation of beta-secretase. Nat Med 2008; 14:723-730.
30. Yu W, Gius D, Onyango P, Muldoon-Jacobs K, Karp J, Feinberg AP, Cui H. Epigenetic silencing of tumour suppressor gene p15 by its antisense RNA. Nature 2008; 451:202-206.
31. Roberts TC, Morris KV, Weinberg MS. Perspectives on the mechanism of transcriptional regulation by long non-coding RNAs. Epigenetics 2014; 9:13-20.
32. Modarresi F, Faghihi MA, Lopez-Toledano MA, Fatemi RP, Magistri M, Brothers SP, van der Brug MP, Wahlestedt C. Inhibition of natural antisense transcripts in vivo results in gene-specific transcriptional upregulation. Nat Biotechnol 2012; 30:453-459.
33. Rougeulle C, Heard E. Antisense RNA in imprinting: spreading silence through Air. Trends Genet 2002; 18:434-437.
34. Panning B, Jaenisch R. RNA and the epigenetic regulation of X chromosome inactivation. Cell 1998; 93:305-308.
35. Feng J, Bi C, Clark BS, Mady R, Shah P, Kohtz JD. The Evf-2 noncoding RNA is transcribed from the Dlx-5/6 ultraconserved region and functions as a Dlx-2 transcriptional coactivator. Genes & development 2006; 20:1470-1484.
36. Tripathi V, Ellis JD, Shen Z, Song DY, Pan Q, Watt AT, Freier SM, Bennett CF, Sharma A, Bubulya PA, Blencowe BJ, Prasanth SG, Prasanth KV. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Molecular cell 2010; 39:925-938.
37. Yan MD, Hong CC, Lai GM, Cheng AL, Lin YW, Chuang SE. Identification and characterization of a novel gene Saf transcribed from the opposite strand of Fas. Hum Mol Genet 2005; 14:1465-1474.
38. Yang L, Lin C, Liu W, Zhang J, Ohgi KA, Grinstein JD, Dorrestein PC, Rosenfeld MG. ncRNA- and Pc2 methylation-dependent gene relocation between nuclear structures mediates gene activation programs. Cell 2011; 147:773-788.
39. Clemson CM, Hutchinson JN, Sara SA, Ensminger AW, Fox AH, Chess A, Lawrence JB. An architectural role for a nuclear noncoding RNA: NEAT1 RNA is essential for the structure of paraspeckles. Molecular cell 2009; 33:717-726.
40. Sunwoo H, Dinger ME, Wilusz JE, Amaral PP, Mattick JS, Spector DL. MEN epsilon/beta nuclear-retained non-coding RNAs are up-regulated upon muscle differentiation and are essential components of paraspeckles. Genome Res 2009; 19:347-359.
41. Willingham AT, Orth AP, Batalov S, Peters EC, Wen BG, Aza-Blanc P, Hogenesch JB, Schultz PG. A strategy for probing the function of noncoding RNAs finds a repressor of NFAT. Science 2005; 309:1570-1573.
42. Yin QF, Yang L, Zhang Y, Xiang JF, Wu YW, Carmichael GG, Chen LL. Long noncoding RNAs with snoRNA ends. Molecular cell 2012; 48:219-230.
43. Augoff K, McCue B, Plow EF, Sossey-Alaoui K. miR-31 and its host gene lncRNA LOC554202 are regulated by promoter hypermethylation in triple-negative breast cancer. Mol Cancer 2012; 11:5.
44. Watanabe T, Takeda A, Tsukiyama T, Mise K, Okuno T, Sasaki H, Minami N, Imai H. Identification and characterization of two novel classes of small RNAs in the mouse germline: retrotransposon-derived siRNAs in oocytes and germline small RNAs in testes. Genes & development 2006; 20:1732-1743.
45. Kino T, Hurt DE, Ichijo T, Nader N, Chrousos GP. Noncoding RNA gas5 is a growth arrest- and starvation-associated repressor of the glucocorticoid receptor. Sci Signal 2010; 3:ra8.
46. Blackburn EH, Greider CW, Szostak JW. Telomeres and telomerase: the path from maize, Tetrahymena and yeast to human cancer and aging. Nat Med 2006; 12:1133-1138.
47. Yao H, Brick K, Evrard Y, Xiao T, Camerini-Otero RD, Felsenfeld G. Mediation of CTCF transcriptional insulation by DEAD-box RNA-binding protein p68 and steroid receptor RNA activator SRA. Genes & development 2010; 24:2543-2555.
48. Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nat Rev Genet 2009; 10:155-159.
49. Pajic A, Spitkovsky D, Christoph B, Kempkes B, Schuhmacher M, Staege MS, Brielmeier M, Ellwart J, Kohlhuber F, Bornkamm GW, Polack A, Eick D. Cell cycle activation by c-myc in a burkitt lymphoma model cell line. Int J Cancer 2000; 87:787-793.
50. Steijger T, Abril JF, Engstrom PG, Kokocinski F, Hubbard TJ, Guigo R, Harrow J, Bertone P, Consortium R. Assessment of transcript reconstruction methods for RNA-seq. Nature methods 2013; 10:1177-1184.
51. Engstrom PG, Steijger T, Sipos B, Grant GR, Kahles A, Ratsch G, Goldman N, Hubbard TJ, Harrow J, Guigo R, Bertone P, Consortium R. Systematic evaluation of spliced alignment programs for RNA-seq data. Nature methods 2013; 10:1185-1191.
52. Anders S, Pyl PT, Huber W. HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics 2014.
53. Dib A, Gabrea A, Glebov OK, Bergsagel PL, Kuehl WM. Characterization of MYC translocations in multiple myeloma cell lines. Journal of the National Cancer Institute Monographs 2008:25-31.
54. Hirvonen HE, Salonen R, Sandberg MM, Vuorio E, Vastrik I, Kotilainen E, Kalimo H. Differential expression of myc, max and RB1 genes in human gliomas and glioma cell lines. British journal of cancer 1994; 69:16-25.
55. Uesaka M, Nishimura O, Go Y, Nakashima K, Agata K, Imamura T. Bidirectional promoters are the major source of gene activation-associated non-coding RNAs in mammals. BMC Genomics 2014; 15:35.
56. Kim M, Vasiljeva L, Rando OJ, Zhelkovsky A, Moore C, Buratowski S. Distinct pathways for snoRNA and mRNA termination. Molecular cell 2006; 24:723-734.
57. Yang L, Duff MO, Graveley BR, Carmichael GG, Chen LL. Genomewide characterization of non-polyadenylated RNAs. Genome biology 2011; 12:R16.
58. Oler AJ, Alla RK, Roberts DN, Wong A, Hollenhorst PC, Chandler KJ, Cassiday PA, Nelson CA, Hagedorn CH, Graves BJ, Cairns BR. Human RNA polymerase III transcriptomes and relationships to Pol II promoter chromatin and enhancer-binding factors. Nat Struct Mol Biol 2010; 17:620-628.
59. Russell J, Zomerdijk JC. The RNA polymerase I transcription machinery. Biochem Soc Symp 2006:203-216.
60. Preker P, Nielsen J, Kammler S, Lykke-Andersen S, Christensen MS, Mapendano CK, Schierup MH, Jensen TH. RNA exosome depletion reveals transcription upstream of active human promoters. Science 2008; 322:1851-1854.
61. Bensaude O. Inhibiting eukaryotic transcription: Which compound to choose? How to evaluate its activity? Transcription 2011; 2:103-108.
62. Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, Xue C, Marinov GK, Khatun J, et al. Landscape of transcription in human cells. Nature 2012; 489:101-108.
63. Aguda BD, Kim Y, Piper-Hunter MG, Friedman A, Marsh CB. MicroRNA regulation of a cancer network: consequences of the feedback loops involving miR-17-92, E2F, and Myc. Proceedings of the National Academy of Sciences of the United States of America 2008; 105:19678-19683.
64. Dews M, Homayouni A, Yu D, Murphy D, Sevignani C, Wentzel E, Furth EE, Lee WM, Enders GH, Mendell JT, Thomas-Tikhonenko A. Augmentation of tumor angiogenesis by a Myc-activated microRNA cluster. Nature genetics 2006; 38:1060-1065.
65. Venturini L, Battmer K, Castoldi M, Schultheis B, Hochhaus A, Muckenthaler MU, Ganser A, Eder M, Scherr M. Expression of the miR-17-92 polycistron in chronic myeloid leukemia (CML) CD34+ cells. Blood 2007; 109:4399-4405.
66. Jin HY, Oda H, Lai M, Skalsky RL, Bethel K, Shepherd J, Kang SG, Liu WH, Sabouri-Ghomi M, Cullen BR, Rajewsky K, Xiao C. MicroRNA-17~92 plays a causative role in lymphomagenesis by coordinating multiple oncogenic pathways. EMBO J 2013; 32:2377-2391.
67. Sacco JJ, Coulson JM, Clague MJ, Urbe S. Emerging roles of deubiquitinases in cancer-associated pathways. IUBMB life 2010; 62:140-157.
68. Zhu L, Xu PC. Downregulated LncRNA-ANCR promotes osteoblast differentiation by targeting EZH2 and regulating Runx2 expression. Biochemical and biophysical research communications 2013; 432:612-617.
69. Kretz M, Webster DE, Flockhart RJ, Lee CS, Zehnder A, Lopez-Pajares V, Qu K, Zheng GX, Chow J, Kim GE, Rinn JL, Chang HY, Siprashvili Z, et al. Suppression of progenitor differentiation requires the long noncoding RNA ANCR. Genes & development 2012; 26:338-343.
70. Hart JR, Garner AL, Yu J, Ito Y, Sun M, Ueno L, Rhee JK, Baksh MM, Stefan E, Hartl M, Bister K, Vogt PK, Janda KD. Inhibitor of MYC identified in a Krohnke pyridine library. Proceedings of the National Academy of Sciences of the United States of America 2014; 111:12556-12561.
71. Wiita AP, Ziv E, Wiita PJ, Urisman A, Julien O, Burlingame AL, Weissman JS, Wells JA. Global cellular response to chemotherapy-induced apoptosis. eLife 2013; 2:e01236.
72. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013; 29:15-21.
73. Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010; 26:841-842.
74. Kent WJ, Zweig AS, Barber G, Hinrichs AS, Karolchik D. BigWig and BigBed: enabling browsing of large distributed datasets. Bioinformatics 2010; 26:2204-2207.
75. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009; 25:1754-1760.
76. Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010; 26:589-595.
77. Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS. Model-based analysis of ChIP-Seq (MACS). Genome biology 2008; 9:R137.
78. Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Molecular cell 2010; 38:576-589.
79. Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome biology 2002; 3:RESEARCH0034.