
INTRODUCTION
Cytogenetically normal acute myeloid leukemia (CN-AML) constituting 40–50% of all AML patients is the largest cytogenetic group [1, 2]. It is characterized with the rapid accumulation of abnormal white blood cell in the bone marrow and the interface with normal blood cells’ production without any chromosomal aberrations. CN-AML patients are usually categorized to be intermediate risk, yet their clinical and molecular characteristics are sharply heterogeneous. Finding proper markers has been an active area in order to obtain a more refined stratification and deeper understanding for CN-AML. Mutations of NPM1 [3] and CEBPA [4] have been used as favorably prognostic biomarkers, while FLT3-ITD [5] and MLL [6] mutations have been associated with worse prognosis in the European Leukemia Net (ELN) reporting system [7]. Also, a recent report combined mutations of DNMT3A, FLT3 and NPM1 to predict clinical features for CN-AML [8]. Besides, high expression of several genes/microRNAs have been associated with adverse prognosis, including BAALC [9], ERG [9], WT1 [10], MN1 [11], DNMT3B [12], TCF4 [13], miR-155 [14] and miR-3151 [15], while high expression of LEF1 [16] has been regarded as favorable prognostic factors. Because the mechanisms of leukemogenesis are still unknown, finding new prognostic biomarkers is critical for obtaining refined risk-stratification and designing novel therapeutic strategies of CN-AML.
Activity of calcium ion transmembrane transportation is a critical biological process for maintenance and regulation of cell cycle, and plays an important role in cell proliferation, differentiation and senescence [17]. Inositol 1,4,5-trisphosphate receptor, type 2 (ITPR2) is a an essential regulator for mediating the mobilization of intracellular Ca2+ stores, and acts as an pivotal role in intracellular Ca2+ signaling in a variety of cell types. Early report showed that ITPR2 participated in Ca2+-calpain and Caspase-mitochondria dependent pathways and regulated the apoptosis of U937 cell [18], also ITPR2 might act as target of CEBPB and all-trans retinoic acid (ATRA) in NB4 cells [19]. Recent reports identified ITPR2 variations as novel susceptibility loci for renal cell carcinoma via a genome-wide association study [20]. Another report identified ITPR2 as a susceptiable gene for Kashin-Beck disease in Han Chinese [21]. However, the prognostic impact of ITPR2 expression has not been reported in CN-AML.
This manuscript provided consolidated evidence for the first time that, high ITPR2 expression (ITPR2high) was associated with worse prognosis in CN-AML. Firstly, ITPR2high was shown in CN-AML patients compared to normal bone marrow (NBM) measured with microarrays. These microarray data was confirmed by qPCR [13, 16]. Secondly, the prognostic value of ITPR2 was determined with 2 independent, relative large CN-AML cohorts, with respect to clinical, molecular characters and analysis of OS and EFS. Multivariable analysis further confirmed ITPR2high as a worse prognostic marker. Finally, underlying mechanisms of why ITPR2 functioned as a worse prognosticator was investigated using microarray or high-throughput sequencing data of gene/microRNA expression, genome-wide DNA methylation, combining with the known annotation and pathway information. These results may potentially facilitate our understanding of leukemogenesis and provide new criterions for risk-stratification in CN-AML, which will be finally exploited and lead to new treatment strategies.
RESULTS
ITPR2 expression discriminates between CN-AML and normal bone marrow
Microarray analysis was used to analyze expression alteration of ITPR2 in CN-AML (n = 116) and normal bone marrow (NBM) (n = 5). ITPR2 was positive in these two populations. However, there existed a significant difference, ITPR2 showed a remarkably higher expression in CN-AML than NBM (P = 0.014, Figure 1A and 1B). Higher expression in CN-AML made the detection of ITPR2 more easily, and discriminated patients from normal population, these two characteristics were important for clinical applications.
Association of ITPR2 expression with clinical and molecular characteristics
In the 157 de novo CN-AML cohort, we found that more ITPR2high patients fell into M1 (P < 0.001) and M2 (P = 0.018), less fell into M4 (P = 0.044) and M5 (P < 0.001) in FAB subtype, compared to ITPR2low patients. FLT3-ITD mutation occurred more in ITPR2high (P < 0.001) than ITPR2low patients. In addition, ITPR2high patients seemed more likely to have high expression of ERG (P < 0.001), BAALC (P = 0.004), WT1 (P < 0.001) and DNMT3B (P < 0.001). Based on the European Leukemia Net (ELN) genetic categories, more Intermediate-I patients fell into ITPR2high group (P < 0.001) (Table 1).
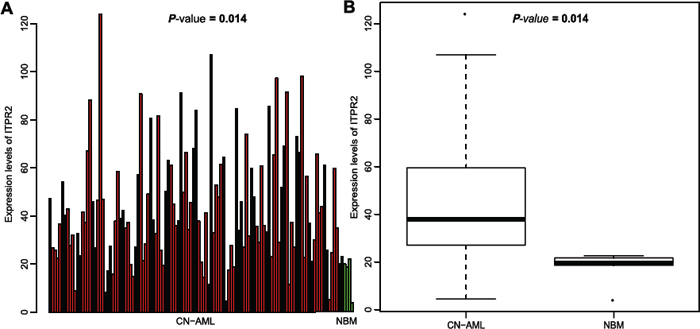
Figure 1: Differential expression between CN-AML and normal bone marrow. (A) Barplot and (B) Boxplot of ITPR2 expression in 116 CN-AML patients and 5 normal bone marrow samples.
Table 1: Patients’ characteristics in the primary cohort of 157 CN-AML patients according to ITPR2 expression levels
Variable |
ITPR2high, n = 78 |
ITPR2low, n = 79 |
P |
Median age. y (range) |
50.5 (18–77) |
49.0 (16–73) |
0.57 |
Female sex, no. (%) |
42 (53.8) |
31 (39.2) |
0.079 |
FAB subtype, no. |
|||
M0 |
1 |
2 |
1 |
M1 |
32 |
13 |
P < 0.001 |
M2 |
22 |
10 |
0.018 |
M3 |
1 |
0 |
0.50 |
M4 |
7 |
17 |
0.044 |
M5 |
10 |
29 |
P < 0.001 |
M6 |
0 |
1 |
1 |
Other |
5 |
8 |
0.56 |
FLT3-ITD, no. |
46 |
20 |
P < 0.001 |
FLT3-TKD, no. |
7 |
13 |
0.23 |
NPM1, mutated no. |
41 |
41 |
1 |
N-RAS, mutated no. |
3 |
10 |
0.079 |
K-RAS, mutated no. |
0 |
1 |
1 |
IDH1, mutated no. |
9 |
10 |
1 |
IDH2, mutated no. |
7 |
6 |
1 |
ELN genetic group, no |
|||
Favorable |
25 |
34 |
0.19 |
Intermediate-I |
72 |
50 |
P < 0.001 |
High ERG, no. |
54 |
24 |
P < 0.001 |
High BAALC, no. |
48 |
30 |
0.004 |
High LEF1, no. |
40 |
38 |
0.75 |
High MN1, no. |
43 |
35 |
0.20 |
High WT1, no. |
51 |
27 |
P < 0.001 |
High DNMT3B, no |
55 |
23 |
P < 0.001 |
High TCF4, no |
44 |
34 |
0.11 |
FAB, French-American-British classification; ITD, internal tandem duplication; TKD, tyrosine kinase domain; ELN, European Leukemia Net.
High ERG, BAALC, LEF1, MN1, WT1, DNMT3B and TCF4 expression were defined as an expression level above the median of all samples, respectively.
High expression of ITPR2 associated with worse prognostic outcomes
Regarding survival time as a continuous variable, ITPR2high patients had lower median overall survival (OS: P = 0.0016) and event-free survival time (EFS: P = 0.002) (Table 2). When all 157 patients were dichotomized according to the ITPR2 expression levels, we found that ITPR2high group showed a significantly shorter OS (P = 0.0039, Figure 2A) and EFS (P = 0.01, Figure 2B) than ITPR2low patient group.
Prognostic value of ITPR2 in ELN genetic subgroups
European Leukemia Net (ELN) divided CN-AML patients into the ELN favorable or ELN intermediate-I genetic categories, according to the mutation of CEBPA, NPM1 and FLT3-ITD [7]. In our analysis to the primary cohort of 157 CN-AML patients, expression of ITPR2 differed dramatically between different ELN genetic groups. More patients of the ELN Intermediate-I genetic category belonged to ITPR2high group (P < 0.001, Table 1), while a trend for more patients of the ELN Favorable category belonged to ITPR2low patient group (34 VS 25, P = 0.19, Table 1). Thus, we further investigated the impact of ITPR2 expression within the two ELN genetic categories separately. ITPR2 expression showed no association with OS (P > 0.9, Figure 3A) or EFS (P > 0.9, Figure 3B) in the ELN favorable category. However, in ELN intermediate-I genetic category, ITPR2high patients showed significantly shorter OS (P = 0.0066, Figure 3C) and EFS (P = 0.009, Figure 3D) than ITPR2low patients.
Table 2: Survival according to ITPR2 expression in the primary cohort of 157 CN-AML patients
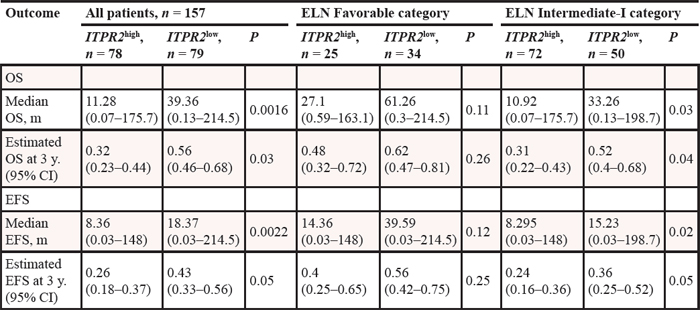
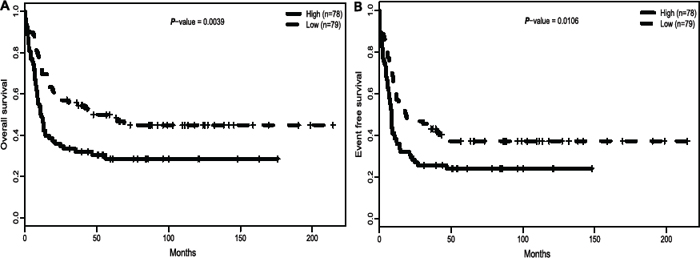
Figure 2: High expression of ITPR2 is associated with worse outcomes. (A) OS and (B) EFS in the primary cohort of 157 CN-AML patients.
Multivariable analysis of ITPR2 expression associated with OS and EFS
To further determine the prognostic value of ITPR2 expression, multivariable analysis was preformed after adjusting for the impact of other known risk factors, including commonly known mutations and several recently published prognostic factors such as ERG [22], BAALC [9], LEF1 [16] and WT1 [10]. In the multiple model for OS, ITPR2high patients had 2.44 times increase of risk to death, other factors associated with longer OS included mutations of NPM1, CEBPA, and high expression of LEF1. In the multiple model of EFS, high expression of ITPR2 remained a dramatically worse prognosticator (P = 0.012) after the adjustment of other risk factors, while mutation of NPM1, CEBPA and high expression of LEF1 were still significantly associated with longer EFS (Table 3).
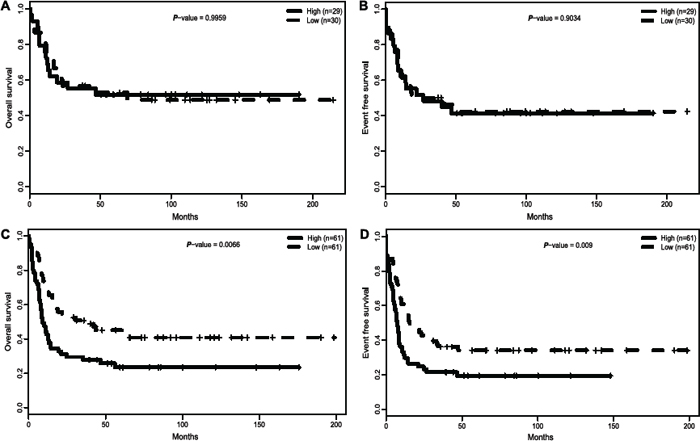
Figure 3: Survival of 157 CN-AML patients according to ELN genetic categories and ITPR2 expression. (A) OS and (B) EFS in the ELN favorable genetic category. (C) OS and (D) EFS in the ELN intermediate-I genetic category.
Validation in an independent cohort of 162 CN-AML patients
An independent cohort of 162 de novo CN-AML patients was exploited to validate our findings. The third quartile (Q3) of ITPR2 expression was used as the cutoff. Patients with FAB M1 were more likely to have a higher expression of ITPR2 (P < 0.001), while FAB M5 seemed to be significantly associated with lower ITPR2 expression levels (P = 0.0015). Further, patients with higher expression of ITPR2 were more likely to have shorter OS (P = 0.011), and have higher expression of ERG, WT1, DNMT3B and TCF4 (All with P < 0.001) and lower expression of LEF1 (P = 0.004). In addition, mean OS showed significant difference between ITPR2high and ITPR2low groups (P = 0.011), and ITPR2high patients seemed to have a shorter OS (P = 0.007, Figure S1.) Noticeably, mean age of ITPR2low patients was older than that of ITPR2high patients (P = 0.034), which consolidated the prognostic value of ITPR2 expression to some extent. (All statistics were listed in Table S1.)
Genome-wide gene-expression profiles associated with high expression of ITPR2
To further investigate the biological role of ITPR2, we performed a genome-wide differential analysis based on the sample division of median ITPR2 expression. 768 up-regulated and 1136 down-regulated genes were identified dramatically associated with high ITPR2 expression (False Discovery Rate, FDR < 0.01; Fold Change, FC >= 1.5, data not shown). Among the 768 up-regulated genes, excitedly, we found WT1, ERG and DNMT3B, high expression of which were all clearly reported as worse prognostic biomarkers in CN-AML [9, 10, 12]. Other up-regulated genes included a cluster of genes that control cell cycle and differentiation (CDK6, CDKN1C, CCND2 et.al), several genes that function as tyrosine kinase (MAP4K3, PTK7 and c-Kit), and genes that previously known to be related with leukemogenesis (MLLT11, MPL, MYCN, MSI2). Noteworthy down-regulated genes included members of LILR family (LILRB1, LILRB3, LILRB4, LILRA1, LILRA2, LILRA6), members of TLR family (TLR2, TLR4, TLR5, TLR7) and the immune molecule CD86, which indicated the possible mechanisms of immune evasion that led to the worse outcome for CN-AML patients with high ITPR2 expression. (See Figure 4A and 4B.)
In addition, cell signaling pathways that were associated with ITPR2 expression alteration were identified. Based on the pathway data provided by MSigDB [23], expressions of genes that participated in a pathway were averaged to represent expression of the pathway. We found that pathways of apoptosis, antigen processing and natural killer cells mediated cytotoxicity were all significantly down-regulated (all P < 0.001), associated with high expression of ITPR2. This result, just being consistent to previously presented dysregulated genes, possibly illustrated why high expression of ITPR2 was associated with adverse outcome in CN-AML. (See Table 4 for the statistics of those pathways.)
Table 3: Multivariable analysis with OS and EFS in the primary cohort of 157 CN-AML patients
Variable |
OS, n = 157 |
EFS, n = 157 |
||
HR(95% CI) |
P |
HR(95% CI) |
P |
|
ITPR2 expression, high VS low |
2.44 (1.39–4.28) |
0.0019 |
1.96 (1.16–3.31) |
0.012 |
Age, per 10-y increase |
1.11 (0.97–1.28) |
0.14 |
1.05 (0.92–1.20) |
0.49 |
Sex male VS female |
0.80 (0.52–1.25) |
0.33 |
0.96 (0.63–1.45) |
0.83 |
NPM1, mutated VS wild type |
0.50 (0.29–0.86) |
0.012 |
0.48 (0.29–0.80) |
0.0047 |
CEBPA, mutated VS wild type |
0.31 (0.14–0.70) |
0.0048 |
0.45 (0.21–0.96) |
0.039 |
FLT3-ITD, mutated VS others |
1.54 (0.92–2.57) |
0.099 |
1.51 (0.92–2.48) |
0.11 |
IDH1, mutated VS wild type |
0.81 (0.40–1.64) |
0.55 |
1.14 (0.60–2.14) |
0.69 |
IDH2, mutated VS wild type |
0.64 (0.28–1.46) |
0.29 |
0.75 (0.32–1.74) |
0.50 |
EVI1, mutated VS wild type |
3.03 (0.38–24.44) |
0.30 |
2.23 (0.28–17.70) |
0.45 |
ERG expression, high VS low |
1.21 (0.73–2.03) |
0.46 |
1.21 (0.74–1.99) |
0.44 |
BAALC expression, high VS low |
1.14 (0.67–1.92) |
0.63 |
0.99 (0.60–1.62) |
0.96 |
LEF1 expression, high VS low |
0.53 (0.33–0.85) |
0.0086 |
0.56 (0.36–0.87) |
0.0098 |
WT1 expression, high VS low |
0.67 (0.39–1.17) |
0.16 |
0.80 (0.48–1.35) |
0.41 |
HR, hazards ratio; CI, confidence interval.
Genome-wide microRNA profiles associated with ITPR2 expression
To gain further insights into biological differences associated with varying ITPR2 expression levels, genome-wide analysis of microRNA expression profiles were exploited using high throughput sequencing of 79 CN-AML patients from The Cancer Genome Atlas (TCGA) [24]. Sixty microRNAs were significantly associated with ITPR2 expression (P < 0.01, data not shown). Of these microRNAs, miR-155 and two members of miR-181 family(miR-181-a-1 and miR-181c) were positively associated with ITPR2 expression, the former of which has been validated to predict worse outcome for CN-AML patients [14], and the latter were recently shown to be potential targets and associated with adverse outcomes for AML patients [25]. MiR-193a was the most significant microRNA negatively associated with ITPR2 expression, which was reported to target c-Kit and up-regulation of miR-193a predicted favorable outcome in our group recently [26, 27]. (See Figure 4C and 4D.)
Genome-wide methylation profiles associated with ITPR2 expression
Because ITPR2 expression was positively correlated with DNMT3B expression, differential analysis for methylation was performed to find different DNA methylation patterns at genome-wide scale and within main cell signaling pathways. But no clear difference was found significantly with respect to ITPR2 expression status at the whole genome-wide level and the 186 known pathways in MSigDB [23]. (Figure S2, pathway results were not shown.)
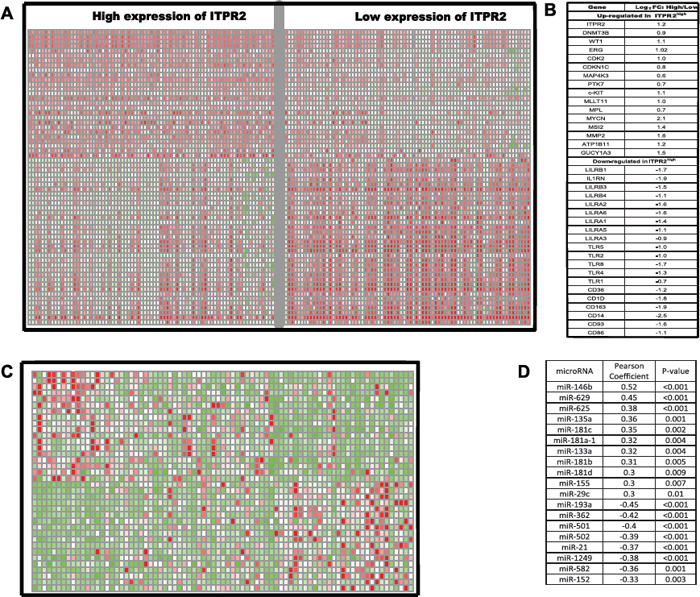
Figure 4: Genes/microRNAs associated with ITPR2 expression. (A) expression heatmap and (B) the list of associated genes. (C) expression heatmap and (D) the list of associated microRNAs.
Table 4: Cell signaling pathways associated with ITPR2 expression levels
Pathway name |
According to ITPR2high |
|
Regulation |
P-value |
|
KEGG_CHEMOKINE_SIGNALING_PATHWAY |
Down |
0.0001 |
KEGG_APOPTOSIS |
Down |
0.00018 |
KEGG_ANTIGEN_PROCESSING_AND_PRESENTATION |
Down |
< 0.0001 |
KEGG_NATURAL_KILLER_CELL_MEDIATED_CYTOTOXICITY |
Down |
< 0.0001 |
KEGG_FC_GAMMA_R_MEDIATED_PHAGOCYTOSIS |
Down |
< 0.0001 |
DISCUSSION
For the first time, we evaluated the prognostic value of ITPR2 expression, high expression of which was associated with shorter OS and EFS in two independent, large cohorts of de novo CN-AML patients. In our study, patients with ITPR2high are significantly more classified in the M1 or M2 FAB subgroups than with ITPR2low, suggesting that the leukemic cells of the ITPR2high patients derive from relatively more immature cells. In addition, high ITPR2 expression was associated with the presence of FLT3-ITD and high expression of ERG, BAALC and WT1, which were all associated with worse outcomes. Besides, more patients of high ITPR2 expression belonged to ELN intermediate-I group (P < 0.001) and less to ELN-favorable group, which also showed high ITPR2 expression as a worse biomarker. This will improve the ELN intermediate-I group risk classification and suggest that these patients may be turned candidates for alternative therapies. Furthermore, the association of high ITPR2 expression with shorter OS and EFS was confirmed in log-rank test and multivariable analyses adjusting for almost all known molecular prognosticators in CN-AML. Considering the fact that these two cohorts of CN-AML patients received uniformed therapeutic treatments separately, these results validated ITPR2 as an independent prognostic factor. Moreover, the fact that ITPR2 showed higher expression in CN-AML than normal bone marrow indicated its power of discrimination and easy access, which were important characteristics for clinical application.
The mechanisms why high ITPR2 expression is associated with adverse treatment response are unknown. However, our exploration in genome-wide gene/microRNA expression analysis provided possible interpretations. We found that expression of WT1, ERG and DNMT3B were significantly associated with ITPR2, and several genes that were active in cell cycle and tyrosine kinase process were all up-regulated, while genes that function as immune factors are down-regulated. In addition, several important cell signaling pathways showed aberrant expression associated with high expression of ITPR2, including the significantly down-regulation of apoptosis, natural killer cell mediated cytotoxicity and antigen processing and presentation. These changes may lead to out of control for cell death and immune escape, which might contribute to worse outcomes.
The microRNA profiles associated with ITPR2 expression also made sense for the worse outcome. MiR-155 was found to be positively, while miR-193a to be the most negatively correlated with ITPR2 expression, because miR-155 was clearly confirmed worse prognosticator in CN-AML and miR-193a was validated as a favorable biomarker by our group.
Epigenetic regulation is an important mechanism that can alter the genomic expression, however we observed no significant association between ITPR2 expression and DNA methylation changes via our analysis of HELP array data. So, these CN-AML patients might show no sensitive to drugs eliminating methylation such as decitabine.
In conclusion, we show that high expression of ITPR2 is associated with shorter OS and EFS in CN-AML patients. ITPR2 shows higher expression in CN-AML than healthy persons, which indicates its easy access by qPCR and potential using in clinical application. However, future studies are needed to establish a standardized protocol of quantification, before it can be used for risk-stratification of CN-AML patients. Furthermore, the derived genome-wide analysis of gene/microRNA expression and DNA methylation shed light on the underlying biologic mechanisms of leukemogenesis, and might help to develop new therapeutic strategies for CN-AML disease.
METHODS
Patients and treatment
A cohort of 157 patients with previously untreated CN-AML (median age, 50 years, range: 16–77 years), who were collected at Erasmus University Medical Center (Rotterdam) between 1990 and 2008 [28], One hundred thirty patients (83%) were aged < 60 years (younger patients) and 27 patients (17%) were ≥ 60 years (older patients). The patients had been treated on study protocols of Dutch-Belgian Hematology-Oncology Cooperative Group (HOVON, http://www.hovon.nl). The detailed therapeutic protocol was shown in Figure S3. All samples were collected at diagnosis, with bone marrow aspirates or peripheral-blood, containing 80–100 percent blast cells after thawing [29]. Conventional cytogenetic examination of more than 20 metaphases from BM was used to determine the diagnosis of a normal karyotype. Patients were assessed for NPM1, CEBPA, IDH1, and IDH2 mutations, FLT3-internaltandem duplications (FLT3-ITD), N-RAS, K-RAS, and FLT3-tyrosine kinase domain mutations (FLT3-TKD [D835]). This research were approved by the institutional review boards at Weill Cornell Medical College and Erasmus University Medical Center, and all subjects provided written informed consent in accordance with the Declaration of Helsinki [30]. To validate our results, another independent cohort of 162 CN-AML patients was exploited, which was provided by the multicenter AMLCG-1999 trial of the German AML Cooperative Group between 1999 and 2003 [31]. These patients received intensive double induction and consolidation chemotherapy [31]. The AMLCG-1999 clinical trials were approved by the local institutional review boards, and informed consent from all patients obtained in accordance with the Declaration of Helsinki.
Microarray for gene expression and methylation, RNA/microRNA sequencing data
For the primary cohort of 157 CN-AML patients, pretreatment samples were studied using Affymetrix HG-U133Plus 2.0 expression GeneChips [28] and HELP methylation arrays [30], while the validating 162 patients were with Affymetrix HG-U133A expression GeneChips [31]. Experimental designs, quality control and normalization of data were carried out according to the standard Affymetrix protocols. Microarray data are available at the (GEO: accession no. GSE1159, GSE6891 and GSE12417 for expression, GSE18700 for methylation) including clinical, cytogenetic and molecular characteristics [32]. To further identify microRNAs correlatively expressed with ITPR2, RNA-Sequencing and microRNA-Sequencing data from The Cancer Genome Atlas (TCGA) were exploited [24], which provided 79 CN-AML patients. Pretreatment and clinical characteristics can be publicly downloaded from TCGA data portal (https://tcga-data.nci.nih.gov/tcga).
Statistical analyses
This study tried to evaluate the prognostic value of ITPR2 expression in CN-AML, and further explored the underlying mechanisms why it functioned, based on the gene/microRNA expression and methylation data. Samples were divided into two groups, high ITPR2 expression (ITPR2high, n = 78) and low ITPR2 expression (ITPR2low, n = 79), based on the median expression value of ITPR2. Also, high and low classifications of ERG, BAALC, WT1, LEF1, MN1, EVI1, DNMT3B and TCF4 were determined according to the median expression of corresponding genes. Pretreatment clinical and molecular characteristics were compared between ITPR2high and ITPR2low patients groups using the Fisher exact test for categories variables and the Wilcoxon rank-sum test for continuous variables. Association between ITPR2 expression and clinical outcomes was analyzed using Kaplan-Meier method, and difference was estimated with log-rank test. Multivariable Cox proportional hazards models were used to study the time-to-event factors associated with survival endpoints.
Differential analysis was conducted with Student’s t-test with multiple hypothesis correction (False Discovery Rate, FDR), to identify genes and pathways whose expression or methylation levels were associated with ITPR2 expression. Pearson correlation test was performed to determine the correlated expression between ITPR2 and microRNA sequencing profiles. All analysis was performed on the platform of R 3.1.1 software package.
ACKNOWLEDGMENTS
The work was supported by grants from the National Natural Science Foundation of China (61372047) and the National High-tech R&D Program of China (2013BAI03B04).
COMPETING INTERESTS
The authors declare that they have no competing interests.
Authors’ contributions
J.L. Shi and L. Fu designed, performed the study and wrote the manuscript. W.D. Wang designed and coordinated the study over the entire period. All authors approved the final manuscript.
REFERENCES
1. Schlenk RF, Dohner K, Krauter J, Frohling S, Corbacioglu A, Bullinger L, Habdank M, Spath D, Morgan M, Benner A, Schlegelberger B, Heil G, Ganser A, et al. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. The New England journal of medicine. 2008; 358:1909–1918.
2. Mrozek K, Heerema NA, Bloomfield CD. Cytogenetics in acute leukemia. Blood reviews. 2004; 18:115–136.
3. Becker H, Marcucci G, Maharry K, Radmacher MD, Mrozek K, Margeson D, Whitman SP, Wu YZ, Schwind S, Paschka P, Powell BL, Carter TH, Kolitz JE, et al. Favorable prognostic impact of NPM1 mutations in older patients with cytogenetically normal de novo acute myeloid leukemia and associated gene- and microRNA-expression signatures: a Cancer and Leukemia Group B study. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2010; 28:596–604.
4. Preudhomme C, Sagot C, Boissel N, Cayuela JM, Tigaud I, de Botton S, Thomas X, Raffoux E, Lamandin C, Castaigne S, Fenaux P, Dombret H, Group A. Favorable prognostic significance of CEBPA mutations in patients with de novo acute myeloid leukemia: a study from the Acute Leukemia French Association (ALFA). Blood. 2002; 100:2717–2723.
5. Sallmyr A, Fan J, Datta K, Kim KT, Grosu D, Shapiro P, Small D, Rassool F. Internal tandem duplication of FLT3 (FLT3/ITD) induces increased ROS production, DNA damage, and misrepair: implications for poor prognosis in AML. Blood. 2008; 111:3173–3182.
6. Dohner K, Tobis K, Ulrich R, Frohling S, Benner A, Schlenk RF, Dohner H. Prognostic significance of partial tandem duplications of the MLL gene in adult patients 16 to 60 years old with acute myeloid leukemia and normal cytogenetics: a study of the Acute Myeloid Leukemia Study Group Ulm. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2002; 20:3254–3261.
7. Dohner H, Estey EH, Amadori S, Appelbaum FR, Buchner T, Burnett AK, Dombret H, Fenaux P, Grimwade D, Larson RA, Lo-Coco F, Naoe T, Niederwieser D, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010; 115:453–474.
8. Loghavi S, Zuo Z, Ravandi F, Kantarjian HM, Bueso-Ramos C, Zhang L, Singh RR, Patel KP, Medeiros L, Stingo F, Routbort M, Cortes J, Luthra R, et al. Clinical features of De Novo acute myeloid leukemia with concurrent DNMT3A, FLT3 and NPM1 mutations. Journal of hematology & oncology. 2014; 7:74.
9. Schwind S, Marcucci G, Maharry K, Radmacher MD, Mrozek K, Holland KB, Margeson D, Becker H, Whitman SP, Wu YZ, Metzeler KH, Powell BL, Kolitz JE, et al. BAALC and ERG expression levels are associated with outcome and distinct gene and microRNA expression profiles in older patients with de novo cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. Blood. 2010; 116:5660–5669.
10. Lyu X, Xin Y, Mi R, Ding J, Wang X, Hu J, Fan R, Wei X, Song Y, Zhao RY. Overexpression of Wilms tumor 1 gene as a negative prognostic indicator in acute myeloid leukemia. PloS one. 2014; 9:e92470.
11. Schwind S, Marcucci G, Kohlschmidt J, Radmacher MD, Mrozek K, Maharry K, Becker H, Metzeler KH, Whitman SP, Wu YZ, Powell BL, Baer MR, Kolitz JE, et al. Low expression of MN1 associates with better treatment response in older patients with de novo cytogenetically normal acute myeloid leukemia. Blood. 2011; 118:4188–4198.
12. C Niederwieser JK, Volinia S, Whitman SP, Metzeler KH, Eisfeld A-K, Maharry K, Yan P, Frankhouser D, Becker H, Schwind S, Carroll AJ, Nicolet D, Mendler JH, Curfman JP, Wu Y-Z, Baer MR, Powell BL, Kolitz JE, Moore JO, Carter TH, Bundschuh R, Larson RA, Stone RM, Mro’ zek K, Marcucci G. Prognostic and biologic significance of DNMT3B expression in older patients with cytogenetically normal primary acutemyeloid leukemia. Leukemia. accepted article preview September 10. 2014; accepted article preview September 10; doi: 10.1038/leu.2014.1267.
13. In ’t Hout FE, van der Reijden BA, Monteferrario D, Jansen JH, Huls G. High expression of transcription factor 4 (TCF4) is an independent adverse prognostic factor in acute myeloid leukemia that could guide treatment decisions. Haematologica. 2014; 99:e257–e259.
14. Marcucci G, Maharry KS, Metzeler KH, Volinia S, Wu YZ, Mrozek K, Nicolet D, Kohlschmidt J, Whitman SP, Mendler JH, Schwind S, Becker H, Eisfeld AK, et al. Clinical role of microRNAs in cytogenetically normal acute myeloid leukemia: miR-155 upregulation independently identifies high-risk patients. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2013; 31:2086–2093.
15. Eisfeld AK, Marcucci G, Maharry K, Schwind S, Radmacher MD, Nicolet D, Becker H, Mrozek K, Whitman SP, Metzeler KH, Mendler JH, Wu YZ, Liyanarachchi S, et al. miR-3151 interplays with its host gene BAALC and independently affects outcome of patients with cytogenetically normal acute myeloid leukemia. Blood. 2012; 120:249–258.
16. Metzeler KH, Heilmeier B, Edmaier KE, Rawat VP, Dufour A, Dohner K, Feuring-Buske M, Braess J, Spiekermann K, Buchner T, Sauerland MC, Dohner H, Hiddemann W, et al. High expression of lymphoid enhancer-binding factor-1 (LEF1) is a novel favorable prognostic factor in cytogenetically normal acute myeloid leukemia. Blood. 2012; 120:2118–2126.
17. Wiel C, Lallet-Daher H, Gitenay D, Gras B, Le Calve B, Augert A, Ferrand M, Prevarskaya N, Simonnet H, Vindrieux D, Bernard D. Endoplasmic reticulum calcium release through ITPR2 channels leads to mitochondrial calcium accumulation and senescence. Nature communications. 2014; 5:3792.
18. Li M, Kondo T, Zhao QL, Li FJ, Tanabe K, Arai Y, Zhou ZC, Kasuya M. Apoptosis induced by cadmium in human lymphoma U937 cells through Ca2+-calpain and caspase-mitochondria-dependent pathways. The Journal of biological chemistry. 2000; 275:39702–39709.
19. Yu L, Zhang YD, Zhou J, Yao DM, Li X. Identification of target genes of transcription factor CEBPB in acute promyelocytic leukemia cells induced by all-trans retinoic acid. Asian Pacific journal of tropical medicine. 2013; 6:473–480.
20. Wu X, Scelo G, Purdue MP, Rothman N, Johansson M, Ye Y, Wang Z, Zelenika D, Moore LE, Wood CG, Prokhortchouk E, Gaborieau V, Jacobs KB, et al. A genome-wide association study identifies a novel susceptibility locus for renal cell carcinoma on 12p11.23. Human molecular genetics. 2012; 21:456–462.
21. Zhang F, Wen Y, Guo X, Zhang Y, Wang X, Yang T, Shen H, Chen X, Tian Q, Deng HW. Genome-wide association study identifies ITPR2 as a susceptibility gene for Kashin-Beck disease in Han Chinese. Arthritis & rheumatology. 2014; 67:176–181.
22. Metzeler KH, Dufour A, Benthaus T, Hummel M, Sauerland MC, Heinecke A, Berdel WE, Buchner T, Wormann B, Mansmann U, Braess J, Spiekermann K, Hiddemann W, et al. ERG expression is an independent prognostic factor and allows refined risk stratification in cytogenetically normal acute myeloid leukemia: a comprehensive analysis of ERG, MN1, and BAALC transcript levels using oligonucleotide microarrays. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2009; 27:5031–5038.
23. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005; 102:15545–15550.
24. Cancer Genome Atlas Research N. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. The New England journal of medicine. 2013; 368:2059–2074.
25. Su R, Lin HS, Zhang XH, Yin XL, Ning HM, Liu B, Zhai PF, Gong JN, Shen C, Song L, Chen J, Wang F, Zhao HL, et al. MiR-181 family: regulators of myeloid differentiation and acute myeloid leukemia as well as potential therapeutic targets. Oncogene advance online publication, September 1, 2014; doi:10.1038/onc.2014.274.
26. Li Y, Gao L, Luo X, Wang L, Gao X, Wang W, Sun J, Dou L, Li J, Xu C, Wang L, Zhou M, Jiang M, et al. Epigenetic silencing of microRNA-193a contributes to leukemogenesis in t(8,21) acute myeloid leukemia by activating the PTEN/PI3K signal pathway. Blood. 2013; 121:499–509.
27. Gao XN, Lin J, Li YH, Gao L, Wang XR, Wang W, Kang HY, Yan GT, Wang LL, Yu L. MicroRNA-193a represses c-kit expression and functions as a methylation-silenced tumor suppressor in acute myeloid leukemia. Oncogene. 2011; 30:3416–3428.
28. Verhaak RG, Wouters BJ, Erpelinck CA, Abbas S, Beverloo HB, Lugthart S, Lowenberg B, Delwel R, Valk PJ. Prediction of molecular subtypes in acute myeloid leukemia based on gene expression profiling. Haematologica. 2009; 94:131–134.
29. Valk PJ, Verhaak RG, Beijen MA, Erpelinck CA, Barjesteh van Waalwijk van Doorn-Khosrovani S, Boer JM, Beverloo HB, Moorhouse MJ, van der Spek PJ, Lowenberg B, Delwel R. Prognostically useful gene-expression profiles in acute myeloid leukemia. The New England journal of medicine. 2004; 350:1617–1628.
30. Figueroa ME, Lugthart S, Li Y, Erpelinck-Verschueren C, Deng X, Christos PJ, Schifano E, Booth J, van Putten W, Skrabanek L, Campagne F, Mazumdar M, Greally JM, et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer cell. 2010; 17:13–27.
31. Metzeler KH, Hummel M, Bloomfield CD, Spiekermann K, Braess J, Sauerland MC, Heinecke A, Radmacher M, Marcucci G, Whitman SP, Maharry K, Paschka P, Larson RA, et al. An 86-probe-set gene-expression signature predicts survival in cytogenetically normal acute myeloid leukemia. Blood. 2008; 112:4193–4201.
32. Barrett T, Edgar R. Gene expression omnibus: microarray data storage, submission, retrieval, and analysis. Methods in enzymology. 2006; 411:352–369.